
EASY-FIA: A Readably Usable Standalone Tool for High-Resolution Mass Spectrometry Metabolomics Data Pre-Processing
 , and
, and
Abstract
:1. Introduction
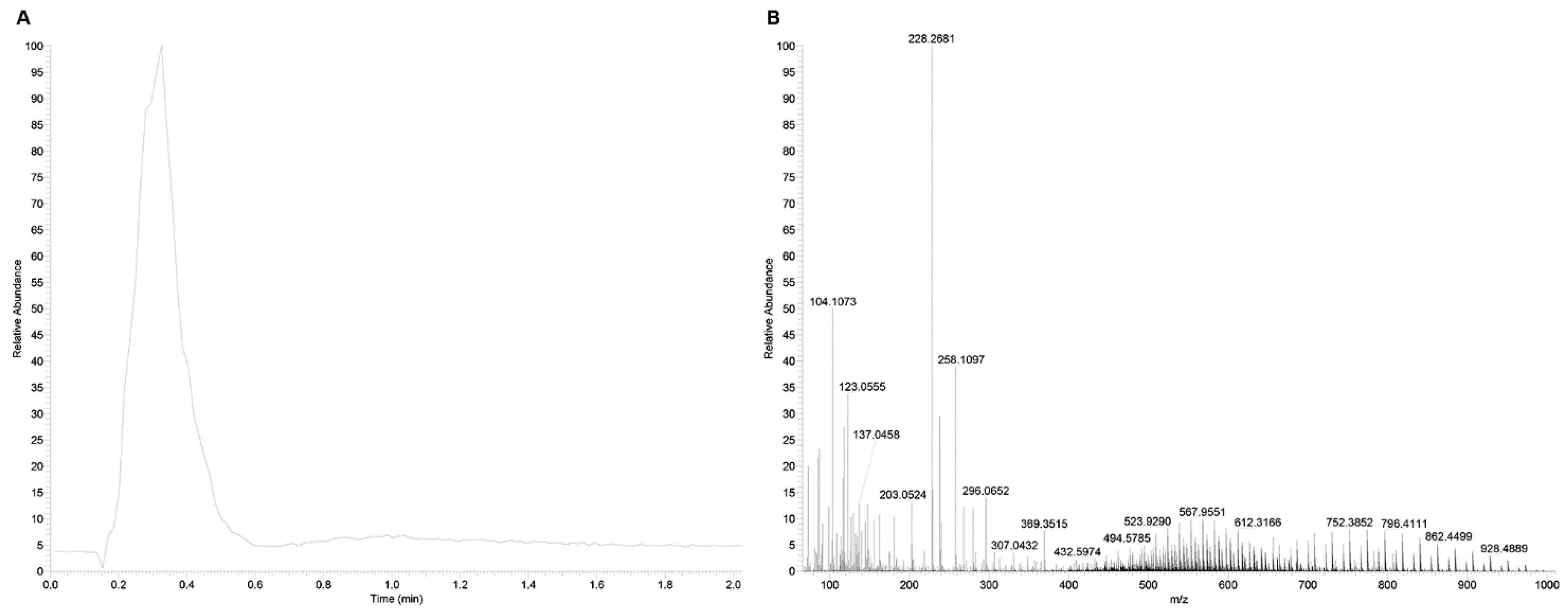
EASY-FIA Approach
2. Materials and Methods
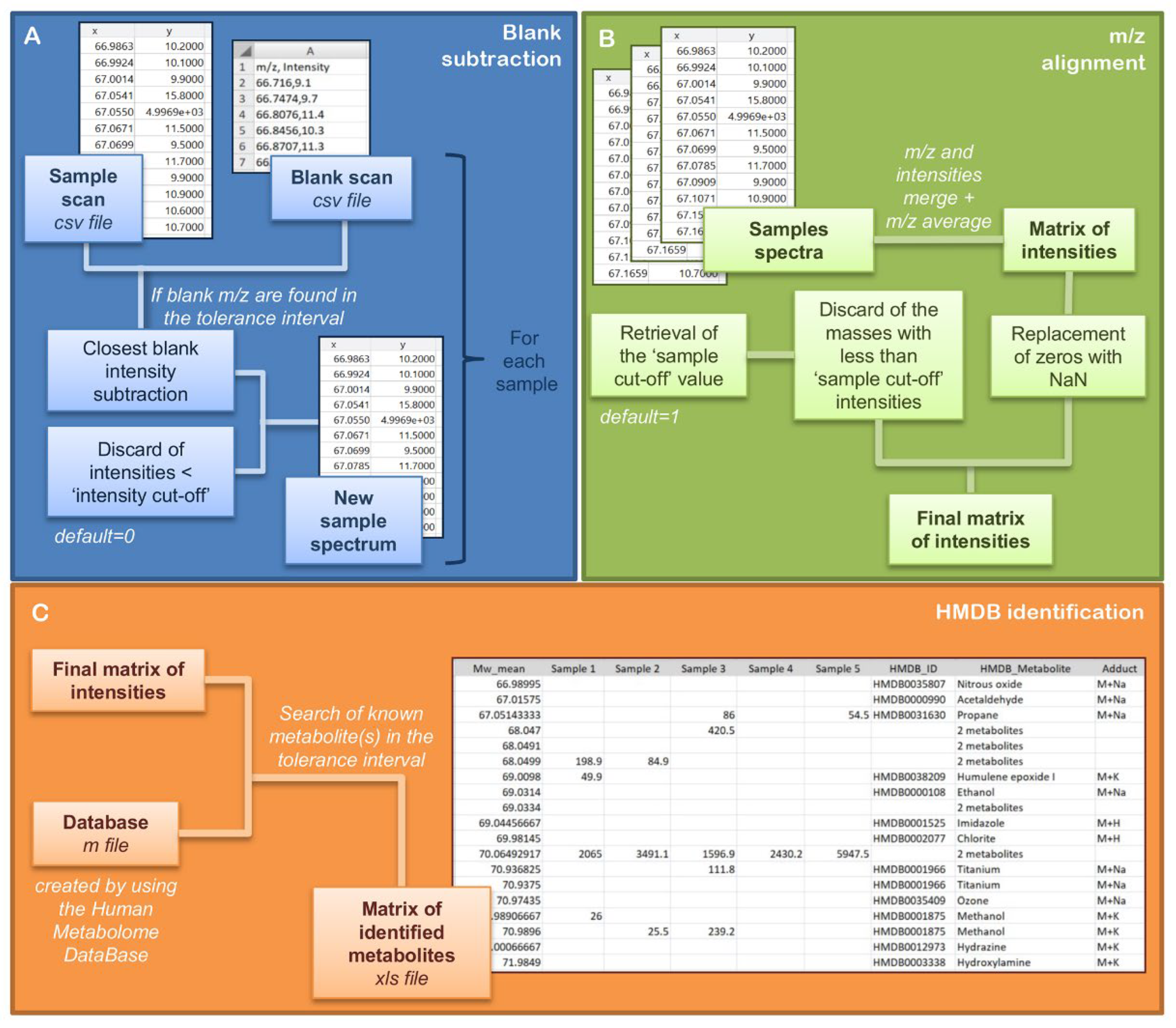
2.1. EASY-FIA Pre-Processing Workflow
2.1.1. Blank Subtraction
2.1.2. Alignment of m/z
2.1.3. Human Metabolome Database (HMDB) Annotation
2.2. Case Studies
2.3. Statistical Analysis
3. Results and Discussion
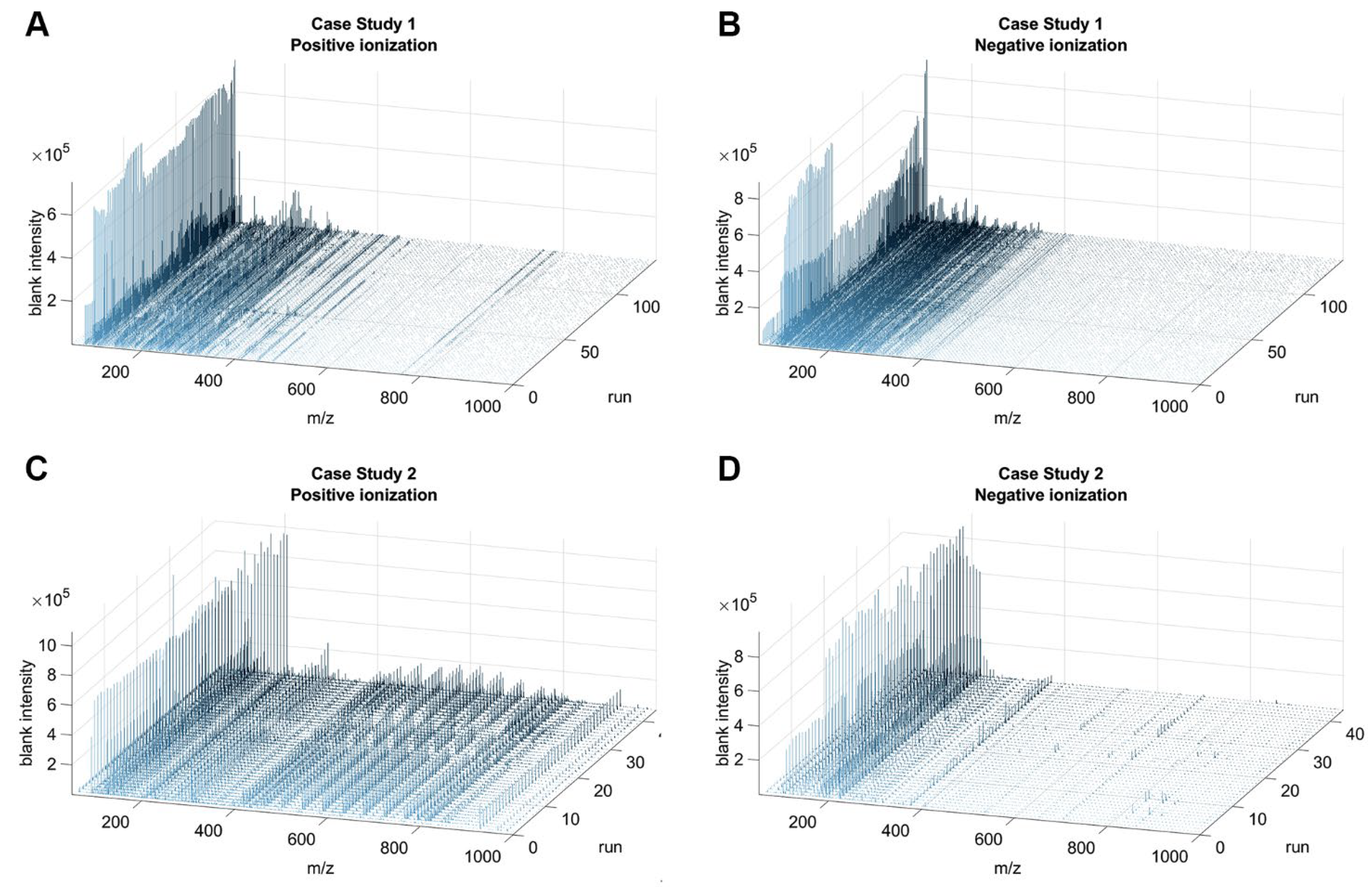
3.1. EASY-FIA Performance on FIA-HRMS Clinical Metabolomics Case Studies
3.2. Unbiased Strategies for Limiting the Matrix Size of the Intensities
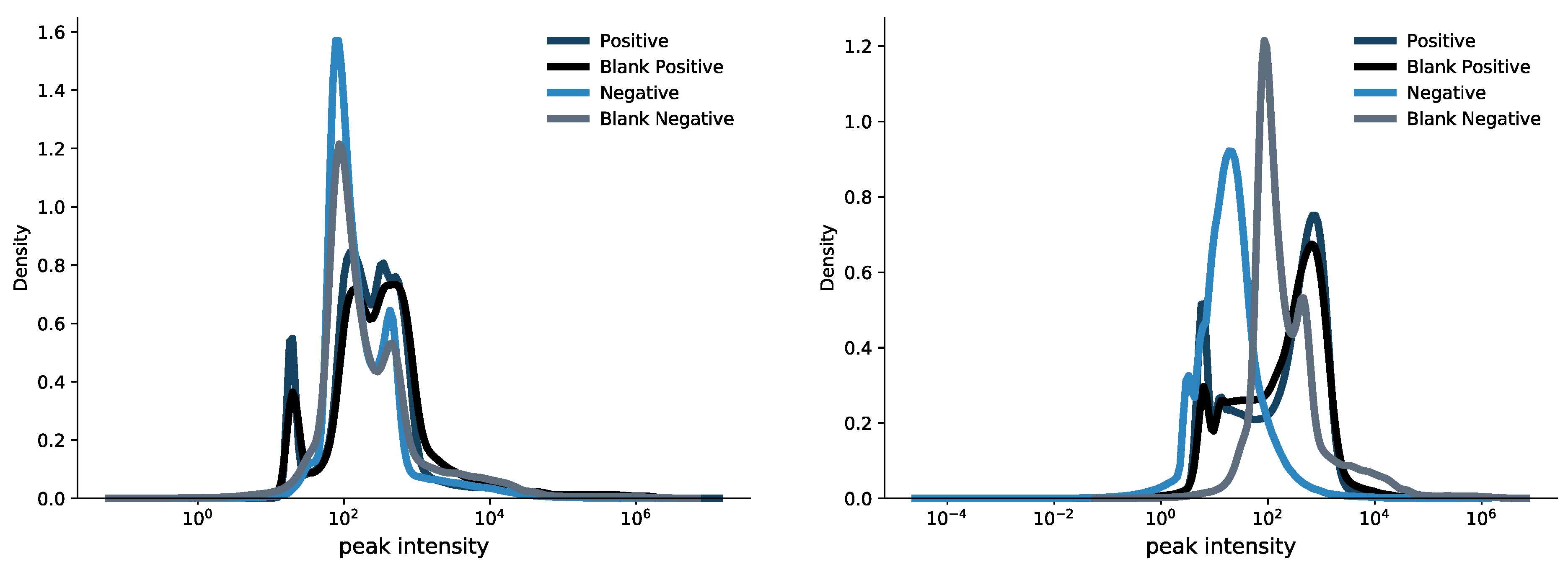
3.3. Assessment of an Unbiased Strategy for Intensity Cut-Off to Remove Blank Spectra Noise
3.4. Assessment of an Unbiased Strategy for m/z Reduction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Patti, G.J.; Yanes, O.; Siuzdak, G. Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, J.K.; Connelly, J.; Lindon, J.; Holmes, E. Metabonomics: A platform for studying drug toxicity and gene function. Nat. Rev. Drug Discov. 2002, 1, 153–161. [Google Scholar] [CrossRef]
- Misra, B.B. New tools and resources in metabolomics: 2016–2017. Electrophoresis 2018, 39, 909–923. [Google Scholar] [CrossRef]
- Goodacre, R.; Vaidyanathan, S.; Dunn, W.B.; Harrigan, G.G.; Kell, D.B. Metabolomics by numbers: Acquiring and understanding global metabolite data. Trends Biotechnol. 2004, 22, 245–252. [Google Scholar] [CrossRef]
- Oliver, S.G.; Winson, M.K.; Kell, D.B.; Baganz, F. Systematic functional analysis of the yeast genome. Trends Biotechnol. 1998, 16, 373–378. [Google Scholar] [CrossRef]
- Jin, Q.; Ma, R.C.W. Metabolomics in Diabetes and Diabetic Complications: Insights from Epidemiological Studies. Cells 2021, 10, 2832. [Google Scholar] [CrossRef]
