Pathogenesis of Osteoarthritis: Risk Factors, Regulatory Pathways in Chondrocytes, and Experimental Models

 , ,
, ,
Abstract
:1. Introduction
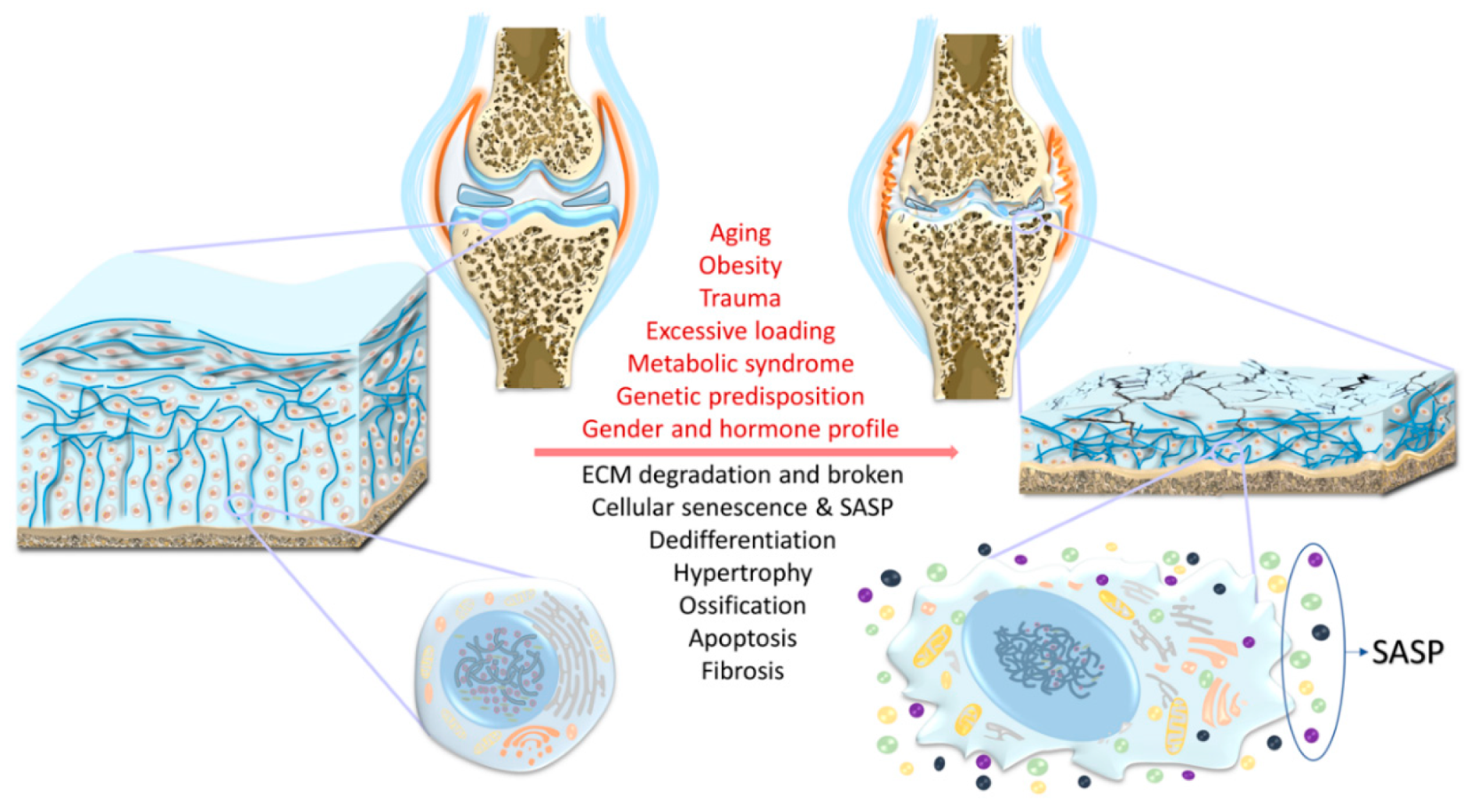
2. Risk Factors
2.1. Aging
2.2. Trauma
2.3. Obesity
2.4. Chronic Mechanical Overloading/Overuse
2.5. Genetics
3. Regulatory Pathways
3.1. Wnt/β-Catenin Signaling
3.2. PI3K/Akt/mTOR Pathway
3.3. Notch Signaling Pathway
3.4. SIRT1/AMPK Pathway
3.5. Hippo Pathway-YAP/TAZ Signaling
3.6. Disruptor of Telomeric Silencing 1-Like (DOT1L) Pathway
3.7. MicroRNAs
3.8. LncRNAs
4. Experimental Models
4.1. In Vivo Models
4.1.1. Aging-Induced Spontaneous OA Models
4.1.2. Trauma-Induced OA Models
4.1.3. Obesity-Induced OA Models
4.1.4. Chemically Induced OA Models
4.1.5. OA Models Involving Genetic Manipulations
4.2. In Vitro Models
4.2.1. Monolayer Culture
4.2.2. 3D Engineered Cartilage Tissues
4.2.3. Tissue Explant Models
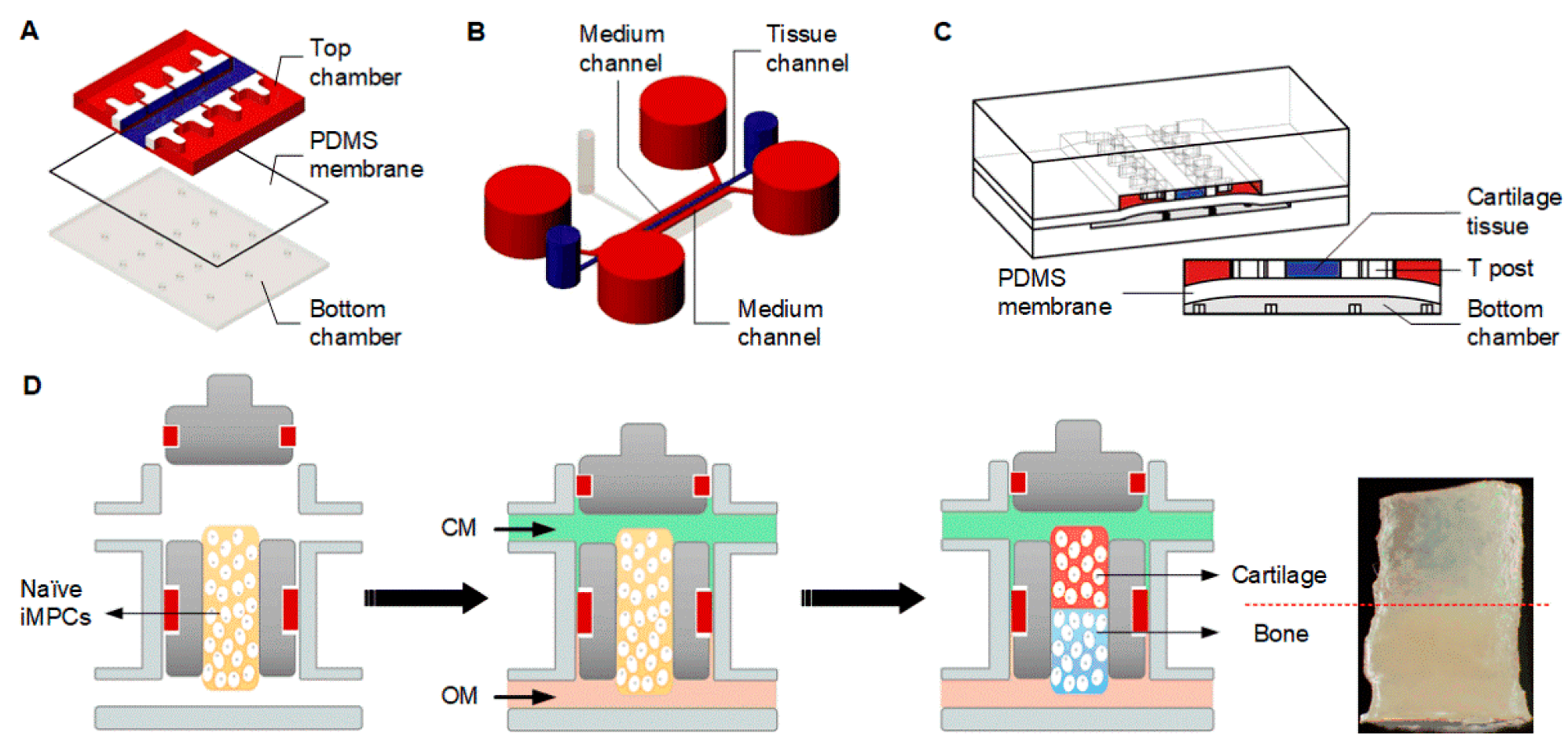
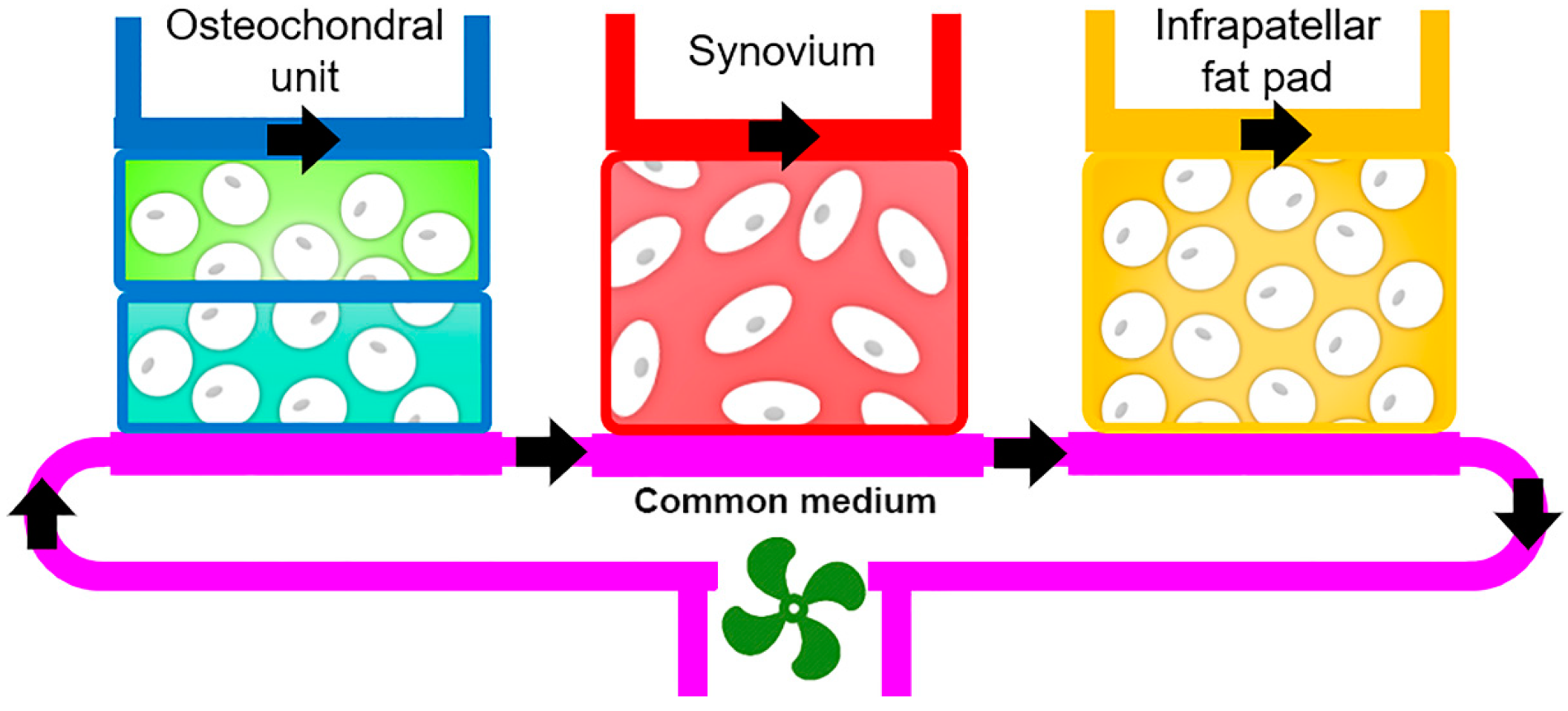
4.2.4. Microphysiological Systems
5. Summary and Future Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Dequeker, J.; Luyten, F.P. The history of osteoarthritis-osteoarthrosis. Ann. Rheum. Dis. 2008, 67, 5. [Google Scholar] [CrossRef] [PubMed]
- Hootman, J.M.; Helmick, C.G.; Barbour, K.E.; Theis, K.A.; Boring, M.A. Updated projected prevalence of self-reported doctor-diagnosed arthritis and arthritis-attributable activity limitation among US adults, 2015–2040. Arthritis Rheumatol. 2016, 68, 1582–1587. [Google Scholar] [CrossRef] [PubMed]
- Cross, M.; Smith, E.; Hoy, D.; Nolte, S.; Ackerman, I.; Fransen, M.; Bridgett, L.; Williams, S.; Guillemin, F.; Hill, C.L.; et al. The global burden of hip and knee osteoarthritis: Estimates from the Global Burden of Disease 2010 study. Ann. Rheum. Dis. 2014, 73, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Vina, E.R.; Kwoh, C.K. Epidemiology of osteoarthritis: Literature update. Curr. Opin. Rheumatol. 2018, 30, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Veronese, N.; Stubbs, B.; Solmi, M.; Smith, T.O.; Noale, M.; Cooper, C.; Maggi, S. Association between lower limb osteoarthritis and incidence of depressive symptoms: Data from the osteoarthritis initiative. Age Ageing 2017, 46, 470–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kye, S.Y.; Park, K. Suicidal ideation and suicidal attempts among adults with chronic diseases: A cross-sectional study. Compr. Psychiatry 2017, 73, 160–167. [Google Scholar] [CrossRef]
- Innes, K.E.; Sambamoorthi, U. The association of perceived memory loss with osteoarthritis and related joint pain in a large Appalachian population. Pain Med. 2018, 19, 1340–1356. [Google Scholar] [CrossRef]
- Litwic, A.; Edwards, M.H.; Dennison, E.M.; Cooper, C. Epidemiology and burden of osteoarthritis. Br. Med. Bull. 2013, 105, 185–199. [Google Scholar] [CrossRef] [Green Version]
- Hunter, D.J.; Bierma-Zeinstra, S. Osteoarthritis. Lancet 2019, 393, 1745–1759. [Google Scholar] [CrossRef]
- Arden, N.; Nevitt, M.C. Osteoarthritis: Epidemiology. Best Pract. Res. Clin. Rheumatol. 2006, 20, 3–25. [Google Scholar] [CrossRef]
- Griffin, T.M.; Guilak, F. The role of mechanical loading in the onset and progression of osteoarthritis. Exerc. Sport Sci. Rev. 2005, 33, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Glyn-Jones, S.; Palmer, A.J.R.; Agricola, R.; Price, A.J.; Vincent, T.L.; Weinans, H.; Carr, A.J. Osteoarthritis. Lancet 2015, 386, 376–387. [Google Scholar] [CrossRef]
- Loeser, R.F. The role of aging in the development of osteoarthritis. Trans. Am. Clin. Climatol. Assoc. 2017, 128, 44. [Google Scholar] [PubMed]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar] [CrossRef] [Green Version]
- Poole, A.R. Osteoarthritis as a whole joint disease. HSS J. 2012, 8, 4–6. [Google Scholar] [CrossRef] [Green Version]
- Camarero-Espinosa, S.; Rothen-Rutishauser, B.; Foster, E.J.; Weder, C.J.B.s. Articular cartilage: From formation to tissue engineering. Biomater. Sci. 2016, 4, 734–767. [Google Scholar] [CrossRef]
- Rim, Y.A.; Nam, Y.; Ju, J.H. The role of chondrocyte hypertrophy and senescence in osteoarthritis initiation and progression. Int. J. Mol. Sci. 2020, 21, 2358. [Google Scholar] [CrossRef] [Green Version]
- Barnett, R. Osteoarthritis. Lancet 2018, 391, 1985. [Google Scholar] [CrossRef]
- Felson, D.T. The course of osteoarthritis and factors that affect it. Rheum. Dis. Clin. N. Am. 1993, 19, 607–615. [Google Scholar]
