
Selective Autophagy as a Potential Therapeutic Target in Age-Associated Pathologies



Abstract
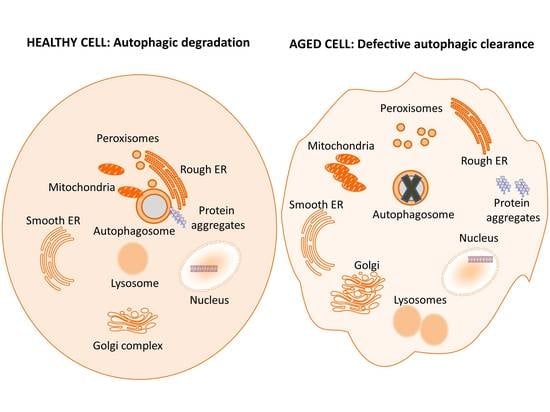
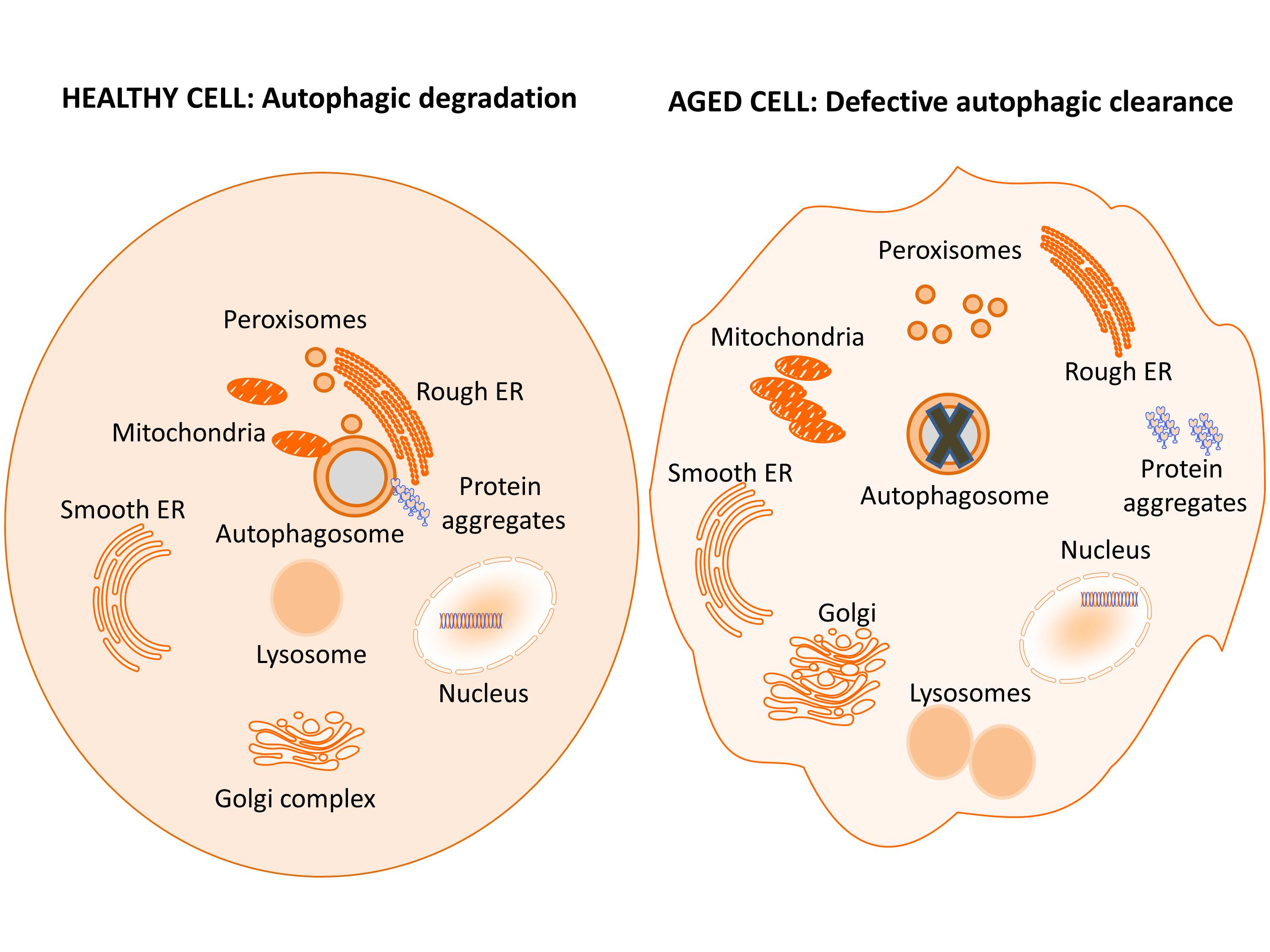
:
1. Introduction
2. Main Text
2.1. General Autophagy
2.2. Selective Autophagy
2.2.1. Mitophagy and Aging
2.2.2. Aggrephagy and Age-Related Disease
2.2.3. Pexophagy and Aging
2.2.4. Nucleophagy and Nuclear Alterations in Aging
2.2.5. Other Types of Selective Autophagy in Age-Related Disease

{kind=link}
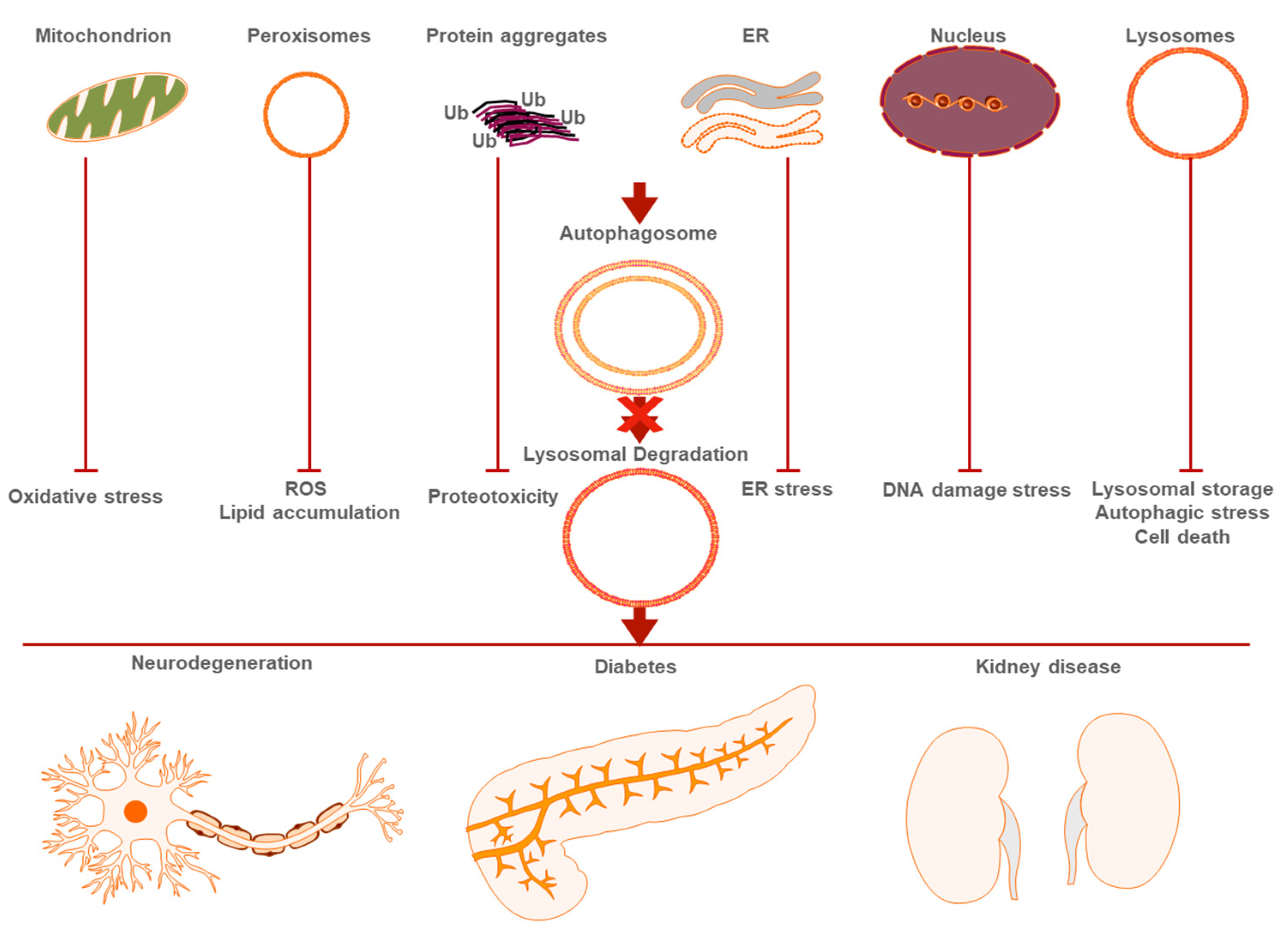{kind=link}
| Autophagy Inducer | Type of Autophagy | Disease/Aging | Organism | Reference |
|---|---|---|---|---|
| Pifithrin | Mitophagy | Diabetes, PD | Mouse | [35] |
| Metformin | Mitophagy | Diabetes, PD | Mouse, Human | [36] |
| Urolithin A | Mitophagy | Aging | Mouse | [40] |
| SB-742457 | Aggrephagy | AD | Human | [46] |
| Lu AE58-54 | Aggrephagy | AD | Human | [47] |
| AVN-211 | Aggrephagy | AD | Mouse | [48] |
| rAAV | Aggrephagy | AD | Mouse, Rat | [50] |
| Resveratrol | Mitophagy, Aggrephagy Lysophagy | AD, Diabetic kidney disease | Human | [38,51,99] |
| Rapamycin | Mitophagy, Aggrephagy | AD | Mouse | [34,51] |
| Lithium | Aggrephagy | AD | Human | [52] |
| Nicotinamide | Aggrephagy | AD | Human | [54] |
| DMF | Aggrephagy | PD | Mouse | [58] |
| Curcumin | Aggrephagy, Lysophagy | PD, Diabetes | Mouse, Rat | [57,104] |
| Beclin-1 and TFEB overexpression | Aggrephagy | PD | Mouse | [55,56] |
| AUTEN-67 | Aggrephagy | AD, PD, Huntington’s disease | Mouse | [53,61] |
| Trehalose | Aggrephagy | PD | Mouse | [62] |
| Calpastatin | Aggrephagy | PD | Mouse | [63] |
| PPI-1011 | Pexophagy | PD | Mouse | [78] |
| Catalase | Lysophagy | Diabetic kidney disease | Human | [100] |
| Tubastatin A | Lysophagy | Diabetic kidney disease | Rat | [101] |
| Torin 1 | Lysophagy | Diabetes | Mouse | [102] |
| Tocopherol | Lysophagy | Diabetes | Rat | [103] |
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dikic, I. Proteasomal and autophagic degradation systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Papandreou, M.-E.; Tavernarakis, N. Tavernarakis, Nucleophagy mediators and mechanisms. Prog. Mol. Biol. Transl. Sci. 2020, 172, 1–14. [Google Scholar]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Nascimbeni, A.C.; Giordano, F.; Dupont, N.; Grasso, D.; Vaccaro, M.I.; Codogno, P.; Morel, E. ER-plasma membrane contact sites contribute to autophagosome biogenesis by regulation of local PI3P synthesis. EMBO J. 2017, 36, 2018–2033. [Google Scholar] [CrossRef]
- Karanasios, E.; Stapleton, E.; Manifava, M.; Kaizuka, T.; Mizushima, N.; Walker, S.; Ktistakis, N.T. Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J. Cell Sci. 2013, 126, 5224–5238. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.-I.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef] [Green Version]