- Schmidt, D.R.; Patel, R.; Kirsch, D.G.; Lewis, C.A.; Vander Heiden, M.G.; Locasale, J.W. Metabolomics in cancer research and emerging applications in clinical oncology. CA Cancer J. Clin. 2021, 71, 333–358. [Google Scholar] [CrossRef]
- Dubin, R.F.; Rhee, E.P. Proteomics and Metabolomics in Kidney Disease, including Insights into Etiology, Treatment, and Prevention. Clin. J. Am. Soc. Nephrol. 2020, 15, 404–411. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.L.; Davis, D.E.; Codreanu, S.G.; Harrison, F.E.; Sherrod, S.D.; McLean, J.A. Targeted and Untargeted Mass Spectrometry Reveals the Impact of High-Fat Diet on Peripheral Amino Acid Regulation in a Mouse Model of Alzheimer’s Disease. J. Proteome Res. 2021, 20, 4405–4414. [Google Scholar] [CrossRef]
- He, Z.; Liu, Z.; Gong, L. Biomarker identification and pathway analysis of rheumatoid arthritis based on metabolomics in combination with ingenuity pathway analysis. Proteomics 2021, 21, e2100037. [Google Scholar] [CrossRef]
- Wei, Y.; Jasbi, P.; Shi, X.; Turner, C.; Hrovat, J.; Liu, L.; Rabena, Y.; Porter, P.; Gu, H. Early Breast Cancer Detection Using Untargeted and Targeted Metabolomics. J. Proteome Res. 2021, 20, 3124–3133. [Google Scholar] [CrossRef] [PubMed]
- Moreau, R.; Clària, J.; Aguilar, F.; Fenaille, F.; Lozano, J.J.; Junot, C.; Colsch, B.; Caraceni, P.; Trebicka, J.; Pavesi, M.; et al. Blood metabolomics uncovers inflammation-associated mitochondrial dysfunction as a potential mechanism underlying ACLF. J. Hepatol. 2020, 72, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Chen, Y.; Li, K.; Zhan, R.; Zhao, M.; Xu, Y.; Lin, Z.; Fu, Y.; He, Q.; Tang, P.C.; et al. Untargeted metabolomics identifies succinate as a biomarker and therapeutic target in aortic aneurysm and dissection. Eur. Heart J. 2021, 42, 4373–4385. [Google Scholar] [CrossRef] [PubMed]
