
Phytochemical Characterization of Pterocephalus frutescens with In-Silico Evaluation as Chemotherapeutic Medicine and Oral Pharmacokinetics Prediction Study

 , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Experimental Design
2.1. Plant Material
2.2. Extraction and Isolation of Pure Compounds
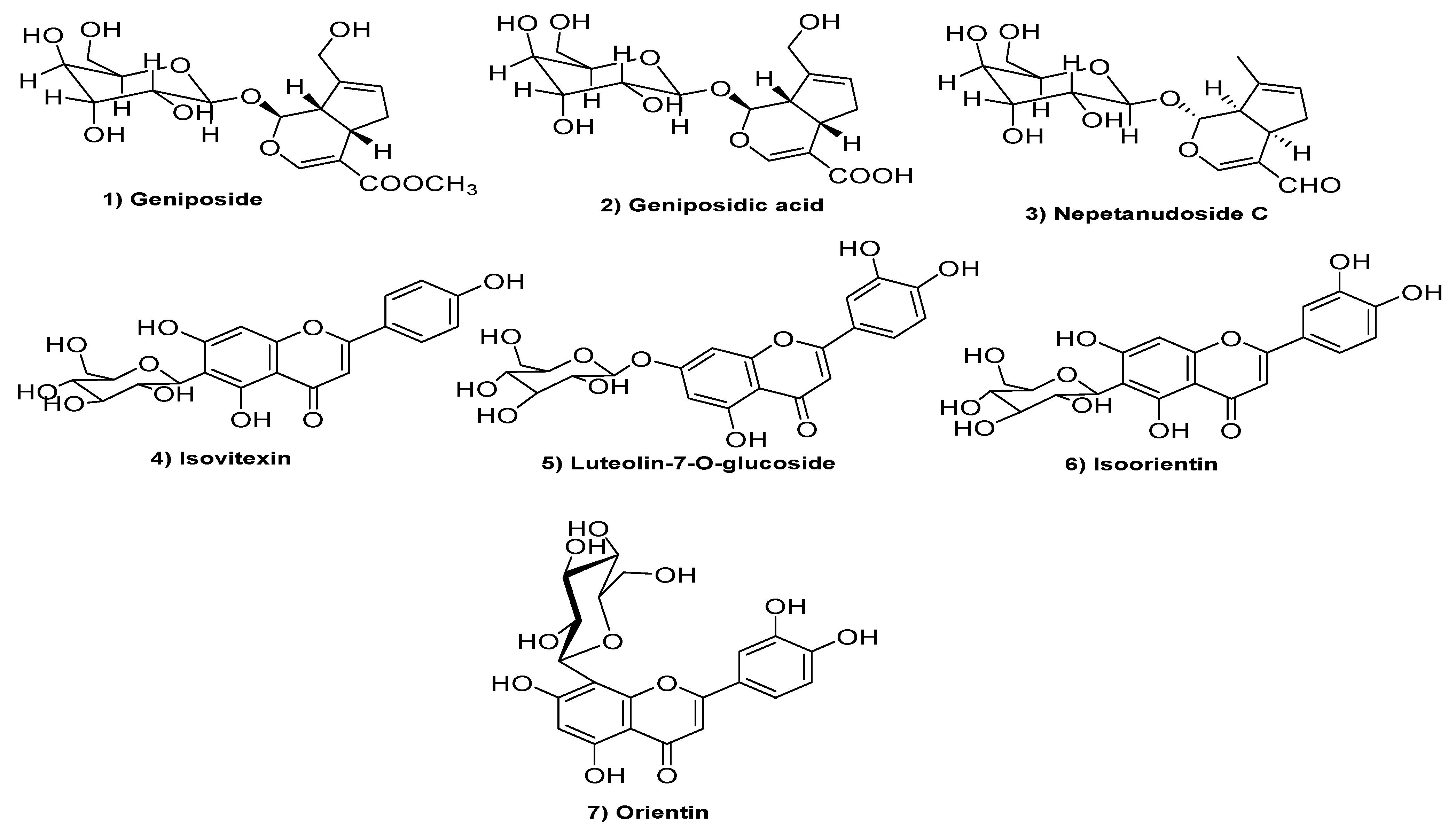
2.3. Structure Elucidation of Isolated Pure Compounds
2.4. Molecular Docking Analysis
2.4.1. Preparation of the Investigated Compounds
2.4.2. Preparation of Helicobacter Specific Virulence Proteins
3. Results and Discussion
3.1. Strutural Elucidation of Isolated Pure Compounds
3.2. Results of In Silico Studies
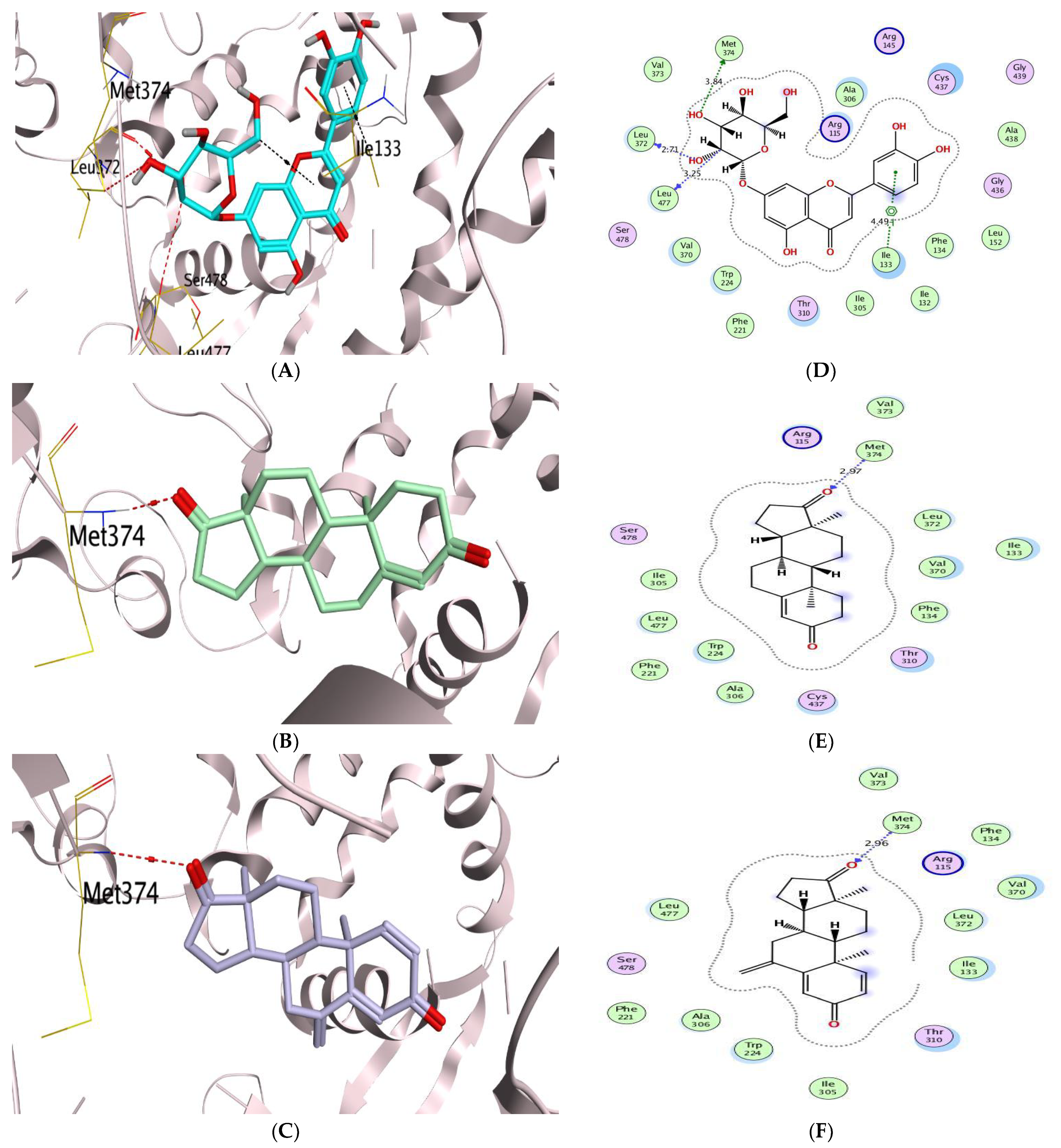
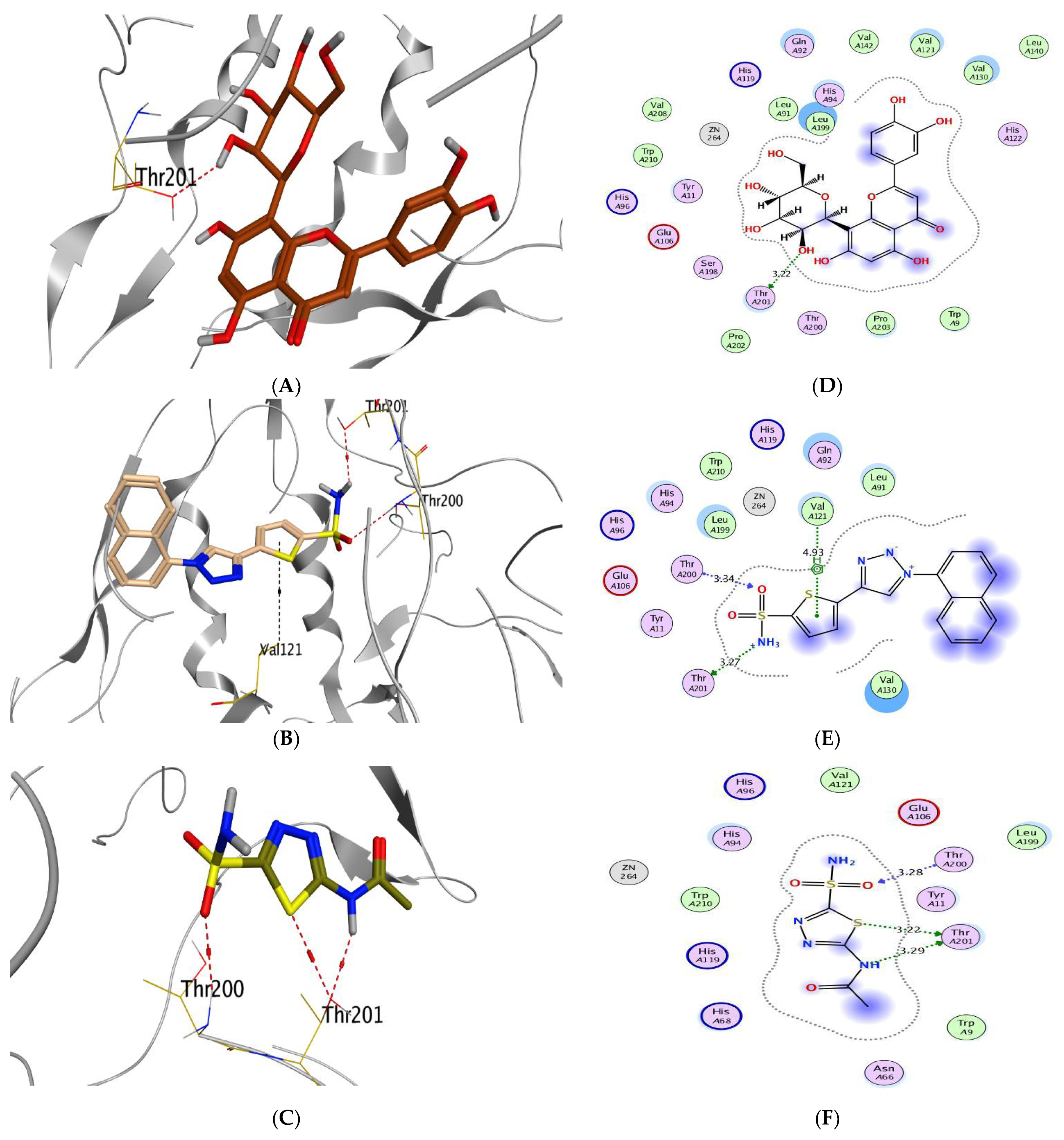
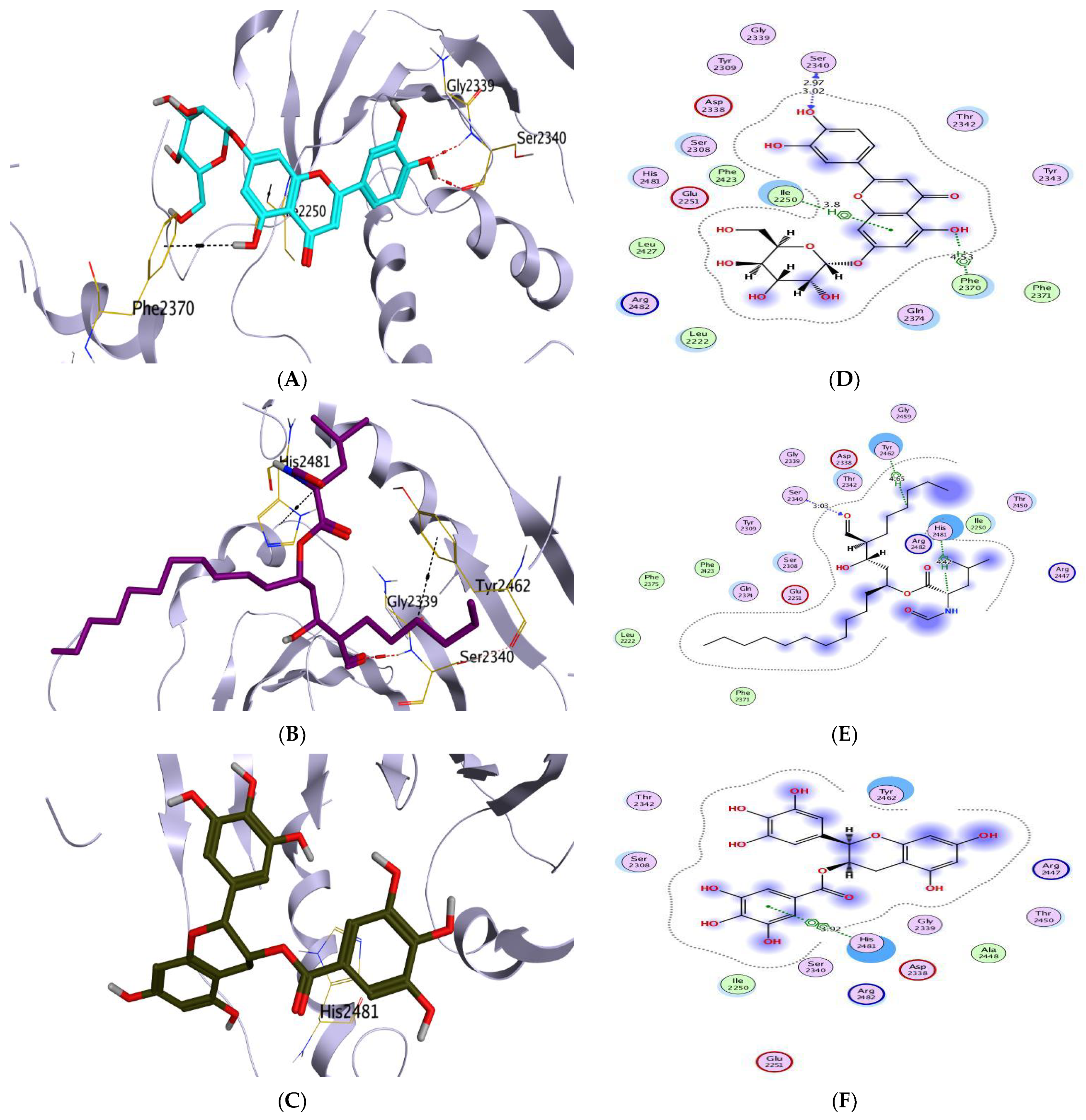
Molecular Docking Analysis
3.3. Prediction of Physicochemical and Pharmacokinetic Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kletter, C.; Kriechbaum, M. Tibetan Medicinal Plants; CRC Press: Boca Raton, FL, USA, 2001. [Google Scholar]
- El-Hela, A.A.; Hegazy, M.M.; Bakr, M.S.A.; Abbass, H.S. Profiling of antiviral and antioxidant phytochemicals of Pterocephalus frutescens hochist. using high-resolution ultra-performance liquid chromatography/quadrupole time-of-flight mass spectrometer. Pharmacogn. Mag. 2020, 16, 592–599. [Google Scholar]
- Guo, C.; Wu, Y.; Zhu, Y.; Wang, Y.; Tian, L.; Lu, Y.; Han, C.; Zhu, G. In vitro and in vivo antitumor effects of n-butanol extracts of Pterocephalus hookeri on Hep3B cancer cell. Evid. Based Complement. Altern. Med. 2015, 2015, 159132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahem, A.; Ahmed, A.; Maha, M.; Soltan, A.; Hussein, A.; Zaki, A. Anti-hepatotoxic effect of Pterocephalus sanctus growing in Egypt. JASRM 2008, 3, 83–87. [Google Scholar]
- Vahedi, H.; Nasrabadi, M.; Lari, J.; Halimi, M. Volatile constituents and antimicrobial activities of Pterocephalus canus. J. Med. Plants Res. 2011, 5, 5646–5648. [Google Scholar]
- Wu, Y.-C.; Guo, C.-X.; Zhu, Y.-Z.; Li, Y.-M.; Guo, F.-J.; Zhu, G.-F. Four new bis-iridoids isolated from the traditional Tibetan herb Pterocephalus hookeri. Fitoterapia 2014, 98, 104–109. [Google Scholar] [CrossRef]
- Ahmed, F.A.; Shahat, A.A. Flavonoid C-Glycosides from Pterocephalus Sanctus Growing in Egypt. Nat. Prod. Commun. 2006, 1, 1934578X0600100605. [Google Scholar] [CrossRef]
- Gülcemal, D.; Masullo, M.; Alankuş-Çalışkan, O.; Karayıldırım, T.; Şenol, S.G.; Piacente, S.; Bedir, E. Monoterpenoid glucoindole alkaloids and iridoids from Pterocephalus pinardii. Magn. Reson. Chem. 2010, 48, 239–243. [Google Scholar] [CrossRef]