- Tsuboyama, K.; Koyama-Honda, I.; Sakamaki, Y.; Koike, M.; Morishita, H.; Mizushima, N. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science 2016, 354, 1036–1041. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [Green Version]
- Lamark, T.; Kirkin, V.; Dikic, I.; Johansen, T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009, 8, 1986–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rui, Y.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Harper, J.W.; Ordureau, A.; Heo, J.-M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Youle, R.J. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. 2018, 16, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pryde, K.R.; Smith, H.L.; Chau, K.; Schapira, A. PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J. Cell Biol. 2016, 213, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.W.; Yang, W.Y. Omegasome-proximal PtdIns(4,5)P2 couples F-actin mediated mitoaggregate disassembly with autophagosome formation during mitophagy. Nat. Commun. 2019, 10, 969. [Google Scholar] [CrossRef]
- Quinsay, M.N.; Thomas, R.L.; Lee, Y.; Gustafsson, B. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 2010, 6, 855–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Esteban-Martínez, L.; Sierra-Filardi, E.; McGreal, R.S.; Salazar-Roa, M.; Mariño, G.; Seco, E.; Durand, S.; Enot, D.; Graña, O.; Malumbres, M.; et al. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J. 2017, 36, 1688–1706. [Google Scholar] [CrossRef]
- Ding, W.X.; Ni, H.M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 2010, 285, 27879–27890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palikaras, K.; Daskalaki, I.; Markaki, M.; Tavernarakis, N. Mitophagy and age-related pathologies: Development of new therapeutics by targeting mitochondrial turnover. Pharmacol. Ther. 2017, 178, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 1332–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koentjoro, B.; Park, J.-S.; Sue, C.M. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci. Rep. 2017, 7, 44373. [Google Scholar] [CrossRef]
- Yang, Y.; Gehrke, S.; Imai, Y.; Huang, Z.; Ouyang, Y.; Wang, J.; Yang, L.; Beal, M.F.; Vogel, O.H.; Lu, B. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc. Natl. Acad. Sci. USA 2006, 103, 10793–10798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matheoud, D.; Sugiura, A.; Bellemare-Pelletier, A.; Laplante, A.; Rondeau, C.; Chemali, M.; Fazel, A.; Bergeron, J.J.; Trudeau, L.-E.; Burelle, Y.; et al. Parkinson’s disease-related proteins PINK1 and parkin repress mitochondrial antigen presentation. Cell 2016, 166, 314–327. [Google Scholar] [CrossRef] [Green Version]
