
Mucopolysaccharidosis Type I Presenting with Persistent Neonatal Respiratory Distress: A Case Report
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
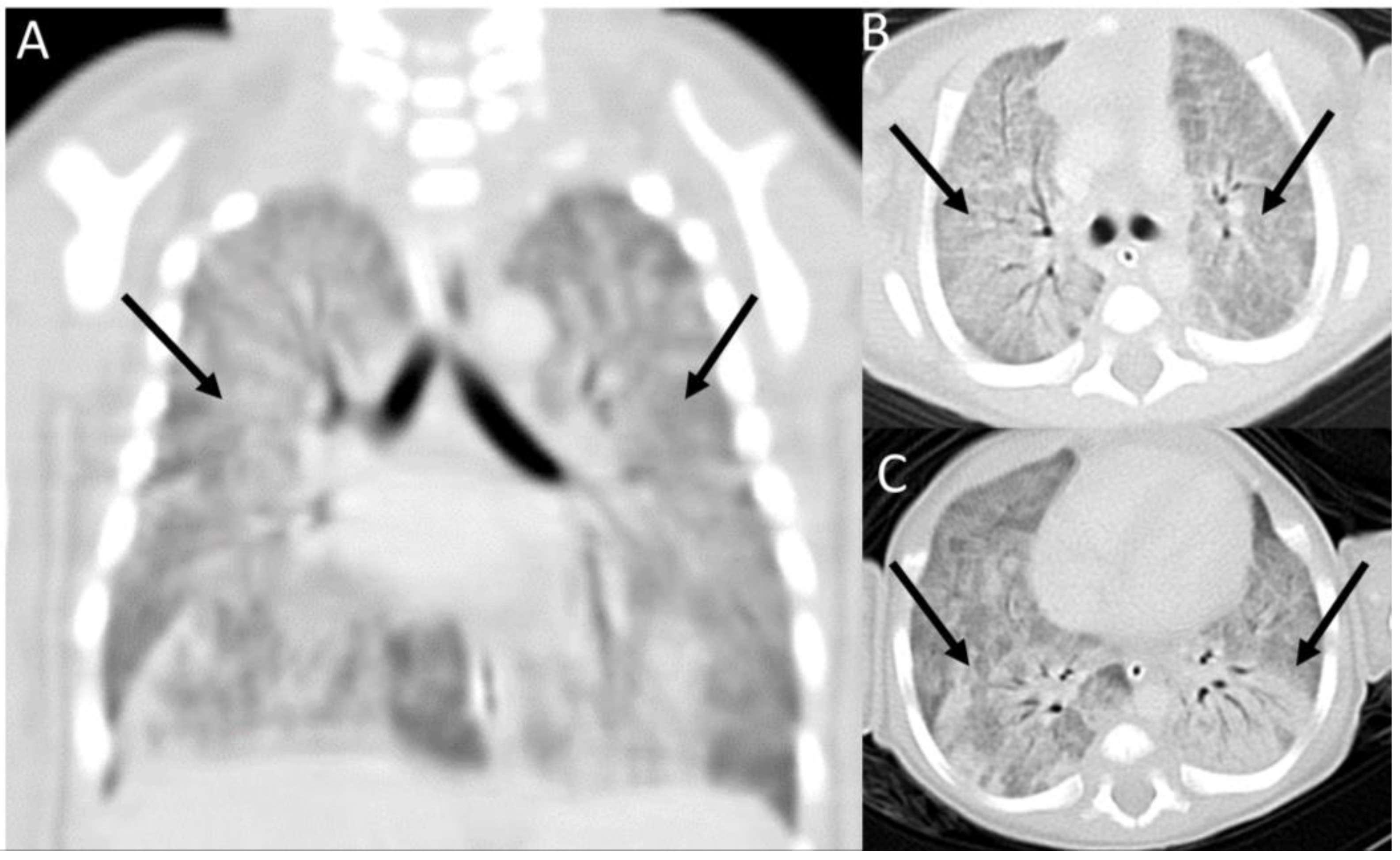
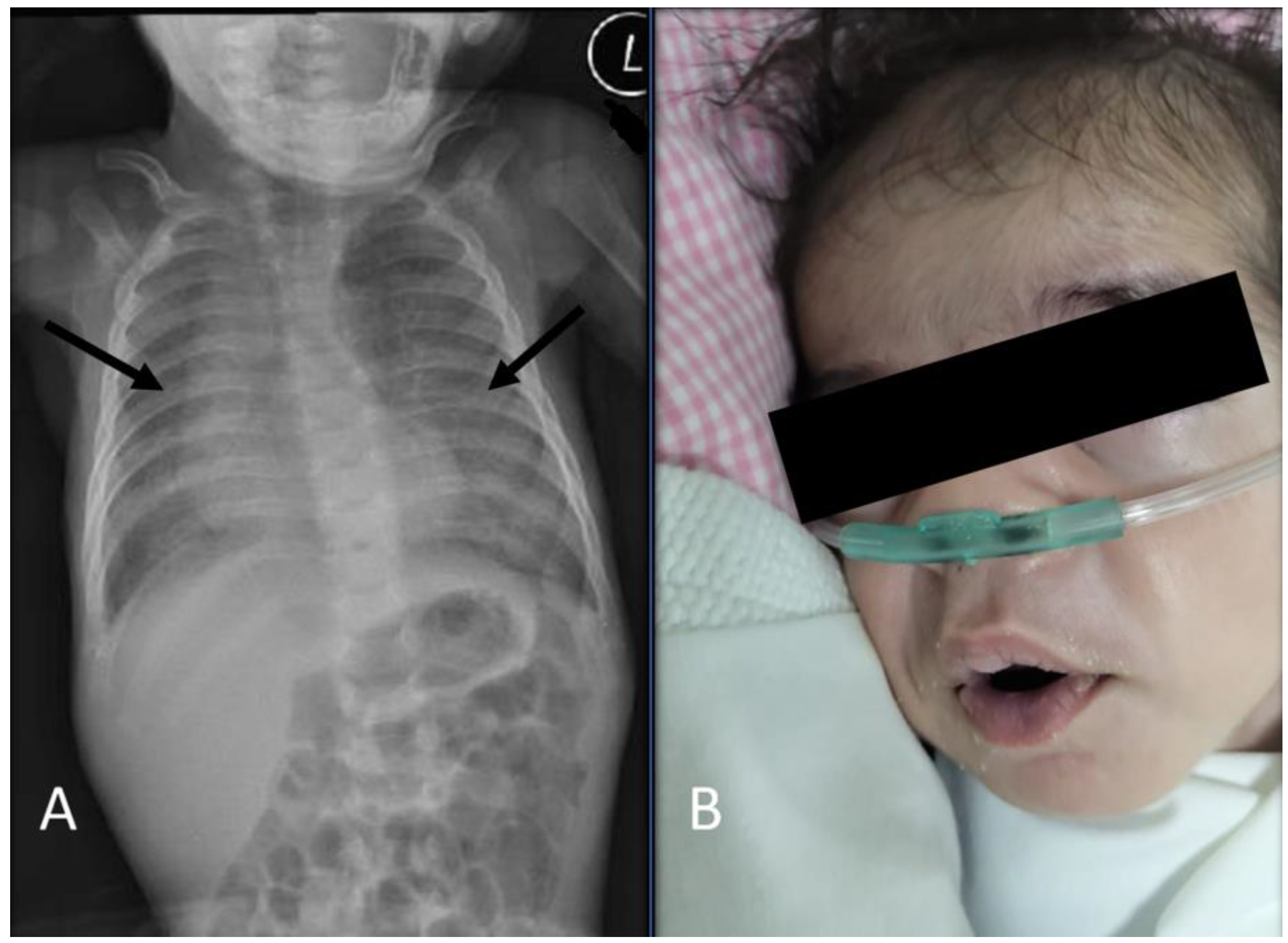
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Węgrzyn, G.; Pierzynowska, K.; Pavone, L.M. Editorial: Molecular Aspects of Mucopolysaccharidoses. Front. Mol. Biosci. 2022, 9, 874267. [Google Scholar] [CrossRef] [PubMed]
- Voskoboeva, E.Y.; Bookina, T.M.; Semyachkina, A.N.; Mikhaylova, S.V.; Vashakmadze, N.D.; Baydakova, G.V.; Kutsev, S.I. Mucopoly-saccharidosis Type I in the Russian Federation and Other Republics of the Former Soviet Union: Molecular Genetic Analysis and Epidemiology. Front. Mol. Biosci. 2022, 8, 783644. [Google Scholar] [CrossRef] [PubMed]
- Campos, D.; Monaga, M. Mucopolysaccharidosis type I: Current knowledge on its pathophysiological mechanisms. Metab. Brain Dis. 2012, 27, 121–129. [Google Scholar] [CrossRef]
- Clarke, L.A.; Giugliani, R.; Guffon, N.; Jones, S.A.; Keenan, H.A.; Munoz-Rojas, M.V.; Okuyama, T.; Viskochil, D.; Whitley, C.B.; Wijburg, F.A.; et al. Genotype-phenotype relationships in mucopolysaccharidosis type I (MPS I): Insights from the International MPS I Registry. Clin. Genet. 2019, 96, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Hampe, C.; Eisengart, J.; Lund, T.; Orchard, P.; Swietlicka, M.; Wesley, J.; McIvor, R. Mucopolysaccharidosis Type I: A Review of the Natural History and Molecular Pathology. Cells 2020, 9, 1838. [Google Scholar] [CrossRef]
- Khan, S.A.; Peracha, H.; Ballhausen, D.; Wiesbauer, A.; Rohrbach, M.; Gautschi, M.; Mason, R.W.; Giugliani, R.; Suzuki, Y.; Orii, K.E.; et al. Epidemiology of mucopolysaccharidoses. Mol. Genet. Metab. 2017, 121, 227–240. [Google Scholar] [CrossRef]
- Martins, A.M.; Dualibi, A.P.; Norato, D.; Takata, E.T.; Santos, E.S.; Valadares, E.R.; Guedes, Z.C.F. Guidelines for the Management of Mucopoly-saccharidosis Type I. J. Pediatr. 2009, 155 (Suppl. S4), S32–S46. [Google Scholar] [CrossRef]
- Moore, D.; Connock, M.J.; Wraith, E.; Lavery, C. The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK. Orphanet J. Rare Dis. 2008, 3, 24. [Google Scholar] [CrossRef]