- Gülcemal, D.; Bedir, E.; Karayıldırım, T.; Milena, M.; Piacente, S.; Şenol, S.; Alankuş-Çalışkan, Ö. Constituents of Pterocephalus pinardii Boiss. Planta Med. 2009, 75, PJ103. [Google Scholar] [CrossRef]
- Li, M.-Y.; Deng, H.; Zhao, J.-M.; Dai, D.; Tan, X.-Y. Peroxisome proliferator-activated receptor gamma ligands inhibit cell growth and induce apoptosis in human liver cancer BEL-7402 cells. World J. Gastroenterol. 2003, 9, 1683–1688. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Maloney, D.J.; Hecht, S.M.; Lannigan, D.A. Structural basis for the activity of the RSK-specific inhibitor, SL0101. Bioorganic Med. Chem. 2007, 15, 5018–5034. [Google Scholar] [CrossRef]
- Gordaliza, M. Natural products as leads to anticancer drugs. Clin. Transl. Oncol. 2007, 9, 767–776. [Google Scholar] [CrossRef]
- Ren, W.; Qiao, Z.; Wang, H.; Zhu, L.; Zhang, L. Flavonoids: Promising anticancer agents. Med. Res. Rev. 2003, 23, 519–534. [Google Scholar] [CrossRef] [PubMed]
- El-Hela, A.A.; Hegazy, M.M.; Abbass, H.S.; Ahmed, A.H.; Bakr, M.S.A.; Elkousy, R.H.; Ibrahim, A.E.; El Deeb, S.; Sayed, O.M.; Gad, E.S. Dinebra retroflexa Herbal Phytotherapy: A Simulation Study Based on Bleomycin-Induced Pulmonary Fibrosis Retraction Potential in Swiss Albino Rats. Medicina 2022, 58, 1719. [Google Scholar] [CrossRef]
- Chumsri, S.; Howes, T.; Bao, T.; Sabnis, G.; Brodie, A. Aromatase, aromatase inhibitors, and breast cancer. J. Steroid Biochem. Mol. Biol. 2011, 125, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, L.; Saha, S.T.; Thomas, J.; Kaur, M. Targeting cellular cholesterol for anticancer therapy. FEBS J. 2019, 286, 4192–4208. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.F.; Teixeira, N.; Oliveira, A.; Augusto, T.V.; Correia-da-Silva, G.; Ramos, M.J.; Fernandes, P.A.; Amaral, C. Discovery of a multi-target compound for estrogen receptor-positive (ER+) breast cancer: Involvement of aromatase and ERs. Biochimie 2021, 181, 65–76. [Google Scholar] [CrossRef]
- Sanderson, J.T.; Hordijk, J.; Denison, M.S.; Springsteel, M.F.; Nantz, M.H.; Berg, M.V.D. Induction and Inhibition of Aromatase (CYP19) Activity by Natural and Synthetic Flavonoid Compounds in H295R Human Adrenocortical Carcinoma Cells. Toxicol. Sci. 2004, 82, 70–79. [Google Scholar] [CrossRef] [Green Version]
- Kuhajda, F.P.; Pizer, E.S.; Li, J.N.; Mani, N.S.; Frehywot, G.L.; Townsend, C.A. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc. Natl. Acad. Sci. USA 2000, 97, 3450–3454. [Google Scholar] [CrossRef]