- Davis, C.-H.O.; Kim, K.-Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, M.; Saito, T.; Zhai, P.; Oka, S.-I.; Mizushima, W.; Nakamura, M.; Ikeda, S.; Shirakabe, A.; Sadoshima, J. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ. Res. 2019, 124, 1360–1371. [Google Scholar] [CrossRef]
- Ahier, A.; Dai, C.-Y.; Kirmes, I.; Cummins, N.; Hung, G.C.C.; Götz, J.; Zuryn, S. PINK1 and parkin shape the organism-wide distribution of a deleterious mitochondrial genome. Cell Rep. 2021, 35, 109203. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Yanos, M.E.; Kayser, E.-B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 2013, 342, 1524–1528. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Ariyoshi, M.; Okawa, Y.; Kaimoto, S.; Uchihashi, M.; Fukai, K. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic beta-cell function in diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 3116–3121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.M.; Lee, W.K.; Lee, Y.-H.; Kang, E.S.; Cha, B.-S.; Lee, B.-W. Metformin restores parkin-mediated mitophagy, suppressed by cytosolic p53. Int. J. Mol. Sci. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Bhansali, S.; Bhansali, A.; Dutta, P.; Walia, R.; Dhawan, V. Metformin upregulates mitophagy in patients with T2DM: A randomized placebo-controlled study. J. Cell. Mol. Med. 2020, 24, 2832–2846. [Google Scholar] [CrossRef]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos, N.D.; Wells, G.; Campanella, N.D.G.M. The pharmacological regulation of cellular mitophagy. Nat. Chem. Biol. 2017, 13, 136–146. [Google Scholar] [CrossRef]
- Andreux, P.A.; Blanco-Bose, W.; Ryu, D.; Burdet, F.; Ibberson, M.; Aebischer, P.; Auwerx, J.; Singh, A.; Rinsch, C. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat. Metab. 2019, 1, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Malampati, S.; Song, J.-X.; Tong, B.C.-K.; Nalluri, A.; Yang, C.-B.; Wang, Z.; Sreenivasmurthy, S.G.; Zhu, Z.; Liu, J.; Su, C.; et al. Targeting aggrephagy for the treatment of alzheimer’s disease. Cells 2020, 9, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan-Or, Z.; Dion, P.A.; Rouleau, G.A. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 2015, 11, 1443–1457. [Google Scholar] [CrossRef]
- Moors, T.; Paciotti, S.; Chiasserini, D.; Calabresi, P.; Parnetti, L.; Beccari, T.; van de Berg, W.D. Lysosomal dysfunction and alpha-synuclein aggregation in parkinson’s disease: Diagnostic links. Mov. Disord. 2016, 31, 791–801. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- De, S.; Wirthensohn, D.C.; Flagmeier, P.; Hughes, C.; Aprile, F.A.; Ruggeri, F.S. Different soluble aggregates of Abeta42 can give rise to cellular toxicity through different mechanisms. Nat. Commun. 2019, 10, 1541. [Google Scholar] [CrossRef] [Green Version]