- Buentzel, J.; Klemp, H.G.; Kraetzner, R.; Schulz, M.; Dihazi, G.H.; Streit, F.; Bleckmann, A.; Menck, K.; Wlochowitz, D.; Binder, C. Metabolomic Profiling of Blood-Derived Microvesicles in Breast Cancer Patients. Int. J. Mol. Sci. 2021, 22, 13540. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, X.; Wei, L.; Wei, D.; Chen, Z.; Cao, Z.; Zhao, Q.; Chang, C. Molecular biological methods in environmental engineering. Water Environ. Res. 2020, 92, 1786–1793. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, L.V.; Fraser, P.; Stewart, D. Metabolomics: A second-generation platform for crop and food analysis. Bioanalysis 2011, 3, 1143–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Saand, M.A.; Huang, L.; Abdelaal, W.B.; Zhang, J.; Wu, Y.; Li, J.; Sirohi, M.H.; Wang, F. Applications of Multi-Omics Technologies for Crop Improvement. Front. Plant Sci. 2021, 12, 563953. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Patti, G.J.; Rinehart, D.; Siuzdak, G. XCMS Online: A Web-Based Platform to Process Untargeted Metabolomic Data. Anal. Chem. 2012, 84, 5035–5039. [Google Scholar] [CrossRef] [Green Version]
- Nanita, S.C.; Kaldon, L.G. Emerging flow injection mass spectrometry methods for high-throughput quantitative analysis. Anal. Bioanal. Chem. 2016, 408, 23–33. [Google Scholar] [CrossRef]
- Cambiaghi, A.; Pinto, B.B.; Brunelli, L.; Falcetta, F.; Aletti, F.; Bendjelid, K.; Pastorelli, R.; Ferrario, M. Characterization of a metabolomic profile associated with responsiveness to therapy in the acute phase of septic shock. Sci. Rep. 2017, 7, 9748. [Google Scholar] [CrossRef]
- La Nasa, J.; Nardella, F.; Andrei, L.; Giani, M.; Degano, I.; Colombini, M.P.; Ribechini, E. Profiling of high molecular weight esters by flow injection analysis-high resolution mass spectrometry for the characterization of raw and archaeological beeswax and resinous substances. Talanta 2020, 212, 120800. [Google Scholar] [CrossRef]
