
Lipidomics in Understanding Pathophysiology and Pharmacologic Effects in Inflammatory Diseases: Considerations for Drug Development

 , , ,
, , ,
Abstract
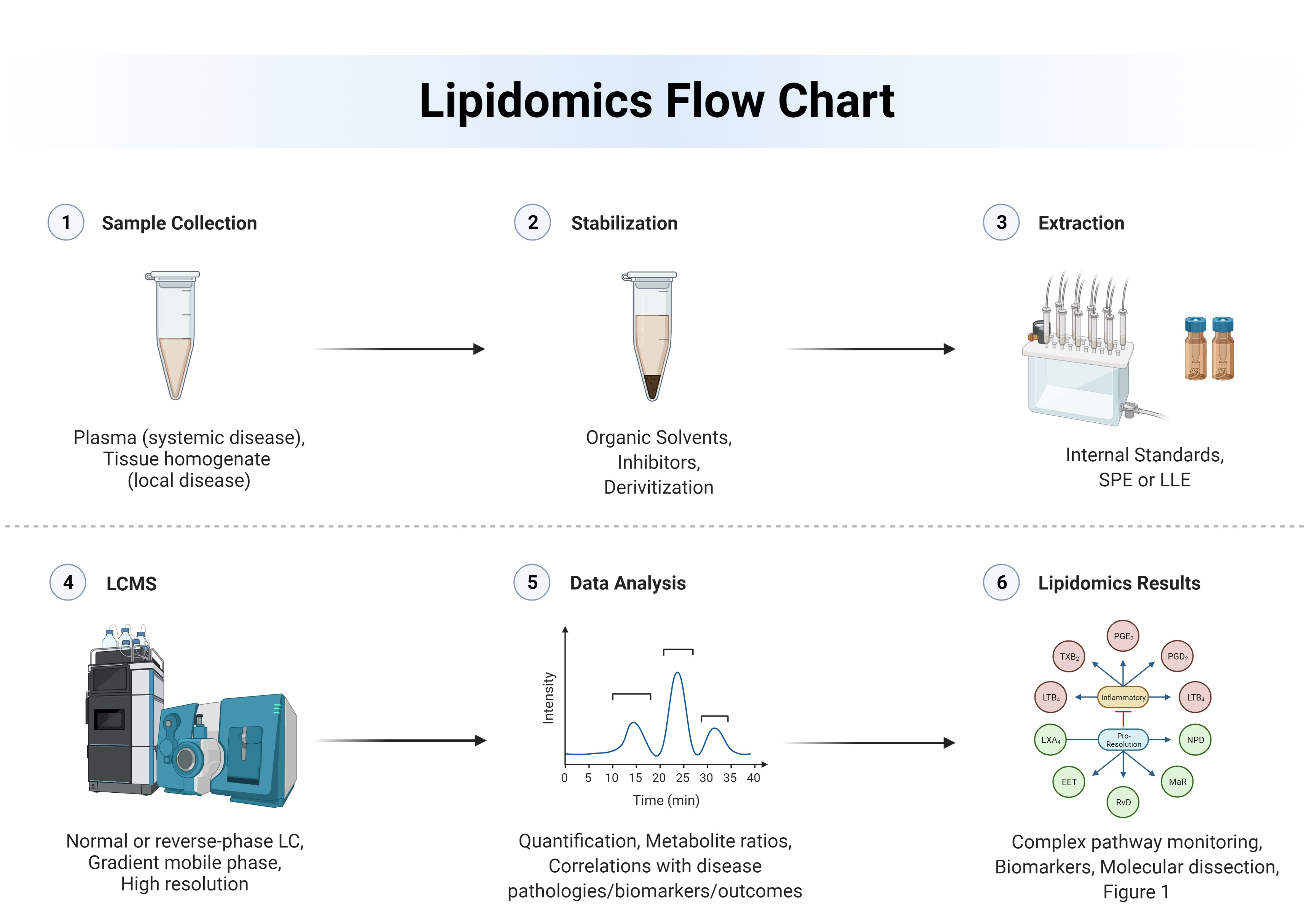
: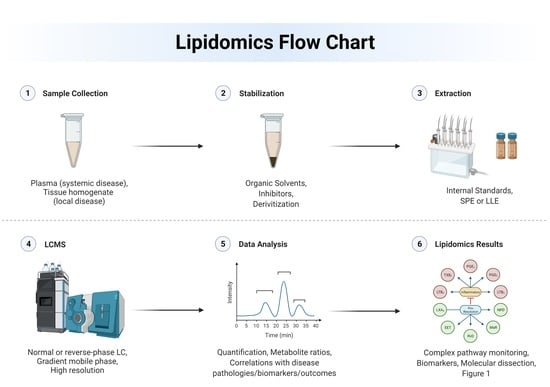
1. Introduction
2. Techniques and Approaches to Lipidomics
2.1. Instrumentation

{kind=link}
{kind=link}
| Method | Advantages | Limitations | References |
|---|---|---|---|
| LC-QTOF/MS |
|
| [16] |
| LC-Orbitrap |
|
| [17] |
| LC-Triple Quadrupole |
|
| [17,18,19,20,21,22,23,24] |
| Nano-ESI-MS |
|
| [25,26] |
| Acoustic Ejection MS |
|
| [27,28] |
| GC-MS |
|
| [21] |
| MALDI-TOF |
|
| [17,19,20] |
2.2. Lipidome Stabilization
2.3. Data Validation
2.4. Data Analysis
3. Lipidomics in Disease Research and Pharmacology
3.1. Cardiovascular Disease and Stroke
3.1.1. Lipoprotein Profiling of CVD
3.1.2. Sphingolipids in CVD
3.1.3. Atherosclerotic Plaques
3.1.4. Stroke Research
3.1.5. Therapeutic Applications
3.2. Neurodegenerative Diseases
3.2.1. Alzheimer’s Disease
3.2.2. Parkinson’s Disease
3.2.3. Therapeutic Applications
3.3. Inflammatory Lung Diseases
3.3.1. Biomarker Identification
3.3.2. Lung Toxicology
3.3.3. Therapeutic Applications
3.4. Autoimmune Diseases
3.4.1. Systemic Lupus Erythematosus
3.4.2. GI Autoimmune Diseases
4. Limitations
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Farooqui, A.A.; Liss, L.; Horrocks, L.A. Neurochemical Aspects of Alzheimer’s Disease: Involvement of Membrane Phospholipids. Metab. Brain Dis. 1988, 3, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Koal, T.; Klavins, K.; Seppi, D.; Kemmler, G.; Humpel, C. Sphingomyelin SM(D18:1/18:0) is Significantly Enhanced in Cerebrospinal Fluid Samples Dichotomized by Pathological Amyloid-Β42, Tau, and Phospho-Tau-181 Levels. J. Alzheimer’s Dis. 2015, 44, 1193–1201. [Google Scholar] [CrossRef] [Green Version]
- Byeon, S.K.; Madugundu, A.K.; Jain, A.P.; Bhat, F.A.; Jung, J.H.; Renuse, S.; Darrow, J.; Bakker, A.; Albert, M.; Moghekar, A.; et al. Cerebrospinal Fluid Lipidomics for Biomarkers of Alzheimer’s Disease. Mol. Omics 2021, 17, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Nevado-Holgado, A.; Whiley, L.; Snowden, S.G.; Soininen, H.; Kloszewska, I.; Mecocci, P.; Tsolaki, M.; Vellas, B.; Thambisetty, M.; et al. Association between Plasma Ceramides and Phosphatidylcholines and Hippocampal Brain Volume in Late Onset Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 60, 809–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowden, J.A.; Heckert, A.; Ulmer, C.Z.; Jones, C.M. Lipid Concentrations in Standard Reference Material (SRM) 1950: Results from an Interlaboratory Comparison Exercise for Lipidomics; NIST: Gaithersburg, MD, USA, 2017.