- Martins, A.M.; Dualibi, A.P.; Norato, D.; Takata, E.T.; Santos, E.S.; Valadares, E.R.; Guedes, Z.C.F. Management of mucopolysaccha-ridosis type I in Saudi Arabia: Insights from Saudi Arabia. Open Access Maced. J. Med. Sci. 2020, 8, 304–309. [Google Scholar]
- Moammar, H.; Cheriyan, G.; Mathew, R.; Al-Sannaa, N. Incidence and patterns of inborn errors of metabolism in the Eastern Province of Saudi Arabia, 1983–2008. Ann. Saudi Med. 2010, 30, 271–277. [Google Scholar] [CrossRef]
- Alfadhel, M.; Benmeakel, M.; Hossain, M.A.; Al Mutairi, F.; Al Othaim, A.; Alfares, A.A.; Al Balwi, M.; Alzaben, A.; Eyaid, W. Thirteen year retrospective review of the spectrum of inborn errors of metabolism presenting in a tertiary center in Saudi Arabia. Orphanet J. Rare Dis. 2016, 11, 126. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.; Arn, P.; Giugliani, R.; Muenzer, J.; Okuyama, T.; Taylor, J.; Fallet, S. The natural history of MPS I: Global perspectives from the MPS I Registry. Genet. Med. 2014, 16, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Bush, D.; Sremba, L.; Lomax, K.; Lipsett, J.; Ketteridge, D.; Bratkovic, D.; Enchautegui-Colon, Y.; Weisfeld-Adams, J.; Galambos, C.; Lummus, S.; et al. Neonatal Onset Interstitial Lung Disease as a Primary Presenting Manifestation of Mucopolysaccharidosis Type I. JIMD Rep. 2019, 43, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Berger, K.I.; Fagondes, S.C.; Giugliani, R.; Hardy, K.A.; Lee, K.S.; McArdle, C.; Rapoport, D.M. Respiratory and sleep disorders in mucopoly-saccharidosis. J. Inherit. Metab. Dis. 2013, 36, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Kiely, B.T.; Kohler, J.L.; Coletti, H.Y.; Poe, M.D.; Escolar, M.L. Early disease progression of Hurler syndrome. Orphanet J. Rare Dis. 2017, 12, 32. [Google Scholar] [CrossRef]
- Al-Kouatly, H.B.; Makhamreh, M.M.; Rice, S.M.; Smith, K.; Harman, C.; Quinn, A.; Valcarcel, B.N.; Firman, B.; Liu, R.; Hegde, M.; et al. High diagnosis rate for nonimmune hydrops fetalis with prenatal clinical exome from the Hydrops-Yielding Diagnostic Results of Prenatal Sequencing (HYDROPS) Study. Anesth. Analg. 2021, 23, 1325–1333. [Google Scholar] [CrossRef]