- Geyer, M.; Schönfeld, C.J.C.R.R. Novel insights into the pathogenesis of osteoarthritis. Curr. Rheumatol. Rep. 2018, 14, 98–107. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Michaelis, M.; Ladel, C.; Siebuhr, A.S.; Bihlet, A.R.; Andersen, J.R.; Guehring, H.; Christiansen, C.; Bay-Jensen, A.C.; Kraus, V.B. Disease-modifying treatments for osteoarthritis (DMOADs) of the knee and hip: Lessons learned from failures and opportunities for the future. Osteoarthr. Cartil. 2016, 24, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Chaganti, R.K.; Lane, N.E. Risk factors for incident osteoarthritis of the hip and knee. Curr. Rev. Musculoskelet. Med. 2011, 4, 99–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidari, B. Knee osteoarthritis prevalence, risk factors, pathogenesis and features: Part I. Caspian J. Intern. Med. 2011, 2, 205–212. [Google Scholar] [PubMed]
- Roos, H.; Adalberth, T.; Dahlberg, L.; Lohmander, L.S. Osteoarthritis of the knee after injury to the anterior cruciate ligament or meniscus: The influence of time and age. Osteoarthr. Cartil. 1995, 3, 261–267. [Google Scholar] [CrossRef] [Green Version]
- Shane Anderson, A.; Loeser, R.F. Why is osteoarthritis an age-related disease? Best Pract. Res. Clin. Rheumatol. 2010, 24, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Felson, D.T.; Zhang, Y.; Hannan, M.T.; Naimark, A.; Weissman, B.; Aliabadi, P.; Levy, D. Risk factors for incident radiographic knee osteoarthritis in the elderly: The Framingham Study. Arthritis Rheum. 1997, 40, 728–733. [Google Scholar] [CrossRef]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [Green Version]
- Kirkwood, T.B. Understanding the odd science of aging. Cell 2005, 120, 437–447. [Google Scholar] [CrossRef] [Green Version]
- Bolduc, J.A.; Collins, J.A.; Loeser, R.F. Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic. Biol. Med. 2019, 132, 73–82. [Google Scholar] [CrossRef]
- Grover, A.K.; Samson, S.E. Benefits of antioxidant supplements for knee osteoarthritis: Rationale and reality. Nutr. J. 2016, 15, 1. [Google Scholar] [CrossRef] [Green Version]
- Rahmati, M.; Nalesso, G.; Mobasheri, A.; Mozafari, M. Aging and osteoarthritis: Central role of the extracellular matrix. Ageing Res. Rev. 2017, 40, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Jeon, O.H.; David, N.; Campisi, J.; Elisseeff, J.H. Senescent cells and osteoarthritis: A painful connection. J. Clin. Investig. 2018, 128, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.S.; Singer, B.D.; Vaughan, D.E. Molecular and physiological manifestations and measurement of aging in humans. Aging Cell 2017, 16, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Buckwalter, J.A. Telomere erosion and senescence in human articular cartilage chondrocytes. J. Gerontol. A Biol. Sci. Med. Sci. 2001, 56, B172–B179. [Google Scholar] [CrossRef] [Green Version]
- Loeser, R.F. Aging and osteoarthritis: The role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr. Cartil. 2009, 17, 971–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsyth, C.B.; Cole, A.; Murphy, G.; Bienias, J.L.; Im, H.-J.; Loeser, R.F., Jr. Increased matrix metalloproteinase-13 production with aging by human articular chondrocytes in response to catabolic stimuli. J. Gerontol. A Biol. 2005, 60, 1118–1124. [Google Scholar] [CrossRef] [Green Version]
- Long, D.; Blake, S.; Song, X.Y.; Lark, M.; Loeser, R.F. Human articular chondrocytes produce IL-7 and respond to IL-7 with increased production of matrix metalloproteinase-13. Arthritis Res. Ther. 2008, 10, R23. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Billinghurst, R.C.; Pidoux, I.; Antoniou, J.; Zukor, D.; Tanzer, M.; Poole, A.R. Sites of collagenase cleavage and denaturation of type II collagen in aging and osteoarthritic articular cartilage and their relationship to the distribution of matrix metalloproteinase 1 and matrix metalloproteinase 13. Arthritis Rheum. 2002, 46, 2087–2094. [Google Scholar] [CrossRef]
- Buckwalter, J.A.; Anderson, D.D.; Brown, T.D.; Tochigi, Y.; Martin, J.A. The roles of mechanical stresses in the pathogenesis of osteoarthritis: Implications for treatment of joint injuries. Cartilage 2013, 4, 286–294. [Google Scholar] [CrossRef]