- Bowden, J.A.; Ulmer, C.Z.; Jones, C.M.; Koelmel, J.P.; Yost, R.A. NIST Lipidomics Workflow Questionnaire: An Assessment of Community-Wide Methodologies and Perspectives. Metabolomics 2018, 14, 53. [Google Scholar] [CrossRef]
- McShane, L.M.; Cavenagh, M.M.; Lively, T.G.; Eberhard, D.A.; Bigbee, W.L.; Williams, P.M.; Mesirov, J.P.; Polley, M.-Y.C.; Kim, K.Y.; Tricoli, J.V.; et al. Criteria for the Use of Omics-Based Predictors in Clinical Trials. Nature 2013, 502, 317–320. [Google Scholar] [CrossRef] [Green Version]
- Abdulkhaleq, L.A.; Assi, M.A.; Abdullah, R.; Zamri-Saad, M.; Taufiq-Yap, Y.H.; Hezmee, M.N.M. The Crucial Roles of Inflammatory Mediators in Inflammation: A Review. Vet. World 2018, 11, 627–635. [Google Scholar] [CrossRef] [Green Version]
- Han, X. Lipidomics for Studying Metabolism. Nat. Rev. Endocrinol. 2016, 12, 668–679. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, K.; Yang, L.; Liu, R.; Chu, Y.; Qin, X.; Yang, P.; Yu, H. Lipid Metabolism in Inflammation-Related Diseases. Analyst 2018, 143, 4526–4536. [Google Scholar] [CrossRef]
- Wu, Z.; Bagarolo, G.I.; Thoröe-Boveleth, S.; Jankowski, J. “Lipidomics”: Mass Spectrometric and Chemometric Analyses of Lipids. Adv. Drug Deliv. Rev. 2020, 159, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Vosegaard, T.; Guo, Z. Applications of Nuclear Magnetic Resonance in Lipid Analyses: An Emerging Powerful Tool for Lipidomics Studies. Prog. Lipid Res. 2017, 68, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Emwas, A.-H.; Roy, R.; McKay, R.T.; Tenori, L.; Saccenti, E.; Gowda, G.A.N.; Raftery, D.; Alahmari, F.; Jaremko, L.; Jaremko, M.; et al. NMR Spectroscopy for Metabolomics Research. Metabolites 2019, 9, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; Bin Nasaruddin, M.; Elliott, C.T.; McGuinness, B.; Passmore, A.P.; Kehoe, P.; Hölscher, C.; McClean, P.L.; Graham, S.F.; Green, B.D. Alzheimer’s Disease–like Pathology Has Transient Effects on the Brain and Blood Metabolome. Neurobiol. Aging 2016, 38, 151–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, J.M.; Trushina, E. Application of Metabolomics in Alzheimer’s Disease. Front. Neurol. 2018, 8, 719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, F.-F. Mass Spectrometry-Based Shotgun Lipidomics—A Critical Review from the Technical Point of View. Anal. Bioanal. Chem. 2018, 410, 6387–6409. [Google Scholar] [CrossRef] [PubMed]
- Züllig, T.; Köfeler, H.C. High resolution mass spectrometry in lipidomics. Mass Spectrom. Rev. 2021, 40, 162–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, H.; Izumi, Y.; Takahashi, M.; Paxton, T.; Tamura, S.; Koike, T.; Yu, Y.; Kato, N.; Nagase, K.; Shiomi, M.; et al. Widely-Targeted Quantitative Lipidomics Method by Supercritical Fluid Chromatography Triple Quadrupole Mass Spectrometry. J. Lipid Res. 2018, 59, 1283–1293. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, C.; Han, X. Enhanced Coverage of Lipid Analysis and Imaging by Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry via a Strategy with an Optimized Mixture of Matrices. Anal. Chim. Acta 2018, 1000, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga, K.; Ishikawa, H.; Taira, S.; Yoshinaga-Kiriake, A.; Usami, Y.; Gotoh, N. Selective Visualization of Administrated Arachidonic and Docosahexaenoic Acids in Brain Using Combination of Simple Stable Isotope-Labeling Technique and Imaging Mass Spectrometry. Anal. Chem. 2020, 92, 8685–8690. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Shon, J.C.; Liu, K.-H. Mass Spectrometry-Based Lipidomics and Its Application ToBiomedical Research. J. Lifestyle Med. 2014, 4, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Tsugawa, H.; Arita, M.; Kanazawa, M.; Ogiwara, A.; Bamba, T.; Fukusaki, E. MRMPROBS: A Data Assessment and Metabolite Identification Tool for Large-Scale Multiple Reaction Monitoring Based Widely Targeted Metabolomics. Anal. Chem. 2013, 85, 5191–5199. [Google Scholar] [CrossRef]
- Zhou, J.; Yin, Y. Strategies for Large-Scale Targeted Metabolomics Quantification by Liquid Chromatography-Mass Spectrometry. Analyst 2016, 141, 6362–6373. [Google Scholar] [CrossRef]
- Köfeler, H.C.; Fauland, A.; Rechberger, G.N.; Trötzmüller, M. Mass Spectrometry Based Lipidomics: An Overview of Technological Platforms. Metabolites 2012, 2, 19–38. [Google Scholar] [CrossRef] [Green Version]
- El-Faramawy, A.; Siu, K.W.M.; Thomson, B.A. Efficiency of Nano-Electrospray Ionization. J. Am. Soc. Mass Spectrom. 2005, 16, 1702–1707. [Google Scholar] [CrossRef] [Green Version]
- Züllig, T.; Trötzmüller, M.; Köfeler, H.C. Lipidomics from Sample Preparation to Data Analysis: A Primer. Anal. Bioanal. Chem. 2020, 412, 2191–2209. [Google Scholar] [CrossRef] [Green Version]
- Pu, F.; Elsen, N.L.; Williams, J.D. Emerging Chromatography-Free High-Throughput Mass Spectrometry Technologies for Generating Hits and Leads. ACS Med. Chem. Lett. 2020, 11, 2108–2113. [Google Scholar] [CrossRef]
- Sinclair, I.; Bachman, M.; Addison, D.; Rohman, M.; Murray, D.C.; Davies, G.; Mouchet, E.; Tonge, M.E.; Stearns, R.G.; Ghislain, L.; et al. Acoustic Mist Ionization Platform for Direct and Contactless Ultrahigh-Throughput Mass Spectrometry Analysis of Liquid Samples. Anal. Chem. 2019, 91, 3790–3794. [Google Scholar] [CrossRef] [Green Version]
- Hyötyläinen, T.; Orešič, M. Optimizing the Lipidomics Workflow for Clinical Studies—Practical Considerations. Anal. Bioanal. Chem. 2015, 407, 4973–4993. [Google Scholar] [CrossRef] [PubMed]
- Asante, I.; Pei, H.; Zhou, E.; Liu, S.; Chui, D.; Yoo, E.; Louie, S.G. Simultaneous Quantitation of Folates, Flavins and B6 Metabolites in Human Plasma by LC–MS/MS Assay: Applications in Colorectal Cancer. J. Pharm. Biomed. Anal. 2018, 158, 66–73. [Google Scholar] [CrossRef]
- Furse, S.; Egmond, M.R.; Killian, J.A. Isolation of Lipids from Biological Samples. Mol. Membr. Biol. 2015, 32, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Han, X. Lipidomics: Techniques, Applications, and Outcomes Related to Biomedical Sciences. Trends Biochem. Sci. 2016, 41, 954–969. [Google Scholar] [CrossRef] [Green Version]
- CLSI. Evaluation of Precision of Quantitative Measurement Procedures, 3rd ed.; CLSI: Wayne, PA, USA, 2014. [Google Scholar]
- Ulmer, C.Z.; Koelmel, J.P.; Jones, C.M.; Garrett, T.J.; Aristizabal-Henao, J.J.; Vesper, H.W.; Bowden, J.A. A Review of Efforts to Improve Lipid Stability during Sample Preparation and Standardization Efforts to Ensure Accuracy in the Reporting of Lipid Measurements. Lipids 2021, 56, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Wieling, J. LC-MS-MS Experiences with Internal Standards. Chromatographia 2002, 55, S107–S113. [Google Scholar] [CrossRef]
- Rubakhin, S.S.; Romanova, E.V.; Nemes, P.; Sweedler, J.V. Profiling Metabolites and Peptides in Single Cells. Nat. Methods 2011, 8, S20–S29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for Linking Genomes to Life and the Environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef] [PubMed]
- Liebal, U.W.; Phan, A.N.T.; Sudhakar, M.; Raman, K.; Blank, L.M. Machine Learning Applications for Mass Spectrometry-Based Metabolomics. Metabolites 2020, 10, 243. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J. The Lipid Bilayer. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
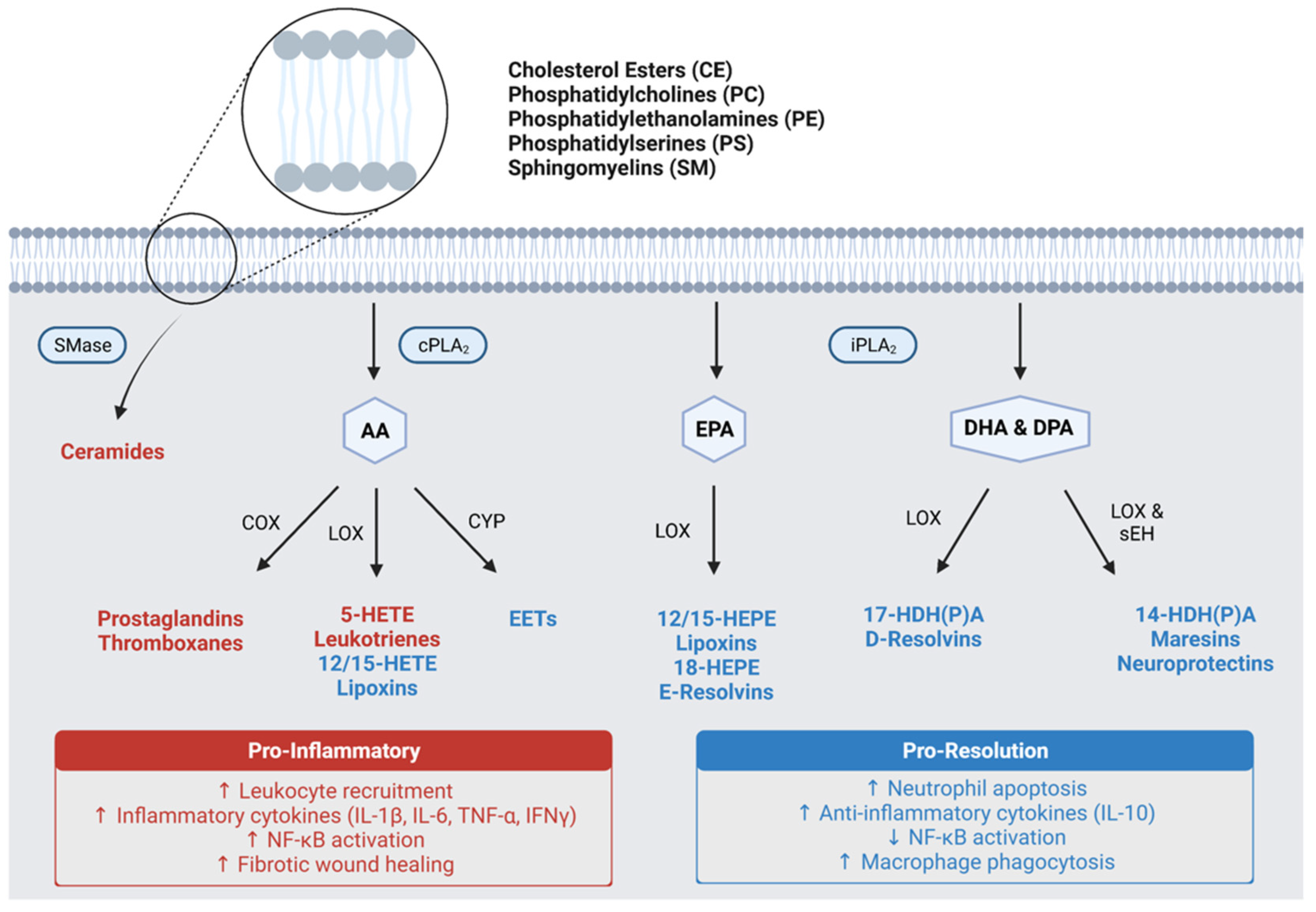
- Fullerton, J.N.; Gilroy, D.W. Resolution of Inflammation: A New Therapeutic Frontier. Nat. Rev. Drug Discov. 2016, 15, 551–567. [Google Scholar] [CrossRef]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism Pathways of Arachidonic Acids: Mechanisms and Potential Therapeutic Targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef]
- Dyall, S.C. Long-Chain Omega-3 Fatty Acids and the Brain: A Review of the Independent and Shared Effects of EPA, DPA and DHA. Front. Aging Neurosci. 2015, 7, 52. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Chiang, N.; van Dyke, T.E. Resolving Inflammation: Dual Anti-Inflammatory and pro-Resolution Lipid Mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Luo, G.; Zhang, L.-F.; Geng, H.-X. Neuroprotective Effects of Epoxyeicosatrienoic Acids. Prostaglandins Other Lipid Mediat. 2018, 138, 9–14. [Google Scholar] [CrossRef]
- Kroetz, D.L.; Zeldin, D.C. Cytochrome P450 Pathways of Arachidonic Acid Metabolism. Curr. Opin. Lipidol. 2002, 13, 273–283. [Google Scholar] [CrossRef] [PubMed]
- López-Vicario, C.; Rius, B.; Alcaraz-Quiles, J.; García-Alonso, V.; Lopategi, A.; Titos, E.; Clària, J. Pro-Resolving Mediators Produced from EPA and DHA: Overview of the Pathways Involved and Their Mechanisms in Metabolic Syndrome and Related Liver Diseases. Eur. J. Pharmacol. 2016, 785, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Colas, R.A.; Shinohara, M.; Dalli, J.; Chiang, N.; Serhan, C.N. Identification and Signature Profiles for Pro-Resolving and Inflammatory Lipid Mediators in Human Tissue. Am. J. Physiol.-Cell Physiol. 2014, 307, C39–C54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centers for Disease Control and Prevention. Underlying Cause of Death, 1999–2018. Available online: https://wonder.cdc.gov/ucd-icd10.html (accessed on 5 November 2020).