- Maher-Edwards, G.; Zvartau-Hind, M.; Hunter, A.; Gold, M.; Hopton, G.; Jacobs, G.; Davy, M.; Williams, P. Double-blind, controlled phase II study of a 5-HT6 receptor antagonist, SB-742457, in Alzheimer’s disease. Curr. Alzheimer Res. 2010, 7, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.; Windfeld, K.; Colding-Jorgensen, E. Safety and efficacy of idalopirdine, a 5-HT6 receptor antagonist, in patients with moderate Alzheimer’s disease (LADDER): A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2014, 13, 1092–1099. [Google Scholar] [CrossRef]
- Ivachtchenko, A.V.; Lavrovsky, Y.; Ivanenkov, Y.A. AVN-211, Novel and highly selective 5-HT6 receptor small molecule antagonist, for the treatment of alzheimer’s disease. Mol. Pharm. 2016, 13, 945–963. [Google Scholar] [CrossRef]
- Wani, A.; Gupta, M.; Ahmad, M.; Shah, A.M.; Ahsan, A.U.; Qazi, P.H. Alborixin clears amyloid-beta by inducing autophagy through PTEN-mediated inhibition of the AKT pathway. Autophagy 2019, 15, 1810–1828. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Giliberto, L.; Zhao, H.; Chandakkar, P.; Wu, Q.; Simon, J.E. Autophagy is involved in oral rAAV/Abeta vaccine-induced Abeta clearance in APP/PS1 transgenic mice. Neurosci. Bull. 2015, 31, 491–504. [Google Scholar]
- Forlenza, O.V.; De-Paula VD, J.R.; Diniz BS, O. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J. Biol. Chem. 2010, 285, 9100–9113. [Google Scholar]
- Forlenza, O.V.; De-Paula, V.J.; Diniz, B.S. Neuroprotective effects of lithium: Implications for the treatment of Alzheimer’s disease and related neurodegenerative disorders. ACS Chem. Neurosci. 2014, 5, 443–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Pitta, M.; Jiang, H.; Lee, J.H.; Zhang, G.; Chen, X. AUTEN-67, an autophagy-enhancing drug candidate with potent antiaging and neuroprotective effects. Autophagy 2016, 12, 273–286. [Google Scholar]
- Liu, D.; Pitta, M.; Jiang, H.; Lee, J.H.; Zhang, G.; Chen, X. Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: Evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol. Aging 2013, 34, 1564–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, B.; Potkar, R.; Trejo, M.; Rockenstein, E.; Patrick, C.; Gindi, R. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 2009, 29, 13578–13588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Bjorklund, A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc. Natl. Acad. Sci. USA 2013, 110, E1817–E1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, H.F.; Shen, L. The multiple pharmaceutical potential of curcumin in Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2014, 13, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; García-Yagüe, A.J.; Scannevin, R.H.; Casarejos, M.J.; Kügler, S.; Rábano, A.; Cuadrado, A. Repurposing the NRF2 activator dimethyl fumarate as therapy against synucleinopathy in parkinson’s disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef] [Green Version]