- Anderson, D.D.; Chubinskaya, S.; Guilak, F.; Martin, J.A.; Oegema, T.R.; Olson, S.A.; Buckwalter, J.A. Post-traumatic osteoarthritis: Improved understanding and opportunities for early intervention. J. Orth. Res. 2011, 29, 802–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez, G.; Cobo-Molinos, J.; Antich, C.; Lopez-Ruiz, E. Osteoarthritis: Trauma vs Disease. Adv. Exp. Med. Biol. 2018, 1059, 63–83. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.C.; Hubbard-Turner, T.; Wikstrom, E.A.; Palmieri-Smith, R.M. Epidemiology of posttraumatic osteoarthritis. J. Athl. Train. 2017, 52, 491–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lübbeke, A.; Salvo, D.; Stern, R.; Hoffmeyer, P.; Holzer, N.; Assal, M. Risk factors for post-traumatic osteoarthritis of the ankle: An eighteen year follow-up study. Int. Orthop. 2012, 36, 1403–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, P.G.; McCarron, J.A.; Levine, M.J.; Melvin, G.M.; Murray, P.J.; Manner, P.A.; Tuan, R.S. An in vivo lapine model for impact-induced injury and osteoarthritic degeneration of articular cartilage. Cartilage 2012, 3, 323–333. [Google Scholar] [CrossRef]
- Chubinskaya, S.; Wimmer, M.A. Key pathways to prevent posttraumatic arthritis for future molecule-based therapy. Cartilage 2013, 4, 13S–21S. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Fan, X.; Xing, L.; Tian, F. Wnt signaling: A promising target for osteoarthritis therapy. Cell Commun. Signal. 2019, 17, 97. [Google Scholar] [CrossRef] [Green Version]
- Deshmukh, V.; Hu, H.; Barroga, C.; Bossard, C.; Kc, S.; Dellamary, L.; Stewart, J.; Chiu, K.; Ibanez, M.; Pedraza, M.; et al. A small-molecule inhibitor of the Wnt pathway (SM04690) as a potential disease modifying agent for the treatment of osteoarthritis of the knee. Osteoarthr. Cartil. 2018, 26, 18–27. [Google Scholar] [CrossRef]
- Held, A.; Glas, A.; Dietrich, L.; Bollmann, M.; Brandstädter, K.; Grossmann, T.N.; Lohmann, C.H.; Pap, T.; Bertrand, J. Targeting β-catenin dependent Wnt signaling via peptidomimetic inhibitors in murine chondrocytes and OA cartilage. Osteoarthr. Cartil. 2018, 26, 818–823. [Google Scholar] [CrossRef] [Green Version]
- Grossmann, T.N.; Yeh, J.T.H.; Bowman, B.R.; Chu, Q.; Moellering, R.E.; Verdine, G.L. Inhibition of oncogenic Wnt signaling through direct targeting of β-catenin. Proc. Natl. Acad. Sci. USA 2012, 109, 17942–17947. [Google Scholar] [CrossRef] [Green Version]
- Bliddal, H.; Leeds, A.R.; Christensen, R. Osteoarthritis, obesity and weight loss: Evidence, hypotheses and horizons—A scoping review. Obes. Rev. 2014, 15, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Silverwood, V.; Blagojevic-Bucknall, M.; Jinks, C.; Jordan, J.L.; Protheroe, J.; Jordan, K.P. Current evidence on risk factors for knee osteoarthritis in older adults: A systematic review and meta-analysis. Osteoarthr. Cartil. 2015, 23, 507–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felson, D.T.; Zhang, Y.; Anthony, J.M.; Naimark, A.; Anderson, J.J. Weight loss reduces the risk for symptomatic knee osteoarthritis in women: The Framingham Study. Ann. Intern. Med. 1992, 116, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: The linking mechanism and the complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef]
- Xie, D.-x.; Wei, J.; Zeng, C.; Yang, T.; Li, H.; Wang, Y.-l.; Long, H.-z.; Wu, Z.-y.; Qian, Y.-x.; Li, K.-h.; et al. Association between metabolic syndrome and knee osteoarthritis: A cross-sectional study. BMC Musculoskel. Disord. 2017, 18, 533. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, N.; Muraki, S.; Oka, H.; Kawaguchi, H.; Nakamura, K.; Akune, T. Association of knee osteoarthritis with the accumulation of metabolic risk factors such as overweight, hypertension, dyslipidemia, and impaired glucose tolerance in Japanese men and women: The ROAD study. J. Rheumatol. 2011, 38, 921. [Google Scholar] [CrossRef]
- Ogunbona, R.A.; Orimadegun, B.E.; Ogunlade, S.O.; Fasanmade, A.A.; Agbedana, E.O. Dyslipidemia and high adiposity are risk factors for osteoarthritis in adults in Nigeria. Am. J. Biomed. Res. 2020, 8, 19–24. [Google Scholar] [CrossRef]
- Afifi, A.E.L.M.A.; Shaat, R.M.; Gharbia, O.M.; Boghdadi, Y.E.L.; Eshmawy, M.M.E.L.; El-Emam, O.A. Osteoarthritis of knee joint in metabolic syndrome. Clin. Rheumatol. 2018, 37, 2855–2861. [Google Scholar] [CrossRef]
- Francisco, V.; Pérez, T.; Pino, J.; López, V.; Franco, E.; Alonso, A.; Gonzalez-Gay, M.A.; Mera, A.; Lago, F.; Gómez, R.; et al. Biomechanics, obesity, and osteoarthritis. The role of adipokines: When the levee breaks. J. Orth. Res. 2018, 36, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; He, C. Pro-inflammatory cytokines: The link between obesity and osteoarthritis. Cytokine Growth Factor Rev. 2018, 44, 38–50. [Google Scholar] [CrossRef]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85. [Google Scholar] [CrossRef] [PubMed]