- Roth, G.A.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, Regional, and National Age-Sex-Specific Mortality for 282 Causes of Death in 195 Countries and Territories, 1980–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef] [Green Version]
- Soehnlein, O.; Libby, P. Targeting Inflammation in Atherosclerosis—From Experimental Insights to the Clinic. Nat. Rev. Drug Discov. 2021, 20, 589–610. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Noels, H. Atherosclerosis: Current Pathogenesis and Therapeutic Options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef]
- Mortensen, M.B.; Dzaye, O.; Steffensen, F.H.; Bøtker, H.E.; Jensen, J.M.; Rønnow Sand, N.P.; Kragholm, K.H.; Sørensen, H.T.; Leipsic, J.; Mæng, M.; et al. Impact of Plaque Burden Versus Stenosis on Ischemic Events in Patients with Coronary Atherosclerosis. J. Am. Coll. Cardiol. 2020, 76, 2803–2813. [Google Scholar] [CrossRef]
- Lau, A.Y.; Wong, K.L.; Lev, M.; Furie, K.; Smith, W.; Kim, A.S. Burden of Intracranial Steno-Occlusive Lesions on Initial Computed Tomography Angiography Predicts Poor Outcome in Patients with Acute Stroke. Stroke 2013, 44, 1310–1316. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.-S.; Chung, P.-W.; Park, K.-Y.; Won, H.-H.; Bang, O.Y.; Chung, C.-S.; Lee, K.H.; Kim, G.-M. Burden of Intracranial Atherosclerosis Is Associated with Long-Term Vascular Outcome in Patients with Ischemic Stroke. Stroke 2017, 48, 2819–2826. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-Density Lipoproteins Cause Atherosclerotic Cardiovascular Disease. 1. Evidence from Genetic, Epidemiologic, and Clinical Studies. A Consensus Statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [Green Version]
- Abdullah, S.M.; Defina, L.F.; Leonard, D.; Barlow, C.E.; Radford, N.B.; Willis, B.L.; Rohatgi, A.; McGuire, D.K.; de Lemos, J.A.; Grundy, S.M.; et al. Long-Term Association of Low-Density Lipoprotein Cholesterol with Cardiovascular Mortality in Individuals at Low 10-Year Risk of Atherosclerotic Cardiovascular Disease. Circulation 2018, 138, 2315–2325. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.B.; Nordestgaard, B.G. Elevated LDL Cholesterol and Increased Risk of Myocardial Infarction and Atherosclerotic Cardiovascular Disease in Individuals Aged 70–100 Years: A Contemporary Primary Prevention Cohort. Lancet 2020, 396, 1644–1652. [Google Scholar] [CrossRef]
- Schaefer, E.J.; Tsunoda, F.; Diffenderfer, M.; Polisecki, E.; Thai, N.; Asztalos, B. The Measurement of Lipids, Lipoproteins, Apolipoproteins, Fatty Acids, and Sterols, and Next Generation Sequencing for the Diagnosis and Treatment of Lipid Disorders; NCBI: Bethesda, MA, USA, 2000.
- Redgrave, T.G.; Roberts, D.C.K.; West, C.E. Separation of Plasma Lipoproteins by Density-Gradient Ultracentrifugation. Anal. Biochem. 1975, 65, 42–49. [Google Scholar] [CrossRef]
- Gerl, M.J.; Vaz, W.L.C.; Domingues, N.; Klose, C.; Surma, M.A.; Sampaio, J.L.; Almeida, M.S.; Rodrigues, G.; Araújo-Gonçalves, P.; Ferreira, J.; et al. Cholesterol Is Inefficiently Converted to Cholesteryl Esters in the Blood of Cardiovascular Disease Patients. Sci. Rep. 2018, 8, 14764. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Nakamura, T.; Ohan, J.; Lesèche, G.; Tedgui, A.; Maclouf, J.; Murphy, R.C. The Relationship of Hydroxyeicosatetraenoic Acids and F2-Isoprostanes to Plaque Instability in Human Carotid Atherosclerosis. J. Clin. Investig. 1999, 103, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Stegemann, C.; Drozdov, I.; Shalhoub, J.; Humphries, J.; Ladroue, C.; Didangelos, A.; Baumert, M.; Allen, M.; Davies, A.H.; Monaco, C.; et al. Comparative Lipidomics Profiling of Human Atherosclerotic Plaques. Circ. Cardiovasc. Genet. 2011, 4, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, Y.H.; Hannun, Y.A. Translational Aspects of Sphingolipid Metabolism. Trends Mol. Med. 2007, 13, 327–336. [Google Scholar] [CrossRef]
- Lemaitre, R.N.; Jensen, P.N.; Hoofnagle, A.; McKnight, B.; Fretts, A.M.; King, I.B.; Siscovick, D.S.; Psaty, B.M.; Heckbert, S.R.; Mozaffarian, D.; et al. Plasma Ceramides and Sphingomyelins in Relation to Heart Failure Risk. Circ. Heart Fail. 2019, 12, e005708. [Google Scholar] [CrossRef]
- Laaksonen, R.; Ekroos, K.; Sysi-Aho, M.; Hilvo, M.; Vihervaara, T.; Kauhanen, D.; Suoniemi, M.; Hurme, R.; März, W.; Scharnagl, H.; et al. Plasma Ceramides Predict Cardiovascular Death in Patients with Stable Coronary Artery Disease and Acute Coronary Syndromes beyond LDL-Cholesterol. Eur. Heart J. 2016, 37, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Burrello, J.; Biemmi, V.; Dei Cas, M.; Amongero, M.; Bolis, S.; Lazzarini, E.; Bollini, S.; Vassalli, G.; Paroni, R.; Barile, L. Sphingolipid Composition of Circulating Extracellular Vesicles after Myocardial Ischemia. Sci. Rep. 2020, 10, 16182. [Google Scholar] [CrossRef] [PubMed]
- Schissel, S.L.; Tweedie-Hardman, J.; Rapp, J.H.; Graham, G.; Williams, K.J.; Tabas, I. Rabbit Aorta and Human Atherosclerotic Lesions Hydrolyze the Sphingomyelin of Retained Low-Density Lipoprotein. Proposed Role for Arterial-Wall Sphingomyelinase in Subendothelial Retention and Aggregation of Atherogenic Lipoproteins. J. Clin. Investig. 1996, 98, 1455–1464. [Google Scholar] [CrossRef]
- Predescu, S.; Knezevic, I.; Bardita, C.; Neamu, R.F.; Brovcovych, V.; Predescu, D. Platelet Activating Factor-Induced Ceramide Micro-Domains Drive Endothelial NOS Activation and Contribute to Barrier Dysfunction. PLoS ONE 2013, 8, e75846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turpin-Nolan, S.M.; Brüning, J.C. The Role of Ceramides in Metabolic Disorders: When Size and Localization Matters. Nat. Rev. Endocrinol. 2020, 16, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Münzer, P.; Borst, O.; Walker, B.; Schmid, E.; Feijge, M.A.H.; Cosemans, J.M.E.M.; Chatterjee, M.; Schmidt, E.-M.; Schmidt, S.; Towhid, S.T.; et al. Acid Sphingomyelinase Regulates Platelet Cell Membrane Scrambling, Secretion, and Thrombus Formation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 61–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Qadri, S.M.; Lang, E.; Pelzl, L.; Umbach, A.T.; Leiss, V.; Birnbaumer, L.; Nürnberg, B.; Pieske, B.; Voelkl, J.; et al. Heterotrimeric G-Protein Subunit Gα I2 Contributes to Agonist-Sensitive Apoptosis and Degranulation in Murine Platelets. Physiol. Rep. 2018, 6, e13841. [Google Scholar] [CrossRef]
- Hua, T.; Bao, Q.; He, X.; Cai, W.; He, J. Lipidomics Revealed Alteration of Sphingolipid Metabolism During the Reparative Phase After Myocardial Infarction Injury. Front. Physiol. 2021, 12, 304. [Google Scholar] [CrossRef]
- Biemmi, V.; Milano, G.; Ciullo, A.; Cervio, E.; Burrello, J.; Dei Cas, M.; Paroni, R.; Tallone, T.; Moccetti, T.; Pedrazzini, G.; et al. Inflammatory Extracellular Vesicles Prompt Heart Dysfunction via TRL4-Dependent NF-ΚB Activation. Theranostics 2020, 10, 2773–2790. [Google Scholar] [CrossRef]
- Verderio, C.; Gabrielli, M.; Giussani, P. Role of Sphingolipids in the Biogenesis and Biological Activity of Extracellular Vesicles. J. Lipid Res. 2018, 59, 1325–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchesini, N.; Hannun, Y.A. Acid and Neutral Sphingomyelinases: Roles and Mechanisms of Regulation. Biochem. Cell Biol. 2004, 82, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Saddoughi, S.A.; Song, P.; Ogretmen, B. Roles of Bioactive Sphingolipids in Cancer Biology and Therapeutics. In Lipids in Health and Disease; Springer: Dordrecht, The Netherlands, 2008; pp. 413–440. [Google Scholar]
- Kohno, S.; Keenan, A.L.; Ntambi, J.M.; Miyazaki, M. Lipidomic Insight into Cardiovascular Diseases. Biochem. Biophys. Res. Commun. 2018, 504, 590–595. [Google Scholar] [CrossRef]