- Iwata, A.; Christianson, J.C.; Bucci, M.; Ellerby, L.M.; Nukina, N.; Forno, L.S.; Kopito, R.R. Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc. Natl. Acad. Sci. USA 2005, 102, 13135–13140. [Google Scholar] [CrossRef] [Green Version]
- Pircs, K.; Petri, R.; Madsen, S.; Brattås, P.L.; Vuono, R.; Ottosson, D.R. Huntingtin aggregation impairs autophagy, leading to Argonaute-2 accumulation and global MicroRNA dysregulation. Cell Rep. 2018, 24, 1397–1406. [Google Scholar] [CrossRef] [Green Version]
- Billes, V.; Kovács, T.; Hotzi, B.; Manzéger, A.; Tagscherer, K.; Komlós, M.; Tarnóci, A.; Pádár, Z.; Erdős, A.; Bjelik, A.; et al. AUTEN-67 (Autophagy Enhancer-67) Hampers the Progression of Neurodegenerative Symptoms in a Drosophila model of Huntington’s Disease. J. Huntingt. Dis. 2016, 5, 133–147. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Rubinsztein, D.C. Huntington’s disease: Degradation of mutant huntingtin by autophagy. FEBS J. 2008, 275, 4263–4270. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Garcia-Arencibia, M.; Imarisio, S.; O’Sullivan, N.; Ricketts, T.H.; Kent, B.; Rao, M.; Lam, W.; Green-Thompson, Z.W.; Nixon, R.A.; et al. Calpain inhibition mediates autophagy-dependent protection against polyglutamine toxicity. Cell Death Differ. 2015, 22, 433–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarow, P.B.; De Duve, C. A fatty acyl-CoA oxidizing system in rat liver peroxisomes; enhancement by clofibrate, a hypolipidemic drug. Proc. Natl. Acad. Sci. USA 1976, 73, 2043–2046. [Google Scholar] [CrossRef] [Green Version]
- Germain, K.; Kim, P.K. Pexophagy: A model for selective autophagy. Int. J. Mol. Sci. 2020, 21, 578. [Google Scholar] [CrossRef] [Green Version]
- Kunau, W.H.; Hartig, A. Peroxisome biogenesis in Saccharomyces cerevisiae. Antonie Van Leeuwenhoek 1992, 62, 63–78. [Google Scholar] [CrossRef]
- Tuttle, D.L.; Lewin, A.; Dunn, W.A. Selective autophagy of peroxisomes in methylotrophic yeasts. Eur. J. Cell Biol. 1993, 60, 283–290. [Google Scholar] [PubMed]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, S.-I.; Abe, K.; Tatemichi, Y.; Fujiki, Y. The membrane peroxin PEX3 induces peroxisome-ubiquitination-linked pexophagy. Autophagy 2014, 10, 1549–1564. [Google Scholar] [CrossRef] [Green Version]
- Deosaran, E.; Larsen, K.B.; Hua, R.; Sargent, G.; Wang, Y.; Kim, S.; Lamark, T.; Jauregui, M.; Law, K.; Lippincott-Schwartz, J.; et al. NBR1 acts as an autophagy receptor for peroxisomes. J. Cell Sci. 2012, 126, 939–952. [Google Scholar] [CrossRef] [Green Version]
- Sargent, G.; Van Zutphen, T.; Shatseva, T.; Zhang, L.; Di Giovanni, V.; Bandsma, R.; Kim, P.K. PEX2 is the E3 ubiquitin ligase required for pexophagy during starvation. J. Cell Biol. 2016, 214, 677–690. [Google Scholar] [CrossRef]
- Legakis, J.E.; Koepke, J.I.; Jedeszko, C.; Barlaskar, F.; Terlecky, L.J.; Edwards, H.J.; Walton, P.A.; Terlecky, S.R. Peroxisome Senescence in Human Fibroblasts. Mol. Biol. Cell 2002, 13, 4243–4255. [Google Scholar] [CrossRef] [PubMed]