- Sekar, S.; Shafie, S.R.; Prasadam, I.; Crawford, R.; Panchal, S.K.; Brown, L.; Xiao, Y. Saturated fatty acids induce development of both metabolic syndrome and osteoarthritis in rats. Sci. Rep. 2017, 7, 46457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.B. Mechanical loading, cartilage degradation, and arthritis. Ann. N. Y. Acad. Sci. 2010, 1211, 37–50. [Google Scholar] [CrossRef]
- Tetsunaga, T.; Nishida, K.; Furumatsu, T.; Naruse, K.; Hirohata, S.; Yoshida, A.; Saito, T.; Ozaki, T. Regulation of mechanical stress-induced MMP-13 and ADAMTS-5 expression by RUNX-2 transcriptional factor in SW1353 chondrocyte-like cells. Osteoarthr. Cartil. 2011, 19, 222–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilak, F. Biomechanical factors in osteoarthritis. Best Pract. Res. Clin. Rheumatol. 2011, 25, 815–823. [Google Scholar] [CrossRef] [Green Version]
- Bennell, K.L.; Creaby, M.W.; Wrigley, T.V.; Bowles, K.-A.; Hinman, R.S.; Cicuttini, F.; Hunter, D.J. Bone marrow lesions are related to dynamic knee loading in medial knee osteoarthritis. Ann. Rheum. Dis. 2010, 69, 1151. [Google Scholar] [CrossRef]
- Spector, T.D.; Harris, P.A.; Hart, D.J.; Cicuttini, F.M.; Nandra, D.; Etherington, J.; Wolman, R.L.; Doyle, D.V. Risk of osteoarthritis associated with long-term weight-bearing sports: A radiologic survey of the hips and knees in female ex-athletes and population controls. Arthritis Rheum. 1996, 39, 988–995. [Google Scholar] [CrossRef]
- Sulsky, S.I.; Carlton, L.; Bochmann, F.; Ellegast, R.; Glitsch, U.; Hartmann, B.; Pallapies, D.; Seidel, D.; Sun, Y. Epidemiological evidence for work load as a risk factor for osteoarthritis of the hip: A systematic review. PLoS ONE 2012, 7, e31521. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Adams, J.; Leddy, H.A.; McNulty, A.L.; O’Conor, C.J.; Guilak, F. The mechanobiology of articular cartilage: Bearing the burden of osteoarthritis. Curr. Rheumatol. Rep. 2014, 16, 451. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, J.P.; Raynauld, J.P.; Berthiaume, M.J.; Abram, F.; Choquette, D.; Haraoui, B.; Beary, J.F.; Cline, G.A.; Meyer, J.M.; Martel-Pelletier, J. Risk factors associated with the loss of cartilage volume on weight-bearing areas in knee osteoarthritis patients assessed by quantitative magnetic resonance imaging: A longitudinal study. Arthritis Res. Ther. 2007, 9, R74. [Google Scholar] [CrossRef] [Green Version]
- Dolmetsch, R.E.; Lewis, R.S.; Goodnow, C.C.; Healy, J.I.J.N. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 1997, 386, 855–858. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, T.T.; Salter, D.M.; Bader, D.L.; Lee, D.A. Signal transduction pathways involving p38 MAPK, JNK, NFκB and AP-1 influences the response of chondrocytes cultured in agarose constructs to IL-1β and dynamic compression. J. Inflamm. Res. 2008, 57, 306–313. [Google Scholar] [CrossRef]
- Healy, Z.R.; Zhu, F.; Stull, J.D.; Konstantopoulos, K. Elucidation of the signaling network of COX-2 induction in sheared chondrocytes: COX-2 is induced via a Rac/MEKK1/MKK7/JNK2/c-Jun-C/EBPβ-dependent pathway. Am. J. Physiol. Cell Physiol. 2008, 294, C1146–C1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delco, M.L.; Bonnevie, E.D.; Bonassar, L.J.; Fortier, L.A. Mitochondrial dysfunction is an acute response of articular chondrocytes to mechanical injury. J. Orth. Res. 2018, 36, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Koike, M.; Nojiri, H.; Ozawa, Y.; Watanabe, K.; Muramatsu, Y.; Kaneko, H.; Morikawa, D.; Kobayashi, K.; Saita, Y.; Sasho, T.; et al. Mechanical overloading causes mitochondrial superoxide and SOD2 imbalance in chondrocytes resulting in cartilage degeneration. Sci. Rep. 2015, 5, 11722. [Google Scholar] [CrossRef] [Green Version]
- Cicuttini, F.M.; Spector, T.D. Genetics of osteoarthritis. Ann. Rheum. Dis. 1996, 55, 665–667. [Google Scholar] [CrossRef] [Green Version]
- Spector, T.D.; Cicuttini, F.; Baker, J.; Loughlin, J.; Hart, D. Genetic influences on osteoarthritis in women: A twin study. BMJ (Clinical Research Ed.) 1996, 312, 940–943. [Google Scholar] [CrossRef] [Green Version]
- Sandell, L.J. Etiology of osteoarthritis: Genetics and synovial joint development. Nat. Rev. Rheumatol. 2012, 8, 77–89. [Google Scholar] [CrossRef]