- Depuydt, M.A.C.; Prange, K.H.M.; Slenders, L.; Örd, T.; Elbersen, D.; Boltjes, A.; de Jager, S.C.A.; Asselbergs, F.W.; de Borst, G.J.; Aavik, E.; et al. Microanatomy of the Human Atherosclerotic Plaque by Single-Cell Transcriptomics. Circ. Res. 2020, 127, 1437–1455. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.-F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.M.; Giannarelli, C. Immune Cell Profiling in Atherosclerosis: Role in Research and Precision Medicine. Nat. Rev. Cardiol. 2022, 19, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Cheng, S.; Lin, Q.; Cao, W.; Yang, J.; Zhang, M.; Shen, A.; Zhang, W.; Xia, Y.; Ma, X.; et al. Single-Cell Lipidomics with High Structural Specificity by Mass Spectrometry. Nat. Commun. 2021, 12, 2869. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthi, R.V.; Ikeda, T.; Feigin, V.L. Global, Regional and Country-Specific Burden of Ischaemic Stroke, Intracerebral Haemorrhage and Subarachnoid Haemorrhage: A Systematic Analysis of the Global Burden of Disease Study 2017. Neuroepidemiology 2020, 54, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, e596–e646. [Google Scholar] [CrossRef] [PubMed]
- Kleindorfer, D.O.; Towfighi, A.; Chaturvedi, S.; Cockroft, K.M.; Gutierrez, J.; Lombardi-Hill, D.; Kamel, H.; Kernan, W.N.; Kittner, S.J.; Leira, E.C.; et al. 2021 Guideline for the Prevention of Stroke in Patients with Stroke and Transient Ischemic Attack: A Guideline from the American Heart Association/American Stroke Association. Stroke 2021, 52. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G.; Hellmann, J.; Proto, J.D.; Kuriakose, G.; Colas, R.A.; Dorweiler, B.; Connolly, E.S.; Solomon, R.; Jones, D.M.; Heyer, E.J.; et al. An Imbalance between Specialized Pro-Resolving Lipid Mediators and pro-Inflammatory Leukotrienes Promotes Instability of Atherosclerotic Plaques. Nat. Commun. 2016, 7, 12859. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, L.; Sun, W.; Pei, L.; Tian, M.; Liang, J.; Liu, X.; Zhang, R.; Fang, H.; Wu, J.; et al. Changes of Metabolites in Acute Ischemic Stroke and Its Subtypes. Front. Neurosci. 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Xie, C.; Zheng, J.; Dong, Q.; Si, T.; Zhang, J.; Hou, S.-T. An Imbalanced Ratio between PC(16:0/16:0) and LPC(16:0) Revealed by Lipidomics Supports the Role of the Lands Cycle in Ischemic Brain Injury. J. Biol. Chem. 2021, 296, 100151. [Google Scholar] [CrossRef] [PubMed]
- Sheth, S.A.; Iavarone, A.T.; Liebeskind, D.S.; Won, S.J.; Swanson, R.A. Targeted Lipid Profiling Discovers Plasma Biomarkers of Acute Brain Injury. PLoS ONE 2015, 10, e0129735. [Google Scholar] [CrossRef]
- Bazan, N.G.; Eady, T.N.; Khoutorova, L.; Atkins, K.D.; Hong, S.; Lu, Y.; Zhang, C.; Jun, B.; Obenaus, A.; Fredman, G.; et al. Novel Aspirin-Triggered Neuroprotectin D1 Attenuates Cerebral Ischemic Injury after Experimental Stroke. Exp. Neurol. 2012, 236, 122–130. [Google Scholar] [CrossRef] [Green Version]
- Miao, Z.; Schultzberg, M.; Wang, X.; Zhao, Y. Role of Polyunsaturated Fatty Acids in Ischemic Stroke—A Perspective of Specialized pro-Resolving Mediators. Clin. Nutr. 2021, 40, 2974–2987. [Google Scholar] [CrossRef]
- Ellero-Simatos, S.; Beitelshees, A.L.; Lewis, J.P.; Yerges-Armstrong, L.M.; Georgiades, A.; Dane, A.; Harms, A.C.; Strassburg, K.; Guled, F.; Hendriks, M.M.W.B.; et al. Oxylipid Profile of Low-Dose Aspirin Exposure: A Pharmacometabolomics Study. J. Am. Heart Assoc. 2015, 4, e002203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.F.; Vickery, T.W.; Serhan, C.N. Chiral Lipidomics of E-Series Resolvins: Aspirin and the Biosynthesis of Novel Mediators. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2011, 1811, 737–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N.; Maddox, J.F.; Petasis, N.A.; Akritopoulou-Zanze, I.; Papayianni, A.; Brady, H.R.; Colgan, S.P.; Madara, J.L. Design of Lipoxin A4 Stable Analogs That Block Transmigration and Adhesion of Human Neutrophils. Biochemistry 1995, 34, 14609–14615. [Google Scholar] [CrossRef]
- Oh, S.F.; Pillai, P.S.; Recchiuti, A.; Yang, R.; Serhan, C.N. Pro-Resolving Actions and Stereoselective Biosynthesis of 18S E-Series Resolvins in Human Leukocytes and Murine Inflammation. J. Clin. Investig. 2011, 121, 569–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, P.R.; Norling, L.V. Implications for Eicosapentaenoic Acid- and Docosahexaenoic Acid-Derived Resolvins as Therapeutics for Arthritis. Eur. J. Pharmacol. 2016, 785, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.-S.; Rezaie, A.R. Thrombin Inhibits Nuclear Factor KappaB and RhoA Pathways in Cytokine-Stimulated Vascular Endothelial Cells When EPCR Is Occupied by Protein C. Thromb. Haemost. 2009, 101, 513–520. [Google Scholar] [PubMed]
- Buddenkotte, J.; Stroh, C.; Engels, I.H.; Moormann, C.; Shpacovitch, V.M.; Seeliger, S.; Vergnolle, N.; Vestweber, D.; Luger, T.A.; Schulze-Osthoff, K.; et al. Agonists of Proteinase-Activated Receptor-2 Stimulate Upregulation of Intercellular Cell Adhesion Molecule-1 in Primary Human Keratinocytes via Activation of NF-Kappa B. J. Investig. Dermatol. 2005, 124, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Jin, H.; Hua, Y.; Keep, R.F.; Xi, G. Role of Protease-Activated Receptor-1 in Brain Injury After Experimental Global Cerebral Ischemia. Stroke 2012, 43, 2476–2482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, J.; Zhou, Y.; Zhao, H.; Chen, Y.; Yan, G.; Wu, L.; Xu, Y.; Zhang, J.; Zhang, X.; Wang, J.; et al. Dabigatran Activates Inflammation Resolution by Promoting Fibrinogen-like Protein 2 Shedding and RvD5 n-3 DPA Production. Theranostics 2021, 11, 4251–4261. [Google Scholar] [CrossRef]
- Lee, I.-O.; Kratz, M.T.; Schirmer, S.H.; Baumhäkel, M.; Böhm, M. The Effects of Direct Thrombin Inhibition with Dabigatran on Plaque Formation and Endothelial Function in Apolipoprotein E-Deficient Mice. J. Pharmacol. Exp. Ther. 2012, 343, 253–257. [Google Scholar] [CrossRef] [Green Version]
- Montecucco, F.; Burger, F.; Pelli, G.; Poku, N.K.; Berlier, C.; Steffens, S.; Mach, F. Statins Inhibit C-Reactive Protein-Induced Chemokine Secretion, ICAM-1 Upregulation and Chemotaxis in Adherent Human Monocytes. Rheumatology 2009, 48, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Chapman, M.J.; Orsoni, A.; Tan, R.; Mellett, N.A.; Nguyen, A.; Robillard, P.; Giral, P.; Thérond, P.; Meikle, P.J. LDL Subclass Lipidomics in Atherogenic Dyslipidemia: Effect of Statin Therapy on Bioactive Lipids and Dense LDL. J. Lipid Res. 2020, 61, 911–932. [Google Scholar] [CrossRef] [Green Version]
- Link, A.; Ayadhi, T.; Bohm, M.; Nickenig, G. Rapid Immunomodulation by Rosuvastatin in Patients with Acute Coronary Syndrome. Eur. Heart J. 2006, 27, 2945–2955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FISCHETTI, F.; CARRETTA, R.; BOROTTO, G.; DURIGUTTO, P.; BULLA, R.; MERONI, P.L.; TEDESCO, F. Fluvastatin Treatment Inhibits Leucocyte Adhesion and Extravasation in Models of Complement-Mediated Acute Inflammation. Clin. Exp. Immunol. 2004, 135, 186–193. [Google Scholar] [CrossRef]
- Birnbaum, Y.; Ye, Y.; Lin, Y.; Freeberg, S.Y.; Nishi, S.P.; Martinez, J.D.; Huang, M.-H.; Uretsky, B.F.; Perez-Polo, J.R. Augmentation of Myocardial Production of 15-Epi-Lipoxin-A 4 by Pioglitazone and Atorvastatin in the Rat. Circulation 2006, 114, 929–935. [Google Scholar] [CrossRef] [Green Version]
- Bergheanu, S.C.; Reijmers, T.; Zwinderman, A.H.; Bobeldijk, I.; Ramaker, R.; Liem, A.-H.; Van Der Greef, J.; Hankemeier, T.; Jukema, J.W. Lipidomic Approach to Evaluate Rosuvastatin and Atorvastatin at Various Dosages: Investigating Differential Effects among Statins. Curr. Med. Res. Opin. 2008, 24, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Kaddurah-Daouk, R.; Baillie, R.A.; Zhu, H.; Zeng, Z.-B.; Wiest, M.M.; Nguyen, U.T.; Watkins, S.M.; Krauss, R.M. Lipidomic Analysis of Variation in Response to Simvastatin in the Cholesterol and Pharmacogenetics Study. Metabolomics 2010, 6, 191–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biringer, R.G. The Role of Eicosanoids in Alzheimer’s Disease. Int. J. Environ. Res. Public Health 2019, 16, 2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, Y.B.; di Meco, A.; Praticó, D. Modulation of Amyloid-β Production by Leukotriene B4 via the γ-Secretase Pathway. J. Alzheimer’s Dis. 2013, 38, 503–506. [Google Scholar] [CrossRef]
- Bates, K.A.; Fonte, J.; Robertson, T.A.; Martins, R.N.; Harvey, A.R. Chronic Gliosis Triggers Alzheimer’s Disease-like Processing of Amyloid Precursor Protein. Neuroscience 2002, 113, 785–796. [Google Scholar] [CrossRef]