- Büttenbender, S.L.; Carvalho, R.; Barbosa, F.D.S.; Ortiz, R.S.; Limberger, R.P.; Mendez, A.S.L. Fragmentation of Cannabinoids by Flow Injection Analysis Tandem Mass Spectrometry (FIA–MS/MS). J. AOAC Int. 2022, 105, 915–927. [Google Scholar] [CrossRef]
- Vilà, M.; Bedmar, À.; Saurina, J.; Núñez, O.; Sentellas, S. High-Throughput Flow Injection Analysis–Mass Spectrometry (FIA-MS) Fingerprinting for the Authentication of Tea Application to the Detection of Teas Adulterated with Chicory. Foods 2022, 11, 2153. [Google Scholar] [CrossRef]
- Guitton, Y.; Tremblay-Franco, M.; Le Corguillé, G.; Martin, J.-F.; Pétéra, M.; Roger-Mele, P.; Delabrière, A.; Goulitquer, S.; Monsoor, M.; Duperier, C.; et al. Create, run, share, publish, and reference your LC–MS, FIA–MS, GC–MS, and NMR data analysis workflows with the Workflow4Metabolomics 3.0 Galaxy online infrastructure for metabolomics. Int. J. Biochem. Cell Biol. 2017, 93, 89–101. [Google Scholar] [CrossRef] [Green Version]
- Lewis, M.R.; Pearce, J.T.M.; Spagou, K.; Green, M.; Dona, A.C.; Yuen, A.H.Y.; David, M.; Berry, D.J.; Chappell, K.; der Sluis, V.H.-V.; et al. Development and Application of Ultra-Performance Liquid Chromatography-TOF MS for Precision Large Scale Urinary Metabolic Phenotyping. Anal. Chem. 2016, 88, 9004–9013. [Google Scholar] [CrossRef] [Green Version]
- Naz, S.; dos Santos, D.C.M.; García, A.; Barbas, C. Analytical protocols based on LC–MS, GC–MS and CE–MS for nontargeted metabolomics of biological tissues. Bioanalysis 2014, 6, 1657–1677. [Google Scholar] [CrossRef]
- Riquelme, G.; Zabalegui, N.; Marchi, P.; Jones, C.M.; Monge, M.E. A Python-Based Pipeline for Preprocessing LC–MS Data for Untargeted Metabolomics Workflows. Metabolites 2020, 10, 416. [Google Scholar] [CrossRef]