- Panoutsopoulou, K.; Zeggini, E.J.J.O.M.G. Advances in osteoarthritis genetics. J. Med. Genet. 2013, 50, 715–724. [Google Scholar] [CrossRef] [Green Version]
- Zengini, E.; Finan, C.; Wilkinson, J.M. The genetic epidemiological landscape of hip and knee osteoarthritis: Where are we now and where are we going? J. Rheumatol. 2016, 43, 260–266. [Google Scholar] [CrossRef]
- Styrkarsdottir, U.; Lund, S.H.; Thorleifsson, G.; Zink, F.; Stefansson, O.A.; Sigurdsson, J.K.; Juliusson, K.; Bjarnadottir, K.; Sigurbjornsdottir, S.; Jonsson, S.; et al. Meta-analysis of Icelandic and UK data sets identifies missense variants in SMO, IL11, COL11A1 and 13 more new loci associated with osteoarthritis. Nat. Genet. 2018, 50, 1681–1687. [Google Scholar] [CrossRef] [PubMed]
- Tachmazidou, I.; Hatzikotoulas, K.; Southam, L.; Esparza-Gordillo, J.; Haberland, V.; Zheng, J.; Johnson, T.; Koprulu, M.; Zengini, E.; Steinberg, J. Identification of new therapeutic targets for osteoarthritis through genome-wide analyses of UK Biobank data. Nat. Genet. 2019, 51, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Barter, M.J.; Gomez, R.; Hyatt, S.; Cheung, K.; Skelton, A.J.; Xu, Y.; Clark, I.M.; Young, D.A. The long non-coding RNA ROCR contributes to SOX9 expression and chondrogenic differentiation of human mesenchymal stem cells. Development 2017, 144, 4510–4521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, J.C.; Keith, A.; Rice, S.J.; Shepherd, C.; Agarwal, V.; Loughlin, J.; Shendure, J. Functional testing of thousands of osteoarthritis-associated variants for regulatory activity. Nat. Commun. 2019, 10, 2434. [Google Scholar] [CrossRef] [Green Version]
- García-Ibarbia, C.; Pérez-Castrillón, J.L.; Ortiz, F.; Velasco, J.; Zarrabeitia, M.T.; Sumillera, M.; Riancho, J.A. Wnt-related genes and large-joint osteoarthritis: Association study and replication. Rheumatol. Int. 2013, 33, 2875–2880. [Google Scholar] [CrossRef]
- Reynard, L.N.; Loughlin, J. Insights from human genetic studies into the pathways involved in osteoarthritis. Nat. Rev. Rheumatol. 2013, 9, 573. [Google Scholar] [CrossRef]
- Reynard, L.N.; Barter, M.J. Osteoarthritis year in review 2019: Genetics, genomics and epigenetics. Osteoarthr. Cartil. 2020, 28, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Usami, Y.; Gunawardena, A.T.; Iwamoto, M.; Enomoto-Iwamoto, M. Wnt signaling in cartilage development and diseases: Lessons from animal studies. Lab. Invest. 2016, 96, 186–196. [Google Scholar] [CrossRef] [Green Version]
- Xi, Y.; Huang, X.; Tan, G.; Chu, X.; Zhang, R.; Ma, X.; Ni, B.; You, H. Protective effects of Erdosteine on interleukin-1β-stimulated inflammation via inhibiting the activation of MAPK, NF-κB, and Wnt/β-catenin signaling pathways in rat osteoarthritis. Eur. J. Pharmacol. 2020, 873, 172925. [Google Scholar] [CrossRef]
- Blom, A.B.; Brockbank, S.M.; van Lent, P.L.; van Beuningen, H.M.; Geurts, J.; Takahashi, N.; van der Kraan, P.M.; van de Loo, F.A.; Schreurs, B.W.; Clements, K.; et al. Involvement of the Wnt signaling pathway in experimental and human osteoarthritis: Prominent role of Wnt-induced signaling protein 1. Arthritis Rheum. 2009, 60, 501–512. [Google Scholar] [CrossRef]
- Oh, H.; Chun, C.H.; Chun, J.S. Dkk-1 expression in chondrocytes inhibits experimental osteoarthritic cartilage destruction in mice. Arthritis Rheum. 2012, 64, 2568–2578. [Google Scholar] [CrossRef] [PubMed]
- Snelling, S.J.; Davidson, R.K.; Swingler, T.E.; Le, L.T.; Barter, M.J.; Culley, K.L.; Price, A.; Carr, A.J.; Clark, I.M. Dickkopf-3 is upregulated in osteoarthritis and has a chondroprotective role. Osteoarthr. Cartil. 2016, 24, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Ma, L.; Wu, L.; Jin, Q. Wnt-β-catenin signaling pathway inhibition by sclerostin may protect against degradation in healthy but not osteoarthritic cartilage. Mol. Med. Rep. 2017, 15, 2423–2432. [Google Scholar] [CrossRef] [PubMed]
- Lietman, C.; Wu, B.; Lechner, S.; Shinar, A.; Sehgal, M.; Rossomacha, E.; Datta, P.; Sharma, A.; Gandhi, R.; Kapoor, M.; et al. Inhibition of Wnt/beta-catenin signaling ameliorates osteoarthritis in a murine model of experimental osteoarthritis. JCI Insight 2018, 3, e96308. [Google Scholar] [CrossRef] [PubMed]