- Michael, J.; Marschallinger, J.; Aigner, L. The Leukotriene Signaling Pathway: A Druggable Target in Alzheimer’s Disease. Drug Discov. Today 2019, 24, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Colonna, M. Microglia in Alzheimer’s Disease: A Target for Immunotherapy. J. Leukoc. Biol. 2019, 106, 219–227. [Google Scholar] [CrossRef]
- Sastre, M.; Klockgether, T.; Heneka, M.T. Contribution of Inflammatory Processes to Alzheimer’s Disease: Molecular Mechanisms. Int. Dev. Neurosci. 2006, 24, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Hu, X.; Qian, L.; Wilson, B.; Lee, C.; Flood, P.; Langenbach, R.; Hong, J.-S. Prostaglandin E2 Released from Activated Microglia Enhances Astrocyte Proliferation in Vitro. Toxicol. Appl. Pharmacol. 2009, 238, 64–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikonomovic, M.D.; Abrahamson, E.E.; Uz, T.; Manev, H.; DeKosky, S.T. Increased 5-Lipoxygenase Immunoreactivity in the Hippocampus of Patients with Alzheimer’s Disease. J. Histochem. Cytochem. 2008, 56, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Völkel, W.; Sicilia, T.; Pähler, A.; Gsell, W.; Tatschner, T.; Jellinger, K.; Leblhuber, F.; Riederer, P.; Lutz, W.K.; Götz, M.E. Increased Brain Levels of 4-Hydroxy-2-Nonenal Glutathione Conjugates in Severe Alzheimer’s Disease. Neurochem. Int. 2006, 48, 679–686. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Bader Lange, M.L.; Sultana, R. Involvements of the Lipid Peroxidation Product, HNE, in the Pathogenesis and Progression of Alzheimer’s Disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 924–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultana, R.; Butterfield, D.A. Role of Oxidative Stress in the Progression of Alzheimer’s Disease. J. Alzheimer’s Dis. 2010, 19, 341–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Fritsche, K.L.; Beversdorf, D.Q.; Gu, Z.; Lee, J.C.; Folk, W.R.; Greenlief, C.M.; Sun, G.Y. Yin-Yang Mechanisms Regulating Lipid Peroxidation of Docosahexaenoic Acid and Arachidonic Acid in the Central Nervous System. Front. Neurol. 2019, 10, 642. [Google Scholar] [CrossRef]
- Kartavenka, K.; Panuwet, P.; Yakimavets, V.; Jaikang, C.; Thipubon, K.; D’Souza, P.E.; Barr, D.B.; Ryan, P.B. LC-MS Quantification of Malondialdehyde-Dansylhydrazine Derivatives in Urine and Serum Samples. J. Anal. Toxicol. 2020, 44, 470–481. [Google Scholar] [CrossRef]
- Palacios-Pelaez, R.; Lukiw, W.J.; Bazan, N.G. Omega-3 Essential Fatty Acids Modulate Initiation and Progression of Neurodegenerative Disease. Mol. Neurobiol. 2010, 41, 367–374. [Google Scholar] [CrossRef]
- Lukiw, W.J.; Cui, J.-G.; Marcheselli, V.L.; Bodker, M.; Botkjaer, A.; Gotlinger, K.; Serhan, C.N.; Bazan, N.G. A Role for Docosahexaenoic Acid-Derived Neuroprotectin D1 in Neural Cell Survival and Alzheimer Disease. J. Clin. Investig. 2005, 115, 2774–2783. [Google Scholar] [CrossRef] [Green Version]
- Lukiw, W.J.; Bazan, N.G. Survival Signalling in Alzheimer’s Disease. Biochem. Soc. Trans. 2006, 34, 1277–1282. [Google Scholar] [CrossRef]
- Bazan, N.G. Neuroprotectin D1-Mediated Anti-Inflammatory and Survival Signaling in Stroke, Retinal Degenerations, and Alzheimer’s Disease. J. Lipid Res. 2009, 50, S400–S405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desale, S.E.; Chinnathambi, S. Role of Dietary Fatty Acids in Microglial Polarization in Alzheimer’s Disease. J. Neuroinflammation 2020, 17, 93. [Google Scholar] [CrossRef] [Green Version]
- Haag, M. Essential Fatty Acids and the Brain. Can. J. Psychiatry 2003, 48, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Chuang, D.Y.; Simonyi, A.; Kotzbauer, P.T.; Gu, Z.; Sun, G.Y. Cytosolic Phospholipase A2 Plays a Crucial Role in ROS/NO Signaling during Microglial Activation through the Lipoxygenase Pathway. J. Neuroinflamm. 2015, 12, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duro, M.V.; Ebright, B.; Yassine, H.N. Lipids and Brain Inflammation in APOE4-Associated Dementia. Curr. Opin. Lipidol. 2022, 33, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, B.; Solomon, V.; Fonteh, A.; Rapoport, S.I.; Bennett, D.A.; Arvanitakis, Z.; Chui, H.C.; Miller, C.; Sullivan, P.M.; et al. Calcium-Dependent Cytosolic Phospholipase A2 Activation Is Implicated in Neuroinflammation and Oxidative Stress Associated with ApoE4. Mol. Neurodegener. 2021, 16, 26. [Google Scholar] [CrossRef] [PubMed]
- Farooqui, T.; Farooqui, A.A. Lipid-Mediated Oxidative Stress and Inflammation in the Pathogenesis of Parkinson’s Disease. Parkinson’s Dis. 2011, 2011, 247467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Roy, A.; Liu, X.; Kordower, J.H.; Mufson, E.J.; Hartley, D.M.; Ghosh, S.; Mosley, R.L.; Gendelman, H.E.; Pahan, K. Selective Inhibition of NF-ΚB Activation Prevents Dopaminergic Neuronal Loss in a Mouse Model of Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2007, 104, 18754–18759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawada, M.; Imamura, K.; Nagatsu, T. Role of Cytokines in Inflammatory Process in Parkinson’s Disease. In Parkinson’s Disease and Related Disorders; Springer: Vienna, Austria, 2006; pp. 373–381. [Google Scholar]
- Nagatsu, T.; Sawada, M. Inflammatory Process in Parkinsons Disease: Role for Cytokines. Curr. Pharm. Des. 2005, 11, 999–1016. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Horrocks, L.A.; Farooqui, T. Glycerophospholipids in Brain: Their Metabolism, Incorporation into Membranes, Functions, and Involvement in Neurological Disorders. Chem. Phys. Lipids 2000, 106, 1–29. [Google Scholar] [CrossRef]
- Jiang, F.; Wu, Q.; Sun, S.; Bi, G.; Guo, L. Identification of Potential Diagnostic Biomarkers for Parkinson’s Disease. FEBS Open Bio 2019, 9, 1460–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda-Matsuo, Y.; Miyata, H.; Mizoguchi, T.; Ohama, E.; Naito, Y.; Uematsu, S.; Akira, S.; Sasaki, Y.; Tanabe, M. Microsomal Prostaglandin E Synthase-1 Is a Critical Factor in Dopaminergic Neurodegeneration in Parkinson’s Disease. Neurobiol. Dis. 2019, 124, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Seet, R.C.S.; Lee, C.-Y.J.; Lim, E.C.H.; Tan, J.J.H.; Quek, A.M.L.; Chong, W.-L.; Looi, W.-F.; Huang, S.-H.; Wang, H.; Chan, Y.-H. Oxidative Damage in Parkinson Disease: Measurement Using Accurate Biomarkers. Free. Radic. Biol. Med. 2010, 48, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Tramutola, A.; Butterfield, D.A. Role of 4-Hydroxy-2-Nonenal (HNE) in the Pathogenesis of Alzheimer Disease and Other Selected Age-Related Neurodegenerative Disorders. Free Radic. Biol. Med. 2017, 111, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Krashia, P.; Cordella, A.; Nobili, A.; la Barbera, L.; Federici, M.; Leuti, A.; Campanelli, F.; Natale, G.; Marino, G.; Calabrese, V.; et al. Blunting Neuroinflammation with Resolvin D1 Prevents Early Pathology in a Rat Model of Parkinson’s Disease. Nat. Commun. 2019, 10, 3945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponce, J.; Ulu, A.; Hanson, C.; Cameron-Smith, E.; Bertoni, J.; Wuebker, J.; Fisher, A.; Siu, K.-C.; Marmelat, V.; Adamec, J.; et al. Role of Specialized Pro-Resolving Mediators in Reducing Neuroinflammation in Neurodegenerative Disorders. Front. Aging Neurosci. 2022, 14, 780811. [Google Scholar] [CrossRef]
- Alecu, I.; Bennett, S.A.L. Dysregulated Lipid Metabolism and Its Role in α-Synucleinopathy in Parkinson’s Disease. Front. Neurosci. 2019, 13, 328. [Google Scholar] [CrossRef] [PubMed]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of α-Synuclein Secondary Structure upon Binding to Synthetic Membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fotuhi, M.; Mohassel, P.; Yaffe, K. Fish Consumption, Long-Chain Omega-3 Fatty Acids and Risk of Cognitive Decline or Alzheimer Disease: A Complex Association. Nat. Rev. Neurol. 2009, 5, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Bazan, N.G. Lipid-Mediated Cell Signaling Protects against Injury and Neurodegeneration. J. Nutr. 2010, 140, 858–863. [Google Scholar] [CrossRef]