- Sussulini, A. (Ed.) Metabolomics: From Fundamentals to Clinical Applications; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2017; Volume 965. [Google Scholar] [CrossRef]
- The Human Serum Metabolome (HUSERMET) Consortium; Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef]
- Tsai, T.-H.; Wang, M.; Ressom, H.W. Preprocessing and Analysis of LC-MS-Based Proteomic Data. In Statistical Analysis in Proteomics; Jung, K., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; Volume 1362, pp. 63–76. [Google Scholar] [CrossRef]
- Beckonert, O.; Keun, H.C.; Ebbels, T.M.D.; Bundy, J.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat. Protoc. 2007, 2, 2692–2703. [Google Scholar] [CrossRef]
- Pedrioli, P.G.A.; Eng, J.; Hubley, R.; Vogelzang, M.; Deutsch, E.; Raught, B.; Pratt, B.; Nilsson, E.; Angeletti, R.H.; Apweiler, R.; et al. A common open representation of mass spectrometry data and its application to proteomics research. Nat. Biotechnol. 2004, 22, 1459–1466. [Google Scholar] [CrossRef]
- Giacomoni, F.; Le Corguillé, G.; Monsoor, M.; Landi, M.; Pericard, P.; Pétéra, M.; Duperier, C.; Tremblay-Franco, M.; Martin, J.-F.; Jacob, D.; et al. Workflow4Metabolomics: A collaborative research infrastructure for computational metabolomics. Bioinformatics 2015, 31, 1493–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delabrière, A.; Hohenester, U.M.; Colsch, B.; Junot, C.; Fenaille, F.; Thévenot, E.A. proFIA: A data preprocessing workflow for flow injection analysis coupled to high-resolution mass spectrometry. Bioinformatics 2017, 33, 3767–3775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrer, T.; Zamboni, N. High-throughput discovery metabolomics. Curr. Opin. Biotechnol. 