- De Santis, M.; Di Matteo, B.; Chisari, E.; Cincinelli, G.; Angele, P.; Lattermann, C.; Filardo, G.; Vitale, N.D.; Selmi, C.; Kon, E. The role of wnt pathway in the pathogenesis of OA and its potential therapeutic implications in the field of regenerative medicine. Biomed Res. Int. 2018, 2018, 7402947. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Chen, M.; Zuscik, M.; Wu, Q.; Wang, Y.J.; Rosier, R.N.; O’Keefe, R.J.; Chen, D. Inhibition of beta-catenin signaling in articular chondrocytes results in articular cartilage destruction. Arthritis Rheum. 2008, 58, 2053–2064. [Google Scholar] [CrossRef] [Green Version]
- Xuan, F.; Yano, F.; Mori, D.; Chijimatsu, R.; Maenohara, Y.; Nakamoto, H.; Mori, Y.; Makii, Y.; Oichi, T.; Taketo, M.M.; et al. Wnt/β-catenin signaling contributes to articular cartilage homeostasis through lubricin induction in the superficial zone. Arthritis Res. Ther. 2019, 21, 247. [Google Scholar] [CrossRef] [Green Version]
- Theologis, T.; Efstathopoulos, N.; Nikolaou, V.; Charikopoulos, I.; Papapavlos, I.; Kokkoris, P.; Papatheodorou, A.; Nasiri-Ansari, N.; Kassi, E. Association between serum and synovial fluid Dickkopf-1 levels with radiographic severity in primary knee osteoarthritis patients. Clin. Rheumatol. 2017, 36, 1865–1872. [Google Scholar] [CrossRef]
- Zhu, M.; Tang, D.; Wu, Q.; Hao, S.; Chen, M.; Xie, C.; Rosier, R.N.; O’Keefe, R.J.; Zuscik, M.; Chen, D. Activation of beta-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult beta-catenin conditional activation mice. J. Bone Miner. Res. 2009, 24, 12–21. [Google Scholar] [CrossRef]
- Malemud, C.J. The PI3K/Akt/PTEN/mTOR pathway: A fruitful target for inducing cell death in rheumatoid arthritis? Future Med. Chem. 2015, 7, 1137–1147. [Google Scholar] [CrossRef]
- Ersahin, T.; Tuncbag, N.; Cetin-Atalay, R. The PI3K/AKT/mTOR interactive pathway. Mol. Biosyst. 2015, 11, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Luo, J.; Guo, J.; Yao, X.; Jing, X.; Guo, F. The PI3K/AKT/mTOR signaling pathway in osteoarthritis: A narrative review. Osteoarthr. Cartil. 2020, 28, 400–409. [Google Scholar] [CrossRef]
- Huang, C.Y.; Lin, H.J.; Chen, H.S.; Cheng, S.Y.; Hsu, H.C.; Tang, C.H. Thrombin promotes matrix metalloproteinase-13 expression through the PKCδ c-Src/EGFR/PI3K/Akt/AP-1 signaling pathway in human chondrocytes. Mediat. Inflamm. 2013, 2013, 326041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Chu, M.; Wang, F.; Zhao, Y.; Chen, H.; Dai, X. Putative functional variants of PI3K/AKT/mTOR pathway are associated with knee osteoarthritis susceptibility. J. Clin. Lab. Anal. 2020, 34, e23240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.F.; Shi, Z.M.; Zou, J.; Li, X.L. Inhibition of PI3K/AKT/mTOR signaling pathway promotes autophagy of articular chondrocytes and attenuates inflammatory response in rats with osteoarthritis. Biomed. Pharmacother. 2017, 89, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lu, H.; Yao, J.; Zhang, X.; Huang, Y.; Ma, S.; Zou, K.; Wei, Y.; Yang, Z.; Li, J.; et al. GABARAP promotes bone marrow mesenchymal stem cells-based the osteoarthritis cartilage regeneration through the inhibition of PI3K/AKT/mTOR signaling pathway. J. Cell. Physiol. 2019, 234, 21014–21026. [Google Scholar] [CrossRef]
- Qian, Y.Q.; Feng, Z.H.; Li, X.B.; Hu, Z.C.; Xuan, J.W.; Wang, X.Y.; Xu, H.C.; Chen, J.X. Downregulating PI3K/Akt/NF-kappaB signaling with allicin for ameliorating the progression of osteoarthritis: In vitro and vivo studies. Food Funct. 2018, 9, 4865–4875. [Google Scholar] [CrossRef]
- Lin, C.; Shao, Y.; Zeng, C.; Zhao, C.; Fang, H.; Wang, L.; Pan, J.; Liu, L.; Qi, W.; Feng, X.; et al. Blocking PI3K/AKT signaling inhibits bone sclerosis in subchondral bone and attenuates post-traumatic osteoarthritis. J. Cell. Physiol. 2018, 233, 6135–6147. [Google Scholar] [CrossRef]
- Qu, R.; Chen, X.; Wang, W.; Qiu, C.; Ban, M.; Guo, L.; Vasilev, K.; Chen, J.; Li, W.; Zhao, Y. Ghrelin protects against osteoarthritis through interplay with Akt and NF-κB signaling pathways. FASEB J. 2018, 32, 1044–1058. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Tanaka, S. Molecular mechanisms underlying osteoarthritis development: Notch and NF-kappaB. Arthritis Res. Ther. 2017, 19, 94. [Google Scholar] [CrossRef]
- Yoon, K.; Gaiano, N. Notch signaling in the mammalian central nervous system: Insights from mouse mutants. Nat. Neurosci. 2005, 8, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Monteagudo, S.; Lories, R.J. A Notch in the joint that exacerbates osteoarthritis. Nat. Rev. Rheumatol. 2018, 14, 563–564. [Google Scholar] [CrossRef]
- Crowe, R.; Zikherman, J.; Niswander, L. Delta-1 negatively regulates the transition from prehypertrophic to hypertrophic chondrocytes during cartilage formation. Development 1999, 126, 987–998. [Google Scholar]