- Araya-Quintanilla, F.; Gutiérrez-Espinoza, H.; Sánchez-Montoya, U.; Muñoz-Yañez, M.J.; Baeza-Vergara, A.; Petersen-Yanjarí, M.; Fernández-Lecaros, L. Effectiveness of Omega-3 Fatty Acid Supplementation in Patients with Alzheimer Disease: A Systematic Review and Meta-Analysis. Neurología 2020, 35, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Arellanes, I.C.; Choe, N.; Solomon, V.; He, X.; Kavin, B.; Martinez, A.E.; Kono, N.; Buennagel, D.P.; Hazra, N.; Kim, G.; et al. Brain Delivery of Supplemental Docosahexaenoic Acid (DHA): A Randomized Placebo-Controlled Clinical Trial. EBioMedicine 2020, 59, 102883. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.-K.; Zhang, Y.P.; Han, S.; Pei, J.; Xu, L.Y.; Lu, P.-H.; Shields, C.B.; Xu, X.-M. Annexin A1 Reduces Inflammatory Reaction and Tissue Damage Through Inhibition of Phospholipase A2 Activation in Adult Rats Following Spinal Cord Injury. J. Neuropathol. Exp. Neurol. 2007, 66, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mejia, R.O.; Newman, J.W.; Toh, S.; Yu, G.-Q.; Zhou, Y.; Halabisky, B.; Cissé, M.; Scearce-Levie, K.; Cheng, I.H.; Gan, L.; et al. Phospholipase A2 Reduction Ameliorates Cognitive Deficits in a Mouse Model of Alzheimer’s Disease. Nat. Neurosci. 2008, 11, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga, N.; Yasuda, Y.; Murayama, T.; Nomura, Y. Possible Involvement of Cytosolic Phospholipase A2 in Cell Death Induced by 1-Methyl-4-Phenylpyridinium Ion, a Dopaminergic Neurotoxin, in GH3 Cells. Brain Res. 2000, 855, 244–251. [Google Scholar] [CrossRef]
- Li, H.; Li, W.; Zhang, X.; Ma, X.-C.; Zhang, R.-W. Aspirin Use on Incident Dementia and Mild Cognitive Decline: A Systematic Review and Meta-Analysis. Front. Aging Neurosci. 2021, 12, 419. [Google Scholar] [CrossRef] [PubMed]
- Pasqualetti, P.; Bonomini, C.; Dal Forno, G.; Paulon, L.; Sinforiani, E.; Marra, C.; Zanetti, O.; Maria Rossini, P. A Randomized Controlled Study on Effects of Ibuprofen on Cognitive Progression of Alzheimer’s Disease. Aging Clin. Exp. Res. 2009, 21, 102–110. [Google Scholar] [CrossRef] [PubMed]
- De Jong, D.; Jansen, R.; Hoefnagels, W.; Jellesma-Eggenkamp, M.; Verbeek, M.; Borm, G.; Kremer, B. No Effect of One-Year Treatment with Indomethacin on Alzheimer’s Disease Progression: A Randomized Controlled Trial. PLoS ONE 2008, 3, e1475. [Google Scholar] [CrossRef] [PubMed]
- Tariq, M.; Khan, H.; Al Moutaery, K.; Al Deeb, S. Protective Effect of Quinacrine on Striatal Dopamine Levels in 6-OHDA and MPTP Models of Parkinsonism in Rodents. Brain Res. Bull. 2001, 54, 77–82. [Google Scholar] [CrossRef]
- Bitto, A.; Giuliani, D.; Pallio, G.; Irrera, N.; Vandini, E.; Canalini, F.; Zaffe, D.; Ottani, A.; Minutoli, L.; Rinaldi, M.; et al. Effects of COX1-2/5-LOX Blockade in Alzheimer Transgenic 3xTg-AD Mice. Inflamm. Res. 2017, 66, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Klegeris, A.; McGeer, P.L. Toxicity of Human Monocytic THP-1 Cells and Microglia toward SH-SY5Y Neuroblastoma Cells Is Reduced by Inhibitors of 5-Lipoxygenase and Its Activating Protein FLAP. J. Leukoc. Biol. 2003, 73, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-Y.; Wang, X.-R.; Xu, D.-M.; Yu, S.-Y.; Shi, Q.-J.; Zhang, L.-H.; Chen, L.; Fang, S.-H.; Lu, Y.-B.; Zhang, W.-P.; et al. HAMI 3379, a CysLT 2 Receptor Antagonist, Attenuates Ischemia-Like Neuronal Injury by Inhibiting Microglial Activation. J. Pharmacol. Exp. Ther. 2013, 346, 328–341. [Google Scholar] [CrossRef] [Green Version]
- KLEGERIS, A.; MCGEER, P. Cyclooxygenase and 5-Lipoxygenase Inhibitors Protect against Mononuclear Phagocyte Neurotoxicity. Neurobiol. Aging 2002, 23, 787–794. [Google Scholar] [CrossRef]
- Elewa, H.F.; Hilali, H.; Hess, D.C.; Machado, L.S.; Fagan, S.C. Minocycline for Short-Term Neuroprotection. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2006, 26, 515–521. [Google Scholar] [CrossRef] [Green Version]
- Howard, R.; Zubko, O.; Bradley, R.; Harper, E.; Pank, L.; O’Brien, J.; Fox, C.; Tabet, N.; Livingston, G.; Bentham, P.; et al. Minocycline at 2 Different Dosages vs Placebo for Patients with Mild Alzheimer Disease. JAMA Neurol. 2020, 77, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Gyengesi, E.; Münch, G. In Search of an Anti-Inflammatory Drug for Alzheimer Disease. Nat. Rev. Neurol. 2020, 16, 1197. [Google Scholar] [CrossRef] [PubMed]
- Dei Cas, M.; Zulueta, A.; Mingione, A.; Caretti, A.; Ghidoni, R.; Signorelli, P.; Paroni, R. An Innovative Lipidomic Workflow to Investigate the Lipid Profile in a Cystic Fibrosis Cell Line. Cells 2020, 9, 1197. [Google Scholar] [CrossRef]
- Madison, M.C.; Landers, C.T.; Gu, B.H.; Chang, C.Y.; Tung, H.Y.; You, R.; Hong, M.J.; Baghaei, N.; Song, L.Z.; Porter, P.; et al. Electronic Cigarettes Disrupt Lung Lipid Homeostasis and Innate Immunity Independent of Nicotine. J. Clin. Investig. 2019, 129, 4290–4304. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Xu, P.; Gong, F.; Tan, Y.; Han, J.; Tian, L.; Yan, J.; Li, K.; Xi, Z.; Liu, X. Altered Lipidomic Profiles in Lung and Serum of Rat after Sub-Chronic Exposure to Ozone. Sci. Total Environ. 2022, 806, 150630. [Google Scholar] [CrossRef]
- Carter, C.L.; Jones, J.W.; Farese, A.M.; MacVittie, T.J.; Kane, M.A. Lipidomic Dysregulation within the Lung Parenchyma Following Whole-Thorax Lung Irradiation: Markers of Injury, Inflammation and Fibrosis Detected by MALDI-MSI. Sci. Rep. 2017, 7, 10343. [Google Scholar] [CrossRef]
- Telenga, E.; Hoffmann, R.F.; T’Kindt, R.; Hoonhorst, S.J.M.; Willemse, B.W.M.; Van Oosterhout, A.J.M.; Heijink, I.H.; Berge, M.V.D.; Jorge, L.; Sandra, P.; et al. Untargeted Lipidomic Analysis in Chronic Obstructive Pulmonary Disease Uncovering Sphingolipids. Am. J. Respir. Crit. Care Med. 2014, 190, 155–164. [Google Scholar] [CrossRef]
- Dong, Y.; Arif, A.A.; Guo, J.; Ha, Z.; Lee-Sayer, S.S.M.; Poon, G.F.T.; Dosanjh, M.; Roskelley, C.D.; Huan, T.; Johnson, P. CD44 Loss Disrupts Lung Lipid Surfactant Homeostasis and Exacerbates Oxidized Lipid-Induced Lung Inflammation. Front. Immunol. 2020, 11, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debeuf, N.; Lambrecht, B.N. Eicosanoid Control over Antigen Presenting Cells in Asthma. Front. Immunol. 2018, 9, 2006. [Google Scholar] [CrossRef]
- Kiss, L.; Schütte, H.; Padberg, W.; Weissmann, N.; Mayer, K.; Gessler, T.; Voswinckel, R.; Seeger, W.; Grimminger, F. Epoxyeicosatrienoates Are the Dominant Eicosanoids in Human Lungs upon Microbial Challenge. Eur. Respir. J. 2010, 36, 1088–1098. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.K.; Peters-Golden, M. Eicosanoid Lipid Mediators in Fibrotic Lung Diseases. Chest 2008, 133, 1442–1450. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Li, Z.; Dong, L.; Wu, Y.; Shen, H.; Chen, Z. Lipid Metabolism in Chronic Obstructive Pulmonary Disease. Int. J. Chronic Obstr. Pulm. Dis. 2019, 14, 1009–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archambault, A.; Zaid, Y.; Rakotoarivelo, V.; Turcotte, C.; Doré, É.; Dubuc, I.; Martin, C.; Flamand, O.; Amar, Y.; Cheikh, A.; et al. High Levels of Eicosanoids and Docosanoids in the Lungs of Intubated COVID-19 Patients. FASEB J. 2021, 35, e21666. [Google Scholar] [CrossRef]
- McCarthy, M.K.; Weinberg, J.B. Eicosanoids and Respiratory Viral Infection: Coordinators of Inflammation and Potential Therapeutic Targets. Mediat. Inflamm. 2012, 2012, 236345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devine, J.F. Chronic Obstructive Pulmonary Disease: An Overview. Am. Health Drug Benefits 2008, 1, 34–42. [Google Scholar]
- Drozdovszky, O.; Barta, I.; Antus, B. Sputum Eicosanoid Profiling in Exacerbations of Chronic Obstructive Pulmonary Disease. Respiration 2014, 87, 408–415. [Google Scholar] [CrossRef]
- Liu, D.; Meister, M.; Zhang, S.; Vong, C.-I.; Wang, S.; Fang, R.; Li, L.; Wang, P.G.; Massion, P.; Ji, X. Identification of Lipid Biomarker from Serum in Patients with Chronic Obstructive Pulmonary Disease. Respir. Res. 2020, 21, 242. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, B.; Sharma, L.; Roberts, L.; Peng, X.; Bermejo, S.; Leighton, I.; Casanovas-Massana, A.; Minasyan, M.; Farhadian, S.; Ko, A.I.; et al. Cutting Edge: Severe SARS-CoV-2 Infection in Humans Is Defined by a Shift in the Serum Lipidome, Resulting in Dysregulation of Eicosanoid Immune Mediators. J. Immunol. 2021, 206, 329–334. [Google Scholar] [CrossRef]
- Geneva: World Health Organization WHO COVID-19 Dashboard. Available online: https://covid19.who.int/ (accessed on 19 February 2022).