- Fhu, C.W.; Ali, A. Fatty Acid Synthase: An Emerging Target in Cancer. Molecules 2020, 25, 3935. [Google Scholar] [CrossRef]
- Brusselmans, K.; Vrolix, R.; Verhoeven, G.; Swinnen, J.V. Induction of Cancer Cell Apoptosis by Flavonoids Is Associated with Their Ability to Inhibit Fatty Acid Synthase Activity. J. Biol. Chem. 2005, 280, 5636–5645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegazy, M.M.; Afifi, W.M.; Metwaly, A.M.; Radwan, M.M.; Abd-Elraouf, M.; Mehany, A.B.M.; Ahmed, E.; Enany, S.; Ezzeldin, S.; Ibrahim, A.E.; et al. Antitrypanosomal, Antitopoisomerase-I, and Cytotoxic Biological Evaluation of Some African Plants Belonging to Crassulaceae; Chemical Profiling of Extract Using UHPLC/QTOF-MS/MS. Molecules 2022, 27, 8809. [Google Scholar] [CrossRef] [PubMed]
- Bjornsti, M.-A.; Kaufmann, S.H. Topoisomerases and cancer chemotherapy: Recent advances and unanswered questions. F1000Research 2019, 8, 1704. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Lim, K.S.; Blackburn, B.J.; Yun, J.; Putnam, C.W.; Bull, D.A.; Won, Y.-W. The Potential of Topoisomerase Inhibitor-Based Antibody–Drug Conjugates. Pharmaceutics 2022, 14, 1707. [Google Scholar] [PubMed]
- Okura, A.; Arakawa, H.; Oka, H.; Yoshinari, T.; Monden, Y. Effect of genistein on topoisomerase activity and on the growth of [VAL 12]Ha-ras-transformed NIH 3T3 cells. Biochem. Biophys. Res. Commun. 1988, 157, 183–189. [Google Scholar] [CrossRef]
- Hevener, K.; Verstak, T.A.; Lutat, K.E.; Riggsbee, D.L.; Mooney, J.W. Recent developments in topoisomerase-targeted cancer chemotherapy. Acta Pharm. Sin. B 2018, 8, 844–861. [Google Scholar] [CrossRef]
- Pastorekova, S.; Gillies, R.J. The role of carbonic anhydrase IX in cancer development: Links to hypoxia, acidosis, and beyond. Cancer Metastasis Rev. 2019, 38, 65–77. [Google Scholar] [CrossRef]
- Saghafi, T.; Taheri, R.A.; Parkkila, S.; Emameh, R.Z. Phytochemicals as Modulators of Long Non-Coding RNAs and Inhibitors of Cancer-Related Carbonic Anhydrases. Int. J. Mol. Sci. 2019, 20, 2939. [Google Scholar] [CrossRef] [Green Version]
- McDonald, P.C.; Chafe, S.C.; Supuran, C.T.; Dedhar, S. Cancer Therapeutic Targeting of Hypoxia Induced Carbonic Anhydrase IX: From Bench to Bedside. Cancers 2022, 14, 3297. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Elmaaty, A.A.; Darwish, K.M.; Khattab, M.; Elhady, S.S.; Salah, M.; Hamed, M.I.; Al-Karmalawy, A.A.; Saleh, M.M. In a search for potential drug candidates for combating COVID-19: Computational study revealed salvianolic acid B as a potential therapeutic targeting 3CLpro and spike proteins. J. Biomol. Struct. Dyn. 2021, 40, 8866–8893. [Google Scholar] [CrossRef]