- Kohn, A.; Rutkowski, T.P.; Liu, Z.; Mirando, A.J.; Zuscik, M.J.; O’Keefe, R.J.; Hilton, M.J. Notch signaling controls chondrocyte hypertrophy via indirect regulation of Sox9. Bone Res. 2015, 3, 15021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mead, T.J.; Yutzey, K.E. Notch pathway regulation of chondrocyte differentiation and proliferation during appendicular and axial skeleton development. Proc. Natl. Acad. Sci. USA 2009, 106, 14420–14425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.; Nelson, L.; Dowthwaite, G.P.; Evans, D.J.R.; Archer, C.W. Notch receptor and Notch ligand expression in developing avian cartilage. J. Anat. 2009, 215, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Dowthwaite, G.P.; Bishop, J.C.; Redman, S.N.; Khan, I.M.; Rooney, P.; Evans, D.J.; Haughton, L.; Bayram, Z.; Boyer, S.; Thomson, B.; et al. The surface of articular cartilage contains a progenitor cell population. J. Cell Sci. 2004, 117, 889–897. [Google Scholar] [CrossRef] [Green Version]
- Hosaka, Y.; Saito, T.; Sugita, S.; Hikata, T.; Kobayashi, H.; Fukai, A.; Taniguchi, Y.; Hirata, M.; Akiyama, H.; Chung, U.-i.; et al. Notch signaling in chondrocytes modulates endochondral ossification and osteoarthritis development. Proc. Natl. Acad. Sci. USA 2013, 110, 1875–1880. [Google Scholar] [CrossRef] [Green Version]
- Sugita, S.; Hosaka, Y.; Okada, K.; Mori, D.; Yano, F.; Kobayashi, H.; Taniguchi, Y.; Mori, Y.; Okuma, T.; Chang, S.H.; et al. Transcription factor Hes1 modulates osteoarthritis development in cooperation with calcium/calmodulin-dependent protein kinase 2. Proc. Natl. Acad. Sci. USA 2015, 112, 3080–3085. [Google Scholar] [CrossRef] [Green Version]
- Mirando, A.J.; Liu, Z.; Moore, T.; Lang, A.; Kohn, A.; Osinski, A.M.; O’Keefe, R.J.; Mooney, R.A.; Zuscik, M.J.; Hilton, M.J. RBP-Jkappa-dependent Notch signaling is required for murine articular cartilage and joint maintenance. Arthritis Rheum. 2013, 65, 2623–2633. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Ren, Y.; Mirando, A.J.; Wang, C.; Zuscik, M.J.; O’Keefe, R.J.; Hilton, M.J. Notch signaling in postnatal joint chondrocytes, but not subchondral osteoblasts, is required for articular cartilage and joint maintenance. Osteoarthr. Cartil. 2016, 24, 740–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu-Bryan, R. Inflammation and intracellular metabolism: New targets in OA. Osteoarthr. Cartil. 2015, 23, 1835–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.H.; Chiua, Y.C.; Wu, C.H.; Jou, I.M.; Tu, Y.K.; Hung, C.H.; Hsieh, P.L.; Tsai, K.L. Homocysteine causes dysfunction of chondrocytes and oxidative stress through repression of SIRT1/AMPK pathway: A possible link between hyperhomocysteinemia and osteoarthritis. Redox Biol. 2018, 15, 504–512. [Google Scholar] [CrossRef]
- Qiu, L.; Luo, Y.; Chen, X. Quercetin attenuates mitochondrial dysfunction and biogenesis via upregulated AMPK/SIRT1 signaling pathway in OA rats. Biomed. Pharmacother. 2018, 103, 1585–1591. [Google Scholar] [CrossRef]
- Feng, K.; Chen, Z.; Pengcheng, L.; Zhang, S.; Wang, X. Quercetin attenuates oxidative stress-induced apoptosis via SIRT1/AMPK-mediated inhibition of ER stress in rat chondrocytes and prevents the progression of osteoarthritis in a rat model. J. Cell. Physiol. 2019, 234, 18192–18205. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, X.; Lotz, M.; Terkeltaub, R.; Liu-Bryan, R. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor gamma coactivator 1alpha. Arthritis Rheumatol. 2015, 67, 2141–2153. [Google Scholar] [CrossRef]
- Ma, S.; Meng, Z.; Chen, R.; Guan, K.-L. The hippo pathway: Biology and pathophysiology. Annu. Rev. Biochem. 2019, 88, 577–604. [Google Scholar] [CrossRef] [Green Version]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Hu, Y.; Song, M.; Liu, Z.; Zhang, W.; Yu, F.X.; Wu, J.; Wang, S.; Izpisua Belmonte, J.C.; Chan, P.; et al. Up-regulation of FOXD1 by YAP alleviates senescence and osteoarthritis. PLoS Biol. 2019, 17, e3000201. [Google Scholar] [CrossRef]