2015, 31, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Brunelli, L.; Davin, A.; Sestito, G.; Mimmi, M.C.; De Simone, G.; Balducci, C.; Pansarasa, O.; Forloni, G.; Cereda, C.; Pastorelli, R.; et al. Plasmatic Hippuric Acid as a Hallmark of Frailty in an Italian Cohort: The Mediation Effect of Fruit–Vegetable Intake. J. Gerontol. Ser. A 2021, 76, 2081–2089. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, M.; Cambiaghi, A.; Brunelli, L.; Giordano, S.; Caironi, P.; Guatteri, L.; Raimondi, F.; Gattinoni, L.; Latini, R.; Masson, S.; et al. Mortality prediction in patients with severe septic shock: A pilot study using a target metabolomics approach. Sci. Rep. 2016, 6, 20391. [Google Scholar] [CrossRef] [Green Version]
- Boccard, J.; Rudaz, S. Harnessing the complexity of metabolomic data with chemometrics: Metabolomic Data Analysis with Chemometrics. J. Chemom. 2014, 28, 1–9. [Google Scholar] [CrossRef]
- Fuhrer, T.; Heer, D.; Begemann, B.; Zamboni, N. High-Throughput, Accurate Mass Metabolome Profiling of Cellular Extracts by Flow Injection–Time-of-Flight Mass Spectrometry. Anal. Chem. 2011, 83, 7074–7080. [Google Scholar] [CrossRef]
- Beuchel, C.; Kirsten, H.; Ceglarek, U.; Scholz, M. Metabolite-Investigator: An integrated user-friendly workflow for metabolomics multi-study analysis. Bioinformatics 2021, 37, 2218–2220. [Google Scholar] [CrossRef]
- Beuchel, C.; Becker, S.; Dittrich, J.; Kirsten, H.; Toenjes, A.; Stumvoll, M.; Loeffler, M.; Thiele, H.; Beutner, F.; Thiery, J.; et al. Clinical and lifestyle related factors influencing whole blood metabolite levels—A comparative analysis of three large cohorts. Mol. Metab. 2019, 29, 76–85. [Google Scholar] [CrossRef]
- Gatto, L.; Gibb, S.; Rainer, J. MSnbase, Efficient and Elegant R-Based Processing and Visualization of Raw Mass Spectrometry Data. J. Proteome Res. 2021, 20, 1063–1069. [Google Scholar] [CrossRef]