- Chaubey, A.; Shenoy, S.; Mathur, A.; Ma, Z.; Valencia, C.A.; Reddy Nallamilli, B.R.; Szekeres EJr Stansberry, L.; Liu, R.; Hegde, M.R. Low-Pass Genome Sequencing: Validation and Diagnostic Utility from 409 Clinical Cases of Low-Pass Genome Sequencing for the Detection of Copy Number Variants to Replace Constitutional Microarray. J. Mol. Diagn. 2020, 22, 823–840. [Google Scholar] [CrossRef]
- Bunge, S.; Kleijer, W.J.; Steglich, C.; Beck, M.; Zuther, C.; Morris, C.P.; Gal, A. Mucopolysaccharidosis type I: Identification of 8 novel mutations and determination of the frequency of the two common alpha-L-iduronidase mutations (W402X and Q70X) among European patients. Hum. Mol. Genet. 1994, 3, 861–866. [Google Scholar] [CrossRef]
- Yang, X.; Mei, S.; Kong, X.; Zhao, Z.; Cai, A.; Yao, J.; Li, Y.; Qin, Z. IDUA gene mutation analysis and prenatal diagnosis of two families affected with mucopolysaccharidosis type I. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2017, 34, 347–351. [Google Scholar]
- Oussoren, E.; Keulemans, J.; van Diggelen, O.P.; Oemardien, L.F.; Timmermans, R.G.; van der Ploeg, A.T.; Ruijter, G.J. Residual α-l-iduronidase activity in fibroblasts of mild to severe Mucopolysaccharidosis type I patients. Mol. Genet. Metab. 2013, 109, 377–381. [Google Scholar] [CrossRef]
- Lee-Chen, G.-J.; Lin, S.-P.; Chen, I.-S.; Chang, J.-H.; Yang, C.-W.; Chin, Y.-W. Mucopolysaccharidosis type I: Identification and charac-terization of mutations affecting alpha-L-iduronidase activity. J. Formos. Med. Assoc. Taiwan Yi Zhi. 2002, 101, 425–428. [Google Scholar] [PubMed]
- Bertola, F.; Filocamo, M.; Casati, G.; Mort, M.; Rosano, C.; Tylki-Szymanska, A.; Tüysüz, B.; Gabrielli, O.; Grossi, S.; Scarpa, M.; et al. IDUA mutational profiling of a cohort of 102 European patients with mucopolysaccharidosis type I: Identification and characterization of 35 novel α-L-iduronidase (IDUA) alleles. Hum. Mutat. 2011, 32, E2189–E2210. [Google Scholar] [CrossRef] [PubMed]