- Cipolla, C.M.; Lodhi, I.J. Peroxisomal dysfunction in age-related diseases. Trends Endocrinol. Metab. 2017, 28, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O.; Van Veldhoven, P.P. Aging, Age-Related Diseases and Peroxisomes. Mol. Biol. Cell 2013, 69, 45–65. [Google Scholar]
- Ivashchenko, O.; Van Veldhoven, P.P.; Brees, C.; Ho, Y.-S.; Terlecky, S.R.; Fransen, M. Intraperoxisomal redox balance in mammalian cells: Oxidative stress and interorganellar cross-talk. Mol. Biol. Cell 2011, 22, 1440–1451. [Google Scholar] [CrossRef] [PubMed]
- Narayan, V.; Ly, T.; Pourkarimi, E.; Murillo, A.B.; Gartner, A.; Lamond, A.; Kenyon, C. Deep Proteome Analysis Identifies Age-Related Processes in C. elegans. Cell Syst. 2016, 3, 144–159. [Google Scholar] [CrossRef] [Green Version]
- Doria, M.; Maugest, L.; Moreau, T.; Lizard, G.; Vejux, A. Contribution of cholesterol and oxysterols to the pathophysiology of Parkinson’s disease. Free Radic. Biol. Med. 2016, 101, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Miville-Godbout, E.; Bourque, M.; Morissette, M.; Al-Sweidi, S.; Smith, T.; Mochizuki, A.; Senanayake, V.; Jayasinghe, D.; Wang, L.; Goodenowe, D.; et al. Plasmalogen Augmentation Reverses Striatal Dopamine Loss in MPTP Mice. PLoS ONE 2016, 11, e0151020. [Google Scholar] [CrossRef] [PubMed]
- Jo, D.S.; Park, N.Y.; Cho, D.-H. Peroxisome quality control and dysregulated lipid metabolism in neurodegenerative diseases. Exp. Mol. Med. 2020, 52, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, M.E.; Tavernarakis, N. Nucleophagy: From homeostasis to disease. Cell Death Differ. 2019, 26, 630–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostofa, M.G.; Rahman, M.A.; Koike, N.; Yeasmin, A.M.; Islam, N.; Waliullah, T.M. CLIP and cohibin separate rDNA from nucleolar proteins destined for degradation by nucleophagy. J. Cell Biol. 2018, 217, 2675–2690. [Google Scholar] [CrossRef] [Green Version]
- Zlotorynski, E. Ageing: Live longer with small nucleoli. Nat. Rev. Mol. Cell Biol. 2017, 18, 651. [Google Scholar] [CrossRef]
- Tiku, V.; Jain, C.; Raz, Y.; Nakamura, S.; Heestand, B.; Liu, W. Small nucleoli are a cellular hallmark of longevity. Nat. Commun. 2017, 8, 16083. [Google Scholar] [CrossRef]
- Golden, T.R.; Beckman, K.B.; Lee, A.H.J.; Dudek, N.; Hubbard, A.; Samper, E.; Melov, S. Dramatic age-related changes in nuclear and genome copy number in the nematode Caenorhabditis elegans. Aging Cell 2007, 6, 179–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, Z.; Xu, C.; Donahue, G.; Shimi, T.; Pan, J.-A.; Zhu, J.; Ivanov, A.; Capell, B.C.; Drake, A.M.; Shah, P.P.; et al. Autophagy mediates degradation of nuclear lamina. Nature 2015, 527, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Cenni, V.; Capanni, C.; Mattioli, E.; Schena, E.; Squarzoni, S.; Bacalini, M.G.; Garagnani, P.; Salvioli, S.; Franceschi, C.; Lattanzi, G. Lamin A involvement in ageing processes. Ageing Res. Rev. 2020, 62, 101073. [Google Scholar] [CrossRef]