- Elmaaty, A.A.; Alnajjar, R.; Hamed, M.I.; Khattab, M.; Khalifa, M.M.; Al-Karmalawy, A.A. Revisiting activity of some glucocorticoids as a potential inhibitor of SARS-CoV-2 main protease: Theoretical study. RSC Adv. 2021, 11, 10027–10042. [Google Scholar] [CrossRef] [PubMed]
- Elmaaty, A.A.; Hamed, M.I.; Ismail, M.I.; Elkaeed, E.B.; Abulkhair, H.S.; Khattab, M.; Al-Karmalawy, A.A. Computational insights on the potential of some NSAIDs for treating COVID-19: Priority set and lead optimization. Molecules 2021, 26, 3772. [Google Scholar] [CrossRef]
- Hamed, M.I.; Darwish, K.M.; Soltane, R.; Chrouda, A.; Mostafa, A.; Shama, N.M.A.; Elhady, S.S.; Abulkhair, H.S.; Khodir, A.E.; Elmaaty, A.A. β-Blockers bearing hydroxyethylamine and hydroxyethylene as potential SARS-CoV-2 Mpro inhibitors: Rational based design, in silico, in vitro, and SAR studies for lead optimization. RSC Adv. 2021, 11, 35536–35558. [Google Scholar] [CrossRef] [PubMed]
- Elmaaty, A.A.; Darwish, K.M.; Chrouda, A.; Boseila, A.A.; Tantawy, M.A.; Elhady, S.S.; Shaik, A.B.; Mustafa, M.; Al-Karmalawy, A.A. In Silico and In Vitro Studies for Benzimidazole Anthelmintics Repurposing as VEGFR-2 Antagonists: Novel Mebendazole-Loaded Mixed Micelles with Enhanced Dissolution and Anticancer Activity. ACS Omega 2021, 7, 875–899. [Google Scholar] [CrossRef]
- Elebeedy, D.; Badawy, I.; Elmaaty, A.A.; Saleh, M.M.; Kandeil, A.; Ghanem, A.; Kutkat, O.; Alnajjar, R.; El Maksoud, A.I.A.; Al-Karmalawy, A.A. In vitro and computational insights revealing the potential inhibitory effect of Tanshinone IIA against influenza A virus. Comput. Biol. Med. 2021, 141, 105149. [Google Scholar] [CrossRef] [PubMed]
- Hammoud, M.M.; Nageeb, A.S.; Morsi, M.A.; Gomaa, E.A.; Elmaaty, A.A.; Al-Karmalawy, A.A. Design, synthesis, biological evaluation, and SAR studies of novel cyclopentaquinoline derivatives as DNA intercalators, topoisomerase II inhibitors, and apoptotic inducers. New J. Chem. 2022, 46, 11422–11436. [Google Scholar] [CrossRef]
- Hammoud, M.M.; Elmaaty, A.A.; Nafie, M.S.; Abdel-Motaal, M.; Mohamed, N.S.; Tantawy, M.A.; Belal, A.; Alnajjar, R.; Eldehna, W.M.; Al-Karmalawy, A.A. Design and synthesis of novel benzoazoninone derivatives as potential CBSIs and apoptotic inducers: In Vitro, in Vivo, molecular docking, molecular dynamics, and SAR studies. Bioorganic Chem. 2022, 127, 105995. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Egbuta, C.; Lo, J. Testosterone complex and non-steroidal ligands of human aromatase. J. Steroid Biochem. Mol. Biol. 2018, 181, 11–19. [Google Scholar] [CrossRef]