- Terlato, N.J.; Cox, G.F. Can mucopolysaccharidosis type I disease severity be predicted based on a patient’s genotype? A com-prehensive review of the literature. Genet. Med. 2003, 5, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Bach, G.; Moskowitz, S.M.; Tieu, P.T.; Matynia, A.; Neufeld, E.F. Molecular analysis of Hurler syndrome in Druze and Muslim Arab patients in Israel: Multiple allelic mutations of the IDUA gene in a small geographic area. Am. J. Hum. Genet. 1993, 53, 330–338. [Google Scholar]
- Kurland, G.; Deterding, R.R.; Hagood, J.S.; Young, L.R.; Brody, A.S.; Castile, R.G.; Dell, S.; Fan, L.L.; Hamvas, A.; Hilman, B.C.; et al. An Official American Thoracic Society Clinical Practice Guideline: Classification, Evaluation, and Management of Childhood Interstitial Lung Disease in Infancy. Am. J. Respir. Crit. Care Med. 2013, 188, 376–394. [Google Scholar] [CrossRef]
- Pillai, N.R.; Ahmed, A.; Vanyo, T.; Whitley, C.B. Early Neonatal Cardiac Phenotype in Hurler Syndrome: Case Report and Literature Review. Genes 2022, 13, 1293. [Google Scholar] [CrossRef]
- Sheikh, S.S.; Yadav, D.K.; Saeed, A. Case Report: Hurler syndrome (Mucopolysaccharidosis Type 1) in a young female patient. F1000Research 2020, 9, 367. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asseri, A.A.; Alzoani, A.; Almazkary, A.M.; Abdulaziz, N.; Almazkary, M.H.; Alahmari, S.A.; Duraisamy, A.J.; Sureshkumar, S. Mucopolysaccharidosis Type I Presenting with Persistent Neonatal Respiratory Distress: A Case Report. Diseases 2023, 11, 67. https://doi.org/10.3390/diseases11020067
Asseri AA, Alzoani A, Almazkary AM, Abdulaziz N, Almazkary MH, Alahmari SA, Duraisamy AJ, Sureshkumar S. Mucopolysaccharidosis Type I Presenting with Persistent Neonatal Respiratory Distress: A Case Report. Diseases. 2023; 11(2):67. https://doi.org/10.3390/diseases11020067
Chicago/Turabian StyleAsseri, Ali Alsuheel, Ahmad Alzoani, Abdulwahab M. Almazkary, Nisreen Abdulaziz, Mufareh H. Almazkary, Samy Ailan Alahmari, Arul J. Duraisamy, and Shruti Sureshkumar. 2023. "Mucopolysaccharidosis Type I Presenting with Persistent Neonatal Respiratory Distress: A Case Report" Diseases 11, no. 2: 67. https://doi.org/10.3390/diseases11020067