- Lan, Y.Y.; Londoño, D.; Bouley, R.; Rooney, M.S.; Hacohen, N. Dnase2a deficiency uncovers lysosomal clearance of damaged nuclear DNA via autophagy. Cell Rep. 2014, 9, 180–192. [Google Scholar] [CrossRef] [Green Version]
- Baron, O.; Boudi, A.; Dias, C.; Schilling, M.; Nölle, A.; Vizcay-Barrena, G. Stall in canonical autophagy-lysosome pathways prompts nucleophagy-based nuclear breakdown in neurodegeneration. Curr. Biol. 2017, 27, 3626–3642.e6. [Google Scholar] [CrossRef] [Green Version]
- Hubner, C.A.; Dikic, I. ER-phagy and human diseases. Cell Death Differ. 2020, 27, 833–842. [Google Scholar] [CrossRef]
- Kurth, I.; Pamminger, T.; Hennings, J.C.; Soehendra, D.; Huebner, A.K.; Rotthier, A.; Baets, J.; Senderek, J.; Topaloglu, H.; Farrell, S.A.; et al. Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat. Genet. 2009, 41, 1179–1181. [Google Scholar] [CrossRef] [PubMed]
- Khaminets, A.; Heinrich, T.; Mari, M.; Grumati, P.; Huebner, A.K.; Akutsu, M.; Liebmann, L.; Stolz, A.; Nietzsche, S.; Koch, N.; et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015, 522, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.; Kim, Y.; Lee, C. A strong synergistic epistasis between FAM134B and TNFRSF19 on the susceptibility to vascular dementia. Psychiatr. Genet. 2011, 21, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.; Krus, K.L.; Kaushik, S.; Dang, D.; Chopra, R.; Qi, L.; Shakkottai, V.G.; Cuervo, A.M.; Lieberman, A.P. Coordinate regulation of mutant NPC1 degradation by selective ER autophagy and MARCH6-dependent ERAD. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kornak, U.; Mademan, I.; Schinke, M.; Voigt, M.; Krawitz, P.; Hecht, J.; Barvencik, F.; Schinke, T.; Gießelmann, S.; Beil, F.T.; et al. Sensory neuropathy with bone destruction due to a mutation in the membrane-shaping atlastin GTPase 3. Brain 2014, 137, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Ge, Y.; Sharoar, G.; He, W.; Xiang, R.; Zhang, Z.; Hu, X.; Yan, R. Impact of RTN3 Deficiency on Expression of BACE1 and Amyloid Deposition. J. Neurosci. 2014, 34, 13954–13962. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.-Y.; Yao, R.-Q.; Li, Y.-X.; Zhao, P.-Y.; Ren, C.; Du, X.-H.; Yao, Y.-M. Lysosomal quality control of cell fate: A novel therapeutic target for human diseases. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Coffey, E.; Beckel, J.; Laties, A.; Mitchell, C. Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer’s disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience 2014, 263, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Qiao, L.; Hamamichi, S.; Caldwell, K.; Caldwell, G.; Yacoubian, T.A.; Wilson, S.; Xie, Z.-L.; Speake, L.D.; Parks, R.; Crabtree, D.; et al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol. Brain 2008, 1, 17. [Google Scholar] [CrossRef] [Green Version]