- Mayer, B. (Ed.) Bioinformatics for Omics Data: Methods and Protocols; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2011. [Google Scholar] [CrossRef]
- Gullo, F.; Ponti, G.; Tagarelli, A.; Tradigo, G.; Veltri, P. MSPtool: A Versatile Tool for Mass Spectrometry Data Preprocessing. In Proceedings of the 2008 21st IEEE International Symposium on Computer-Based Medical Systems, Jyvaskyla, Finland, 17–19 June 2008; pp. 209–214. [Google Scholar] [CrossRef]
- Sauve, A.C.; Speed, T.P. Normalization, baseline correction and alignment of high-throughput mass spectrometry data. Proc. Gensips 2004, 4, 1–4. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case Study | Acquisition Mode | Missing Values | Intensity Values |
|---|---|---|---|
| 1 | Positive | 32,290,973 | 1,685,027 |
| 1 | Negative | 32,762,205 | 1,952,661 |
| 2 | Positive | 1,168,071 | 519,901 |
| 2 | Negative | 2,343,890 | 937,146 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morabito, A.; De Simone, G.; Ferrario, M.; Falcetta, F.; Pastorelli, R.; Brunelli, L. EASY-FIA: A Readably Usable Standalone Tool for High-Resolution Mass Spectrometry Metabolomics Data Pre-Processing. Metabolites 2023, 13, 13. https://doi.org/10.3390/metabo13010013
Morabito A, De Simone G, Ferrario M, Falcetta F, Pastorelli R, Brunelli L. EASY-FIA: A Readably Usable Standalone Tool for High-Resolution Mass Spectrometry Metabolomics Data Pre-Processing. Metabolites. 2023; 13(1):13. https://doi.org/10.3390/metabo13010013
Chicago/Turabian StyleMorabito, Aurelia, Giulia De Simone, Manuela Ferrario, Francesca Falcetta, Roberta Pastorelli, and Laura Brunelli. 2023. "EASY-FIA: A Readably Usable Standalone Tool for High-Resolution Mass Spectrometry Metabolomics Data Pre-Processing" Metabolites 13, no. 1: 13. https://doi.org/10.3390/metabo13010013