- Bellis, C.; Kulkarni, H.; Mamtani, M.; Kent, J.W.; Wong, G.; Weir, J.M.; Barlow, C.; Diego, V.; de Almeida, M.A.A.; Dyer, T.D.; et al. Human Plasma Lipidome Is Pleiotropically Associated with Cardiovascular Risk Factors and Death. Circ. Cardiovasc. Genet. 2014, 7, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.J.; Hough, G.; Hama, S.; Aboulhosn, J.; Belperio, J.A.; Saggar, R.; van Lenten, B.J.; Ardehali, A.; Eghbali, M.; Reddy, S.; et al. Proinflammatory High-Density Lipoprotein Results from Oxidized Lipid Mediators in the Pathogenesis of Both Idiopathic and Associated Types of Pulmonary Arterial Hypertension. Pulm. Circ. 2015, 5, 640–648. [Google Scholar] [CrossRef] [Green Version]
- Neuhofer, A.; Zeyda, M.; Mascher, D.; Itariu, B.K.; Murano, I.; Leitner, L.; Hochbrugger, E.E.; Fraisl, P.; Cinti, S.; Serhan, C.N.; et al. Impaired Local Production of Proresolving Lipid Mediators in Obesity and 17-HDHA as a Potential Treatment for Obesity-Associated Inflammation. Diabetes 2013, 62, 1945–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, W.L.; Knotts, T.A.; Chavez, J.A.; Wang, L.-P.; Hoehn, K.L.; Summers, S.A. Lipid Mediators of Insulin Resistance. Nutr. Rev. 2008, 65, S39–S46. [Google Scholar] [CrossRef]
- Zaid, Y.; Doré, É.; Dubuc, I.; Archambault, A.S.; Flamand, O.; Laviolette, M.; Flamand, N.; Boilard, É.; Flamand, L. Chemokines and Eicosanoids Fuel the Hyperinflammation within the Lungs of Patients with Severe COVID-19. J. Allergy Clin. Immunol. 2021, 148, 368–380.e3. [Google Scholar] [CrossRef] [PubMed]
- Ford-Hutchinson, A.W.; Bray, M.A.; Doig, M.V.; Shipley, M.E.; Smith, M.J.H. Leukotriene B, a Potent Chemokinetic and Aggregating Substance Released from Polymorphonuclear Leukocytes. Nature 1980, 286, 264–265. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Tanaka, N.; Ikari, J.; Suzuki, M.; Anazawa, R.; Abe, M.; Saito, Y.; Tatsumi, K. Comprehensive Lipid Profiling of Bleomycin-Induced Lung Injury. J. Appl. Toxicol. 2019, 39, 658–671. [Google Scholar] [CrossRef] [PubMed]
- Charbeneau, R.P.; Peters-Golden, M. Eicosanoids: Mediators and Therapeutic Targets in Fibrotic Lung Disease. Clin. Sci. 2005, 108, 479–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, J.H.; Khanna, A.K.; Kethireddy, S.; Yamane, D.; Levine, A.; Jackson, A.M.; McCurdy, M.T.; Tabatabai, A.; Kumar, G.; Park, P.; et al. Aspirin Use Is Associated with Decreased Mechanical Ventilation, Intensive Care Unit Admission, and In-Hospital Mortality in Hospitalized Patients With Coronavirus Disease 2019. Anesth. Analg. 2021, 132, 930–941. [Google Scholar] [CrossRef]
- Zheng, B.-J.; Chan, K.-W.; Lin, Y.-P.; Zhao, G.-Y.; Chan, C.; Zhang, H.-J.; Chen, H.-L.; Wong, S.S.Y.; Lau, S.K.P.; Woo, P.C.Y.; et al. Delayed Antiviral plus Immunomodulator Treatment Still Reduces Mortality in Mice Infected by High Inoculum of Influenza A/H5N1 Virus. Proc. Natl. Acad. Sci. USA 2008, 105, 8091–8096. [Google Scholar] [CrossRef] [Green Version]
- Carey, M.A.; Bradbury, J.A.; Rebolloso, Y.D.; Graves, J.P.; Zeldin, D.C.; Germolec, D.R. Pharmacologic Inhibition of COX-1 and COX-2 in Influenza A Viral Infection in Mice. PLoS ONE 2010, 5, e11610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of COVID-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Wang, G.; Zhao, R.; An, Z.; Liu, L. Eicosanoid Metabolomic Profile of Remdesivir Treatment in Rat Plasma by High-Performance Liquid Chromatography Mass Spectrometry. Front. Pharmacol. 2021, 12, 74750. [Google Scholar] [CrossRef]
- Gupta, A.; Kalantar-Zadeh, K.; Reddy, S.T. Ramatroban as a Novel Immunotherapy for COVID-19. Mol. Genet. Med. 2020, 14. [Google Scholar] [CrossRef] [PubMed]
- Andreakos, E.; Papadaki, M.; Serhan, C.N. Dexamethasone, Pro-resolving Lipid Mediators and Resolution of Inflammation in COVID-19. Allergy 2021, 76, 626–628. [Google Scholar] [CrossRef]
- The RECOVERY Collaborative Group. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Pyrillou, K.; Chairakaki, A.-D.; Tamvakopoulos, C.; Andreakos, E. Dexamethasone Induces Ω3-Derived Immunoresolvents Driving Resolution of Allergic Airway Inflammation. J. Allergy Clin. Immunol. 2018, 142, 691–695.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manickam, M.; Meenakshisundaram, S.; Pillaiyar, T. Activating Endogenous Resolution Pathways by Soluble Epoxide Hydrolase Inhibitors for the Management of COVID-19. Arch. Pharm. 2022, 355, e2100367. [Google Scholar] [CrossRef] [PubMed]
- Hammock, B.D.; Wang, W.; Gilligan, M.M.; Panigrahy, D. Eicosanoids. Am. J. Pathol. 2020, 190, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, H.B.; Pereira, A.M.; Melo, T.; Paiva, A.; Domingues, M.R. Lipidomics in Autoimmune Diseases with Main Focus on Systemic Lupus Erythematosus. J. Pharm. Biomed. Anal. 2019, 174, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Godson, C.; Mitchell, S.; Harvey, K.; Petasis, N.A.; Hogg, N.; Brady, H.R. Cutting Edge: Lipoxins Rapidly Stimulate Nonphlogistic Phagocytosis of Apoptotic Neutrophils by Monocyte-Derived Macrophages. J. Immunol. 2000, 164, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Croxtall, J.D.; Choudhury, Q.; Tokumoto, H.; Flower, R.J. Lipocortin-1 and the Control of Arachidonic Acid Release in Cell Signalling. Biochem. Pharmacol. 1995, 50, 465–474. [Google Scholar] [CrossRef]
- Kobayashi, A.; Ito, A.; Shirakawa, I.; Tamura, A.; Tomono, S.; Shindou, H.; Hedde, P.N.; Tanaka, M.; Tsuboi, N.; Ishimoto, T.; et al. Dietary Supplementation with Eicosapentaenoic Acid Inhibits Plasma Cell Differentiation and Attenuates Lupus Autoimmunity. Front. Immunol. 2021, 12, 650856. [Google Scholar] [CrossRef]
- Baumgart, D.C.; Carding, S.R. Inflammatory Bowel Disease: Cause and Immunobiology. Lancet 2007, 369, 1627–1640. [Google Scholar] [CrossRef]
- Baumgart, D.C.; Sandborn, W.J. Inflammatory Bowel Disease: Clinical Aspects and Established and Evolving Therapies. Lancet 2007, 369, 1641–1657. [Google Scholar] [CrossRef]
- Bazarganipour, S.; Hausmann, J.; Oertel, S.; El-Hindi, K.; Brachtendorf, S.; Blumenstein, I.; Kubesch, A.; Sprinzl, K.; Birod, K.; Hahnefeld, L.; et al. The Lipid Status in Patients with Ulcerative Colitis: Sphingolipids Are Disease-Dependent Regulated. J. Clin. Med. 2019, 8, 971. [Google Scholar] [CrossRef] [Green Version]
- Pearl, D.S.; Masoodi, M.; Eiden, M.; Brümmer, J.; Gullick, D.; Mckeever, T.M.; Whittaker, M.A.; Nitch-Smith, H.; Brown, J.F.; Shute, J.K.; et al. Altered Colonic Mucosal Availability of N-3 and n-6 Polyunsaturated Fatty Acids in Ulcerative Colitis and the Relationship to Disease Activity. J. Crohn’s Colitis 2014, 8, 70–79. [Google Scholar] [CrossRef]
- Marcon, R.; Bento, A.F.; Dutra, R.C.; Bicca, M.A.; Leite, D.F.P.; Calixto, J.B. Maresin 1, a Proresolving Lipid Mediator Derived from Omega-3 Polyunsaturated Fatty Acids, Exerts Protective Actions in Murine Models of Colitis. J. Immunol. 2013, 191, 4288–4298. [Google Scholar] [CrossRef] [Green Version]
- Masterson, J.C.; McNamee, E.N.; Fillon, S.A.; Hosford, L.; Harris, R.; Fernando, S.D.; Jedlicka, P.; Iwamoto, R.; Jacobsen, E.; Protheroe, C.; et al. Eosinophil-Mediated Signalling Attenuates Inflammatory Responses in Experimental Colitis. Gut 2015, 64, 1236–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gewirtz, A.T.; Collier-Hyams, L.S.; Young, A.N.; Kucharzik, T.; Guilford, W.J.; Parkinson, J.F.; Williams, I.R.; Neish, A.S.; Madara, J.L. Lipoxin A4 Analogs Attenuate Induction of Intestinal Epithelial Proinflammatory Gene Expression and Reduce the Severity of Dextran Sodium Sulfate-Induced Colitis. J. Immunol. 2002, 168, 5260–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, T.; Yoshida, M.; Arita, M.; Nishitani, Y.; Nishiumi, S.; Masuda, A.; Mizuno, S.; Takagawa, T.; Morita, Y.; Kutsumi, H.; et al. Resolvin E1, an Endogenous Lipid Mediator Derived from Eicosapentaenoic Acid, Prevents Dextran Sulfate Sodium–Induced Colitis. Inflamm. Bowel Dis. 2010, 16, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobbetti, T.; Dalli, J.; Colas, R.A.; Federici Canova, D.; Aursnes, M.; Bonnet, D.; Alric, L.; Vergnolle, N.; Deraison, C.; Hansen, T.V.; et al. Protectin D1 N-3 DPA and Resolvin D5 n-3 DPA Are Effectors of Intestinal Protection. Proc. Natl. Acad. Sci. USA 2017, 114, 3963–3968. [Google Scholar] [CrossRef] [Green Version]
- Mangino, M.J.; Brounts, L.; Harms, B.; Heise, C. Lipoxin Biosynthesis in Inflammatory Bowel Disease. Prostaglandins Other Lipid Mediat. 2006, 79, 84–92. [Google Scholar] [CrossRef]
- Köhnke, T.; Gomolka, B.; Bilal, S.; Zhou, X.; Sun, Y.; Rothe, M.; Baumgart, D.C.; Weylandt, K.H. Acetylsalicylic Acid Reduces the Severity of Dextran Sodium Sulfate-Induced Colitis and Increases the Formation of Anti-Inflammatory Lipid Mediators. BioMed Res. Int. 2013, 2013, 748160. [Google Scholar] [CrossRef] [PubMed]
- Vong, L.; Ferraz, J.G.P.; Dufton, N.; Panaccione, R.; Beck, P.L.; Sherman, P.M.; Perretti, M.; Wallace, J.L. Up-Regulation of Annexin-A1 and Lipoxin A4 in Individuals with Ulcerative Colitis May Promote Mucosal Homeostasis. PLoS ONE 2012, 7, e39244. [Google Scholar] [CrossRef]
- Ağış, E.R.; Savaş, B.; Melli, M. Impact of Colonic Mucosal Lipoxin A4 Synthesis Capacity on Healing in Rats with Dextran Sodium Sulfate-Induced Colitis. Prostaglandins Other Lipid Mediat. 2015, 121, 63–69. [Google Scholar] [CrossRef]
- Chiu, C.-Y.; Gomolka, B.; Dierkes, C.; Huang, N.R.; Schroeder, M.; Purschke, M.; Manstein, D.; Dangi, B.; Weylandt, K.H. Omega-6 Docosapentaenoic Acid-Derived Resolvins and 17-Hydroxydocosahexaenoic Acid Modulate Macrophage Function and Alleviate Experimental Colitis. Inflamm. Res. 2012, 61, 967–976. [Google Scholar] [CrossRef]