- Leitans, J.; Kazaks, A.; Balode, A.; Ivanova, J.; Zalubovskis, R.; Supuran, C.T.; Tars, K. Efficient Expression and Crystallization System of Cancer-Associated Carbonic Anhydrase Isoform IX. J. Med. Chem. 2015, 58, 9004–9009. [Google Scholar] [CrossRef]
- Pemble, C.W.; Johnson, L.C.; Kridel, S.J.; Lowther, W.T. Crystal structure of the thioesterase domain of human fatty acid synthase inhibited by Orlistat. Nat. Struct. Mol. Biol. 2007, 14, 704–709. [Google Scholar] [CrossRef]
- Wu, C.-C.; Li, T.-K.; Farh, L.; Lin, L.-Y.; Lin, T.-S.; Yu, Y.-J.; Yen, T.-J.; Chiang, C.-W.; Chan, N.-L. Structural Basis of Type II Topoisomerase Inhibition by the Anticancer Drug Etoposide. Science 2011, 333, 459–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Güvenalp, Z.; Kilic, N.; Kazaz, C.; Kaya, Y.; Demirezer, L.Ö. Chemical constituents of Galium tortumense. Turk. J. Chem. 2006, 30, 515–523. [Google Scholar]
- Takeda, Y.; Morimoto, Y.; Matsumoto, T.; Honda, G.; Tabata, M.; Fujita, T.; Otsuka, H.; Sezik, E.; Yesilada, E. Nepetanudoside, an iridoid glucoside with an unusual stereostructure from Nepeta nuda ssp. albiflora. J. Nat. Prod. 1995, 58, 1217–1221. [Google Scholar] [CrossRef]
- Huang, D.; Guo, W.; Gao, J.; Chen, J.; Olatunji, J.O. Clinacanthus nutans (Burm. f.) Lindau Ethanol Extract Inhibits Hepatoma in Mice through Upregulation of the Immune Response. Molecules 2015, 20, 17405–17428. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, D.; Siqueira, E.P.; Nunes, Y.R.F.; Cota, B.B. Flavonoids from leaves of Mauritia flexuosa. Rev. Bras. de Farm. 2013, 23, 614–620. [Google Scholar] [CrossRef]
- Chiruvella, K.K.; Mohammed, A.; Dampuri, G.; Ghanta, R.G.; Raghavan, S.C. Phytochemical and Antimicrobial Studies of Methyl Angolensate and Luteolin-7-O-glucoside Isolated from Callus Cultures of Soymida febrifuga. IJBS 2007, 3, 269–278. [Google Scholar]
- El-Demerdash, A.; Al-Karmalawy, A.A.; Abdel-Aziz, T.M.; Elhady, S.S.; Darwish, K.M.; Hassan, A.H.E. Investigating the structure–activity relationship of marine natural polyketides as promising SARS-CoV-2 main protease inhibitors. RSC Adv. 2021, 11, 31339–31363. [Google Scholar] [CrossRef] [PubMed]
- Elebeedy, D.; Elkhatib, W.F.; Kandeil, A.; Ghanem, A.; Kutkat, O.; Alnajjar, R.; Saleh, M.A.; Abd El Maksoud, A.I.; Badawy, I.; Al-Karmalawy, A.A. Anti-SARS-CoV-2 activities of tanshinone IIA, carnosic acid, rosmarinic acid, salvianolic acid, baicalein, and glycyrrhetinic acid between computational and in vitro insights. RSC Adv. 2021, 11, 29267–29286. [Google Scholar] [CrossRef] [PubMed]