- Chougala, M.B.; Bhaskar, J.J.; Rajan, M.; Salimath, P.V. Effect of curcumin and quercetin on lysosomal enzyme activities in streptozotocin-induced diabetic rats. Clin. Nutr. 2012, 31, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Gan, Y.; Huang, W.F.; Wu, H.-L.; Zhang, X.; Zheng, H.J.; Liu, H.-F. Lysosome restoration to activate podocyte autophagy: A new therapeutic strategy for diabetic kidney disease. Cell Death Dis. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brijmohan, A.S.; Batchu, S.N.; Majumder, S.; Alghamdi, T.A.; Thieme, K.; McGaugh, S.; Liu, Y.; Advani, S.L.; Bowskill, B.B.; Kabir, M.G.; et al. HDAC6 Inhibition Promotes Transcription Factor EB Activation and Is Protective in Experimental Kidney Disease. Front. Pharmacol. 2018, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhang, W.; Jia, Q.; Feng, Z.; Guo, J.; Han, X.; Liu, Y.; Shang, H.; Wang, Y.; Liu, W.J. Advanced glycation end-products suppress autophagic flux in podocytes by activating mammalian target of rapamycin and inhibiting nuclear translocation of transcription factor EB. J. Pathol. 2018, 245, 235–248. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Chen, Y.; Tan, X.; Zhang, L.; Zhang, H.; Li, Z. High dose vitamin E attenuates diabetic nephropathy via alleviation of autophagic stress. Front. Physiol. 2018, 9, 1939. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.J.; Shen, T.T.; Chen, R.H.; Wu, H.-L.; Wang, Y.J.; Deng, J.K.; Chen, Q.H.; Pan, Q.; Fu, C.-M.H.; Tao, J.-L.; et al. Autophagy-Lysosome Pathway in Renal Tubular Epithelial Cells Is Disrupted by Advanced Glycation End Products in Diabetic Nephropathy. J. Biol. Chem. 2015, 290, 20499–20510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.J.; Zhang, X.; Guo, J.; Zhang, W.; Ai, S.; Zhang, F.; Wang, Y.; Liu, W.J. Lysosomal dysfunction-induced autophagic stress in diabetic kidney disease. J. Cell Mol. Med. 2020, 24, 8276–8290. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papandreou, M.-E.; Tavernarakis, N. Selective Autophagy as a Potential Therapeutic Target in Age-Associated Pathologies. Metabolites 2021, 11, 588. https://doi.org/10.3390/metabo11090588
Papandreou M-E, Tavernarakis N. Selective Autophagy as a Potential Therapeutic Target in Age-Associated Pathologies. Metabolites. 2021; 11(9):588. https://doi.org/10.3390/metabo11090588
Chicago/Turabian StylePapandreou, Margarita-Elena, and Nektarios Tavernarakis. 2021. "Selective Autophagy as a Potential Therapeutic Target in Age-Associated Pathologies" Metabolites 11, no. 9: 588. https://doi.org/10.3390/metabo11090588