- Bento, A.F.; Claudino, R.F.; Dutra, R.C.; Marcon, R.; Calixto, J.B. Omega-3 Fatty Acid-Derived Mediators 17(R)-Hydroxy Docosahexaenoic Acid, Aspirin-Triggered Resolvin D1 and Resolvin D2 Prevent Experimental Colitis in Mice. J. Immunol. 2011, 187, 1957–1969. [Google Scholar] [CrossRef] [Green Version]
- Arita, M.; Yoshida, M.; Hong, S.; Tjonahen, E.; Glickman, J.N.; Petasis, N.A.; Blumberg, R.S.; Serhan, C.N. Resolvin E1, an Endogenous Lipid Mediator Derived from Omega-3 Eicosapentaenoic Acid, Protects against 2,4,6-Trinitrobenzene Sulfonic Acid-Induced Colitis. Proc. Natl. Acad. Sci. USA 2005, 102, 7671–7676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marion-Letellier, R.; Savoye, G.; Beck, P.L.; Panaccione, R.; Ghosh, S. Polyunsaturated Fatty Acids in Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2013, 19, 650–661. [Google Scholar] [CrossRef]
- Ueda, Y.; Kawakami, Y.; Kunii, D.; Okada, H.; Azuma, M.; Le, D.S.N.T.; Yamamoto, S. Elevated Concentrations of Linoleic Acid in Erythrocyte Membrane Phospholipids in Patients with Inflammatory Bowel Disease. Nutr. Res. 2008, 28, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Wiese, D.M.; Horst, S.N.; Brown, C.T.; Allaman, M.M.; Hodges, M.E.; Slaughter, J.C.; Druce, J.P.; Beaulieu, D.B.; Schwartz, D.A.; Wilson, K.T.; et al. Serum Fatty Acids Are Correlated with Inflammatory Cytokines in Ulcerative Colitis. PLoS ONE 2016, 11, e0156387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyata, N.; Roman, R.J. Role of 20-Hydroxyeicosatetraenoic Acid (20-HETE) in Vascular System. J. Smooth Muscle Res. 2005, 41, 175–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graber, M.N.; Alfonso, A.; Gill, D.L. Recovery of Ca2+ Pools and Growth in Ca2+ Pool-Depleted Cells Is Mediated by Specific Epoxyeicosatrienoic Acids Derived from Arachidonic Acid. J. Biol. Chem. 1997, 272, 29546–29553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trebble, T.M.; Arden, N.K.; Wootton, S.A.; Calder, P.C.; Mullee, M.A.; Fine, D.R.; Stroud, M.A. Fish Oil and Antioxidants Alter the Composition and Function of Circulating Mononuclear Cells in Crohn Disease. Am. J. Clin. Nutr. 2004, 80, 1137–1144. [Google Scholar] [CrossRef]

| Disease/Injury | Pharmacologic Agent | Mass Detection | Lipid Source | Findings | References |
|---|---|---|---|---|---|
| CVD | None | Orbitrap | Human Plasma | The ratio of CE to free cholesterol is lowered in CVD patients | [61] |
| None | LC-triple quadrupole, Shotgun MS | Patient tissue sections/extracts, plasma | Polyunsaturated CE are largely enriched in carotid plaques | [63] | |
| None | LC- triple quadrupole | Mouse heart tissue, plasma | Upregulated drastically in tissue after myocardial injury to activate cellular regeneration and inhibit pro-inflammatory cytokines | [73,74] | |
| RvD1 | LC- triple quadrupole | Mouse heart tissue, plasma | RvD1 supplementation restored RvD1: LTB4 ratios and reduced markers of oxidative stress and necrosis. | [88] | |
| AT-NPD1 | LC-triple quadrupole | Mouse Brain Tissue | AT-NPD1 administration 3 h post-stroke improved neurologic scores up to 7 days after stroke, reduced radiographic measures of cerebral edema, and decreased histopathologic infarct volume | [93] | |
| Statins | LC- triple quadrupole | Human Serum | Promotes synthesis of pro-resolving SPMs | [105] | |
| Stroke | None | LC- triple quadrupole | Human endarterectomy plaques, mouse artery lesions | SPMs, such as RvD1 is significantly decreased in vulnerable plaque regions | [88] |
| None | LC-quadrupole Orbitrap | Human Serum | FA levels vary greatly post-stroke compared to healthy controls. Phosphoglyceride profiles are distinctly different between small artery and large artery occlusions. | [89] | |
| None | LC-Shotgun MS | Mouse cerebral cortex | PC levels are reduced within first 7 days post-stroke, suppresses microglial secretion of pro-inflammatory cytokines. LPC levels are increased within first 7 days post-stroke, which suppresses neuronal viability. | [90] | |
| None | LC-Orbitrap | Human serum, Rat and Mouse cerebral cortex | plasma ceramide and sphingomyelin are increased 24 h post-stroke | [91] | |
| Healthy | Low-dose Aspirin | LC-triple quadrupole | Human Serum | Global decrease in linoleic acid and oxylipid metabolites produced by cytochrome P450. | [94] |
| Alzheimer’s | DHA | LC-triple quadrupole | Human Neural Cell Line and Human Brain Tissues | DHA and NPD1 were reduced in Alzheimer’s. DHA stimulated NPD1 biosynthesis and attenuates amyloid-β secretion in cells. | [125] |
| DHA | GC-MS | Human Cerebrospinal fluid | DHA increased 28%, EPA increased 43%, and EPA was 3-fold higher in non-APOE4 patients. | [149] | |
| None | LC-triple quadrupole | Mouse Brain Tissue | Increased AA and metabolites indicating activation of group IV isoform of phospholipase A2. | [151] | |
| None | LC-triple quadrupole | Human Brain Tissue | Increased 4HNE-GSH conjugates in patient temporal cortex, frontal cortex, and substantia innominata. | [129] | |
| Parkinson’s | Levodopa | GC-MS | Human Plasma and Urine | Plasma F2-isoprostanes, HETEs, hydroxycholesterols, 7-ketocholesterol, and neuroprostanes were elevated in patients. Total HETEs was negatively correlated with levodopa intake. | [140] |
| COPD | None | LC-triple quadrupole | Human Serum | Identified potential biomarkers and achieved high sensitivity and specificity using a combination of 4 individual lipids and 10 lipid ratios. Increased C16:1 CE and TAG (54:6) 22:6/16:0/16:0. Decreased PI (36:6) and PI (44:6) | [178] |
| SARS-CoV-2 | None | LC- triple quadrupole | Human Serum | Moderate and severe infections can be separated by changes in PUFAs. Changes corresponded with decreased ALOX12 and COX2, specifically loss of RvE3 and prostaglandins, and increased ALOX5 and cytochrome p450 activity in severe patients. | [179] |
| None | LC-triple quadrupole | Human Bronchoalveolar Lavage | Found increased PGE2, TXB2, 12-HHTrE, and LTB4 which correlated with cytokines | [185] | |
| None | LC-triple quadrupole | Human Bronchoalveolar Lavage | Severe patients requiring intubation had elevated eicosanoids including thromboxane, prostaglandins, and leukotrienes (LTB4 and LTE4). SPMs increased including lipoxin A4 and D-series resolvins. | [174] | |
| Remdesivir | LC- triple quadrupole | Rat Plasma | DHA, RvD2, 5-HEPE, and 5-HETE levels decreased following remdesivir while TXB2 increased and PGE2 positively correlated with remdesivir metabolite concentrations in plasma. | [193] | |
| Lung Injury | Bleomycin | LC-Orbitrap | Mouse Plasma and Bronchoalveolar Lavage | Lung samples but not plasma samples revealed changed lipid profiles. Prostaglandins increased by day 2 and ALOX5/15 DHA metabolites increased by day 7 post-injury. | [187] |
| Radiation | MALDI-TOF/TOF, Orbitrap, FT-ICR MS | Rhesus Macaques Lung Tissue | Regardless of pathological findings, lipidomics identified decreased pulmonary surfactant lipids, particularly PC (14:0/16:0), PC (16:0/16:0), PC (16:0/16:1). Tissues with high histological inflammation showed high concentrations of PUFA containing PCs. | [167] | |
| Allergic Airway | Dexamethasone | LC- triple quadrupole | Mouse Serum and Lung Tissue | Ovalbumin sensitization model induced upregulation of PGD2, PGE2, and DHA-derived protectins and 17-HDHA in lung samples but not serum. Dexamethasone activated the 17-HDHA pathway and increased protectins within 6 h. | [197] |
| SLE | None | GC-MS, LC-triple quadrupole | Human Plasma | TG increased, PE and PC decreased. Plasmenyl-PE has an antioxidant role which supports normal cellular functions and hence could be used as a potential biomarker. | [200] |
| None | GC-MS, LC-TOF/MS | Human Plasma | Lower levels of oleic acid and EPA were associated with higher disease severity in SLE patients. | [203] | |
| IBD | None | LC- triple quadrupole and LC-QTOF/MS | Human Plasma | Lowered EPA levels | [206] |
| None | GC-MS | Mucosal membrane | AA, DPA and DHA increased | [207] | |
| None | LC equipped with diode array detector | Colonic Mucosa | Inhibited SPM production leading to worsening of colitis in mice. | [210] | |
| Dextran Sulfate Sodium | LC-triple quadrupole | Mouse Colon Tissue | Better outcome predicted with higher levels of NPD1, NPD1-isomer ((10S,17S)-DiHDoHE) reduced neutrophil infiltration and inflammatory markers | [214] | |
| None | GC-MS and LC-MS | Human Plasma | PUFA and eicosanoids derived from AA corresponded to increased colonic inflammatory cytokines found in the bowel inflammation process | [220,221,222] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahluwalia, K.; Ebright, B.; Chow, K.; Dave, P.; Mead, A.; Poblete, R.; Louie, S.G.; Asante, I. Lipidomics in Understanding Pathophysiology and Pharmacologic Effects in Inflammatory Diseases: Considerations for Drug Development. Metabolites 2022, 12, 333. https://doi.org/10.3390/metabo12040333
Ahluwalia K, Ebright B, Chow K, Dave P, Mead A, Poblete R, Louie SG, Asante I. Lipidomics in Understanding Pathophysiology and Pharmacologic Effects in Inflammatory Diseases: Considerations for Drug Development. Metabolites. 2022; 12(4):333. https://doi.org/10.3390/metabo12040333
Chicago/Turabian StyleAhluwalia, Kabir, Brandon Ebright, Kingsley Chow, Priyal Dave, Andrew Mead, Roy Poblete, Stan G. Louie, and Isaac Asante. 2022. "Lipidomics in Understanding Pathophysiology and Pharmacologic Effects in Inflammatory Diseases: Considerations for Drug Development" Metabolites 12, no. 4: 333. https://doi.org/10.3390/metabo12040333