- Khattab, M.; Al-Karmalawy, A.A. Computational repurposing of benzimidazole anthelmintic drugs as potential colchicine binding site inhibitors. Futur. Med. Chem. 2021, 13, 1623–1638. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, A.; Al-Karmalawy, A.A.; El Maksoud, A.I.A.; Hanafy, S.M.; Emara, H.A.; Saleh, R.M.; Elshal, M.F. Rumex Vesicarius L. extract improves the efficacy of doxorubicin in triple-negative breast cancer through inhibiting Bcl2, mTOR, JNK1 and augmenting p21 expression. Informatics Med. Unlocked 2022, 29, 100869. [Google Scholar] [CrossRef]
- Asmari, M.; Waqas, M.; Ibrahim, A.E.; Halim, S.A.; Khan, A.; Al-Harrasi, A.; Wätzig, H.; El Deeb, S. Microscale Thermophoresis and Molecular Modelling to Explore the Chelating Drug Transportation in the Milk to Infant. Molecules 2022, 27, 4604. [Google Scholar] [CrossRef] [PubMed]
- El-Naggar, A.M.; Abou-El-Regal, M.M.; El-Metwally, S.A.; Sherbiny, F.F.; Eissa, I.H. Synthesis, characterization and molecular docking studies of thiouracil derivatives as potent thymidylate synthase inhibitors and potential anticancer agents. Mol. Divers. 2017, 21, 967–983. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, E.; El Deeb, S.; Ibrahim, A.E.; Faisal, M.M. Bimodal Release Two-In-One Clonazepam Matrix Lozenge Tablets for Managing Anxiety-Related Disorders: Formulation, Optimization and In Vivo Evaluation. Sci. Pharm. 2022, 90, 43. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. iLOGP: A Simple, Robust, and Efficient Description of n-Octanol/Water Partition Coefficient for Drug Design Using the GB/SA Approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aromatase Enzyme | CA IX | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Comp. No | S Score (Kcal/mol) | RMSD (Å) | Binding Interaction | Distance (Å) | Comp. No | S Score (Kcal/mol) | RMSD (Å) | Binding Interaction | Distance (Å) |
| (1) | −8.15 | 0.70 | LEU372/H-donor MET374/H-acceptor | 2.93 3.10 | (1) | −7.41 | 1.43 | GLU106/H-donor THR200/H-acceptor | 3.14 3.07 |
| (2) | −7.70 | 0.89 | CYS437/H-donor MET374/H-donor LEU477/H-donor | 3.30 4.50 3.26 | (2) | −7.05 | 1.45 | THR201/H-donor THR200/H-acceptor | 2.67 3.23 |
| (3) | −7.22 | 1.49 | LEU372/H-donor LEU477/H-donor MET374/H-acceptor ALA306/H-acceptor | 3.12 2.94 3.19 3.38 | (3) | −7.41 | 1.64 | THR201/H-donor THR200/H-acceptor HIS94/H-pi | 2.88 3.17 3.55 |
| (4) | −8.54 | 2.27 | LEU477/H-donor LEU477/H-donor MET374/H-acceptor ILE133/pi-H | 2.83 2.72 3.37 4.22 | (4) | −7.21 | 1.41 | THR201/H-donor GLN71/H-acceptor ZN264/Metal LEU91/pi-H GLN92/pi-H | 3.18 3.03 2.30 3.98 3.85 |
| (5) | −8.73 | 1.74 | MET374/H-donor LEU372/H-donor LEU477/H-donor ILE133/pi-H | 3.84 2.71 3.25 4.49 | (5) | −8.13 | 1.83 | GLU106/H-donor | 3.08 |
| (6) | −8.52 | 1.27 | MET303/H-donor MET303/H-donor LEU477/H-donor LEU477/H-donor MET374/H-acceptor ILE133/pi-H GLY439/pi-H | 4.24 3.91 3.10 2.66 3.03 3.96 4.36 | (6) | −7.41 | 1.31 | ZN264/Metal | 2.30 |
| (7) | −8.67 | 1.81 | MET374/H-donor MET374/H-acceptor | 3.92 3.03 | (7) | −8.92 | 2.13 | THR201/H-donor | 3.22 |
| Co-crystallized | −8.02 | 1.84 | MET374/H-acceptor | 2.97 | Co-crystallized | −5.91 | 1.72 | THR201/H-donor THR200/H-acceptor VAL121/pi-H | 3.27 3.34 4.93 |
| Reference | −7.76 | 1.07 | MET374/ H-acceptor | 2.96 | Reference | −5.83 | 1.31 | THR201/H-donor THR201/H-donor THR200/H-acceptor | 3.29 3.22 3.28 |
| FAS | Topisomerase II-DNA Complex | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Comp. No | S Score | RMSD | Binding Interaction | Distance | Comp. No | S Score | RMSD | Binding Interaction | Distance |
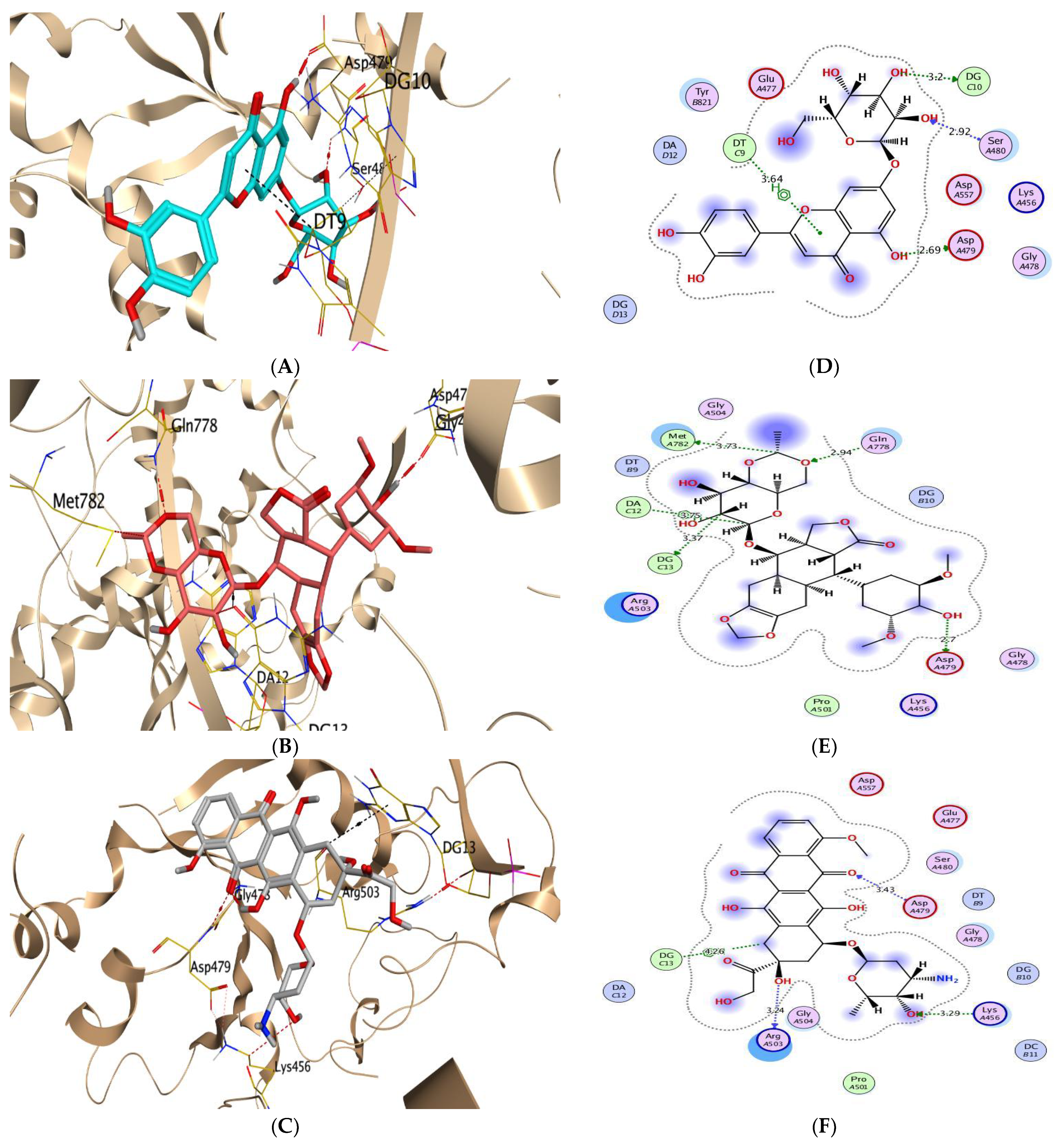
| (1) | −5.86 | 0.93 | SER2340/H-acceptor ARG2482/H-acceptor | 3.12 3.23 | (1) | −7.31 | 1.77 | DG10/H-donor ASP479/H-donor DT9/H-pi | 2.96 2.86 3.78 |
| (2) | −6.05 | 1.84 | ASP2338/H-donor PRO2341/H-donor SER2340/H-acceptor TYR2462/H-pi | 3.01 2.91 3.19 3.54 | (2) | −6.87 | 1.46 | MET782/H-donor ARG503/H-acceptor | 3.16 2.99 |
| (3) | −5.85 | 0.93 | SER2308/H-donor THR2342/H-donor SER2340/H-acceptor HIS2481/H-pi | 2.95 3.23 3.07 3.73 | (3) | −7.16 | 1.44 | ARG503/H-acceptor ASP479/H-acceptor DG13/H-pi DG13/H-pi | 3.01 2.95 4.54 3.68 |
| (4) | −6.22 | 1.09 | THR2342/H-donor SER2340/H-acceptor | 3.05 3.35 | (4) | −7.47 | 1.15 | ASP479/H-donor DT9/pi-H DT9/pi-H | 3.08 4.16 4.21 |
| (5) | −6.82 | 2.26 | SER2340/H-donor SER2340/H-acceptor PHE2370/H-pi ILE2250/pi-H | 2.97 3.02 4.53 3.80 | (5) | −7.99 | 2.15 | ASP479/H-donor DG10/H-donor SER480/H-acceptor DT9/pi-H | 2.69 3.20 2.92 3.64 |
| (6) | −6.49 | 1.10 | SER2340/H-donor | 3.00 | (6) | −7.78 | 1.49 | ASP479/H-donor DT9/H-donor | 2.98 2.97 |
| (7) | −6.40 | 1.61 | HIS2481/H-donor | 3.33 | (7) | −7.69 | 2.18 | ASP479/H-donor DT9/H-donor GLU477/H-donor ARG503/H-acceptor DG13/H-pi DT9/pi-H | 3.00 2.71 3.03 3.11 3.66 4.47 |
| Co-crystallized | −8.09 | 1.39 | SER2340/H-acceptor TYR2462/H-pi HIS2481/H-pi | 3.03 4.65 4.42 | Co-crystallized | −10.52 | 1.42 | ASP479/H-donor MET782/H-donor DG13/H-donor GLN778/H-acceptor DA12/H-pi | 2.70 3.73 3.37 2.94 3.75 |
| Reference | −6.03 | 1.76 | HIS2481/pi-pi | 3.92 | Reference | −8.96 | 1.84 | ARG503/H-donor ASP479/H-acceptor LYS456/H-acceptor DG13/H-pi | 3.24 3.43 3.29 4.26 |
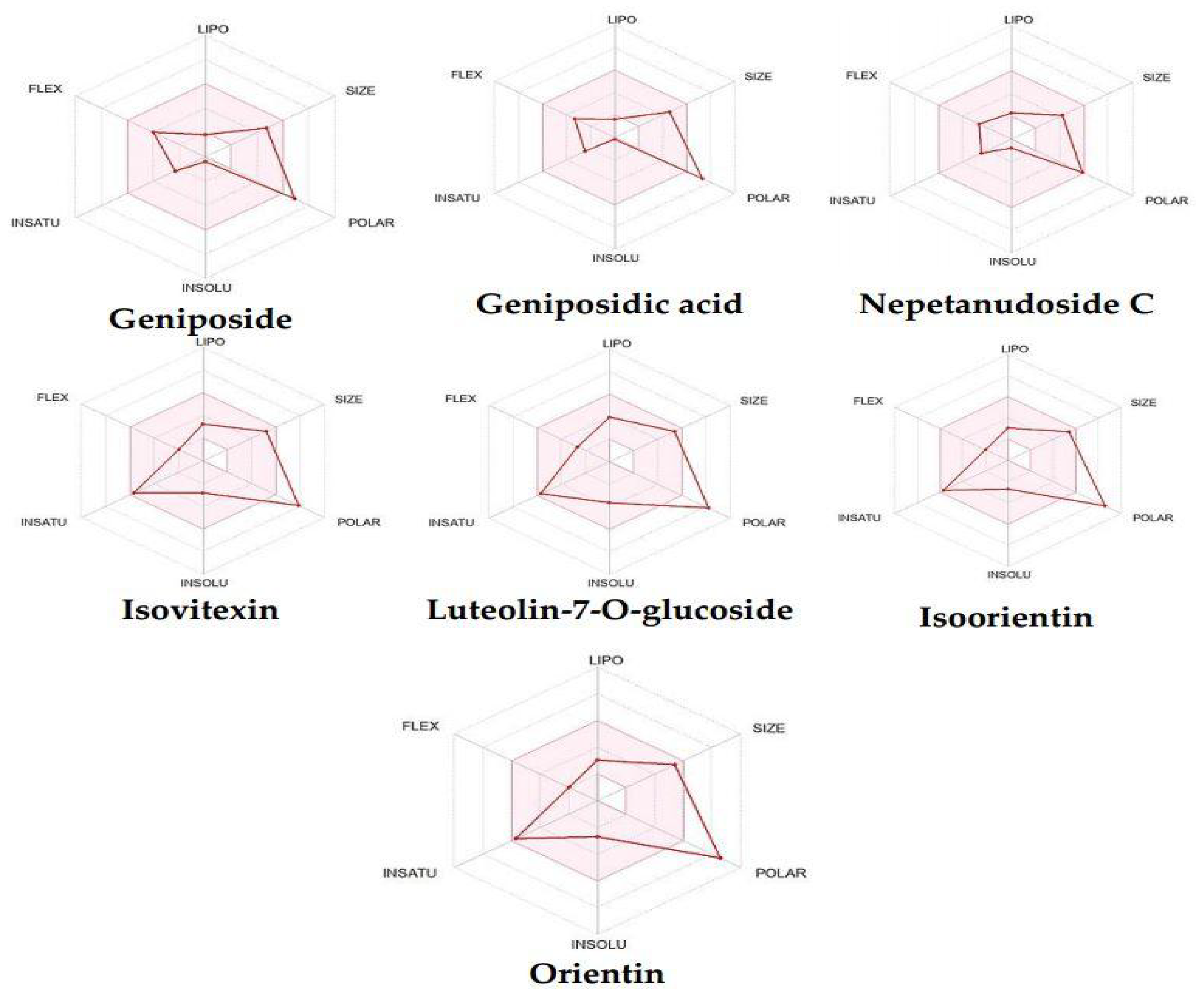
| Parameter | Geniposide | Geniposidic Acid | Nepetanudoside C | Isovitexin | Luteolin-7-O-glucoside | Isoorientin | Orientin |
|---|---|---|---|---|---|---|---|
| Physicochemical properties | |||||||
| Molecular weight | 388.14 | 374.12 | 342.13 | 432.11 | 448.10 | 448.10 | 448.10 |
| LogP | 2.89 | 1.31 | 1.22 | 1.97 | 1.99 | 1.6 | 1 |
| Rotatable bonds | 6 | 5 | 4 | 3 | 4 | 3 | 3 |
| Acceptors | 10 | 10 | 8 | 10 | 11 | 11 | 11 |
| Donors | 5 | 6 | 4 | 7 | 7 | 8 | 8 |
| Surface area square angstrom (Ų) | 155.14 | 166.14 | 125.68 | 181.05 | 190.28 | 201.28 | 201.28 |
| Drug likeness | |||||||
| Lipinski violations | 0 | 1 | 0 | 1 | 2 | 2 | 2 |
| Ghose violations | 1 | 1 | 1 | 0 | 0 | 1 | 1 |
| Veber violations | 1 | 1 | 0 | 1 | 1 | 1 | 1 |
| Pharmacokinetics | |||||||
| GI absorption | Low | Low | High | Low | Low | Low | Low |
| BBB permeant | No | No | No | No | No | No | No |
| CYP1A2 inhibitor | No | No | No | No | No | No | No |
| CYP2C19 inhibitor | No | No | No | No | No | No | No |
| CYP2C9 inhibitor | No | No | No | No | No | No | No |
| CYP2D6 inhibitor | No | No | No | No | No | No | No |
| CYP3A4 inhibitor | No | No | No | No | No | No | No |
| CYP1A2 inhibitor | No | No | No | No | No | No | No |
| CYP2C19 inhibitor | No | No | No | No | No | No | No |
| CYP2C9 inhibitor | No | No | No | No | No | No | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Hela, A.A.; Bakr, M.S.A.; Hegazy, M.M.; Dahab, M.A.; Elmaaty, A.A.; Ibrahim, A.E.; El Deeb, S.; Abbass, H.S. Phytochemical Characterization of Pterocephalus frutescens with In-Silico Evaluation as Chemotherapeutic Medicine and Oral Pharmacokinetics Prediction Study. Sci. Pharm. 2023, 91, 7. https://doi.org/10.3390/scipharm91010007
El-Hela AA, Bakr MSA, Hegazy MM, Dahab MA, Elmaaty AA, Ibrahim AE, El Deeb S, Abbass HS. Phytochemical Characterization of Pterocephalus frutescens with In-Silico Evaluation as Chemotherapeutic Medicine and Oral Pharmacokinetics Prediction Study. Scientia Pharmaceutica. 2023; 91(1):7. https://doi.org/10.3390/scipharm91010007
Chicago/Turabian StyleEl-Hela, Atef A., Marwa S. Abu Bakr, Mostafa M. Hegazy, Mohammed A. Dahab, Ayman Abo Elmaaty, Adel Ehab Ibrahim, Sami El Deeb, and Hatem S. Abbass. 2023. "Phytochemical Characterization of Pterocephalus frutescens with In-Silico Evaluation as Chemotherapeutic Medicine and Oral Pharmacokinetics Prediction Study" Scientia Pharmaceutica 91, no. 1: 7. https://doi.org/10.3390/scipharm91010007