- Deng, Y.; Lu, J.; Li, W.; Wu, A.; Zhang, X.; Tong, W.; Ho, K.K.; Qin, L.; Song, H.; Mak, K.K. Reciprocal inhibition of YAP/TAZ and NF-κB regulates osteoarthritic cartilage degradation. Nat. Commun. 2018, 9, 4564. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zhou, L.; Ling, L.; Meng, X.; Chu, F.; Zhang, S.; Zhou, F. The crosstalk between hippo-YAP pathway and innate immunity. Front. Immunol. 2020, 11, 323. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Cai, D.; Zhou, F.; Yu, J.; Wu, X.; Yu, D.; Zou, Y.; Hong, Y.; Yuan, C.; Chen, Y.; et al. Targeting downstream subcellular YAP activity as a function of matrix stiffness with Verteporfin-encapsulated chitosan microsphere attenuates osteoarthritis. Biomaterials 2020, 232, 119724. [Google Scholar] [CrossRef]
- Gong, Y.; Li, S.-J.; Liu, R.; Zhan, J.-F.; Tan, C.; Fang, Y.-F.; Chen, Y.; Yu, B. Inhibition of YAP with siRNA prevents cartilage degradation and ameliorates osteoarthritis development. Int. J. Mol. Med. 2019, 97, 103–114. [Google Scholar] [CrossRef]
- Zhong, W.; Li, Y.; Li, L.; Zhang, W.; Wang, S.; Zheng, X. YAP-mediated regulation of the chondrogenic phenotype in response to matrix elasticity. J. Mol. Histol. 2013, 44, 587–595. [Google Scholar] [CrossRef]
- Karystinou, A.; Roelofs, A.J.; Neve, A.; Cantatore, F.P.; Wackerhage, H.; De Bari, C. Yes-associated protein (YAP) is a negative regulator of chondrogenesis in mesenchymal stem cells. Arthritis Res. Ther. 2015, 17, 147. [Google Scholar] [CrossRef] [Green Version]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Zhang, Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011, 25, 1345–1358. [Google Scholar] [CrossRef] [Green Version]
- Castaño Betancourt, M.C.; Cailotto, F.; Kerkhof, H.J.; Cornelis, F.M.; Doherty, S.A.; Hart, D.J.; Hofman, A.; Luyten, F.P.; Maciewicz, R.A.; Mangino, M.; et al. Genome-wide association and functional studies identify the DOT1L gene to be involved in cartilage thickness and hip osteoarthritis. Proc. Natl. Acad. Sci. USA 2012, 109, 8218–8223. [Google Scholar] [CrossRef] [Green Version]
- Monteagudo, S.; Cornelis, F.M.F.; Aznar-Lopez, C.; Yibmantasiri, P.; Guns, L.-A.; Carmeliet, P.; Cailotto, F.; Lories, R.J. DOT1L safeguards cartilage homeostasis and protects against osteoarthritis. Nat. Commun. 2017, 8, 15889. [Google Scholar] [CrossRef] [Green Version]
- Cornelis, F.M.F.; de Roover, A.; Storms, L.; Hens, A.; Lories, R.J.; Monteagudo, S. Increased susceptibility to develop spontaneous and post-traumatic osteoarthritis in Dot1l-deficient mice. Osteoarthr. Cartil. 2019, 27, 513–525. [Google Scholar] [CrossRef] [Green Version]
- He, D.; Liu, J.; Hai, Y.; Zhu, Q.; Shen, Y.; Guo, S.; Zhang, W.; Zhou, X. Increased DOT1L in synovial biopsies of patients with OA and RA. Clin. Rheumatol. 2018, 37, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Calin, G.A.; Croce, C.M. MicroRNAs in Cancer. Annu. Rev. Med. 2009, 60, 167–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, T.; Lu, J.; Cobb, B.S.; Rodda, S.J.; McMahon, A.P.; Schipani, E.; Merkenschlager, M.; Kronenberg, H.M. Dicer-dependent pathways regulate chondrocyte proliferation and differentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 1949–1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swingler, T.E.; Niu, L.; Smith, P.; Paddy, P.; Le, L.; Barter, M.J.; Young, D.A.; Clark, I.M. The function of microRNAs in cartilage and osteoarthritis. Clin. Exp. Rheumatol. 2019, 37 (Suppl. S120), 40–47. [Google Scholar] [CrossRef]
- Zhang, W.; Hsu, P.; Zhong, B.; Guo, S.; Zhang, C.; Wang, Y.; Luo, C.; Zhan, Y.; Zhang, C. MiR-34a enhances chondrocyte apoptosis, senescence and facilitates development of osteoarthritis by targeting DLL1 and regulating PI3K/AKT pathway. Cell. Physiol. Biochem. 2018, 48, 1304–1316. [Google Scholar] [CrossRef]
- Philipot, D.; Guérit, D.; Platano, D.; Chuchana, P.; Olivotto, E.; Espinoza, F.; Dorandeu, A.; Pers, Y.-M.; Piette, J.; Borzi, R.M.; et al. p16INK4a and its regulator miR-24 link senescence and chondrocyte terminal differentiation-associated matrix remodeling in osteoarthritis. Arthritis Res. Ther. 2014, 16, R58. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Wang, T.; Cai, B.; Wang, X.; Feng, W.; Han, Y.; Li, D.; Li, S.; Liu, J. MicroRNA-495 enhances chondrocyte apoptosis, senescence and promotes the progression of osteoarthritis by targeting AKT1. Am. J. Transl. Res. 2019, 11, 2232–2244. [Google Scholar]
- Zhong, G.; Long, H.; Ma, S.; Shunhan, Y.; Li, J.; Yao, J. miRNA-335-5p relieves chondrocyte inflammation by activating autophagy in osteoarthritis. Life Sci. 2019, 226, 164–172. [Google Scholar] [CrossRef]
- Zhao, X.; Li, H.; Wang, L. MicroRNA-107 regulates autophagy and apoptosis of osteoarthritis chondrocytes by targeting TRAF3. Int. Immunopharmacol. 2019, 71, 181–187. [Google Scholar] [CrossRef]
- Miyaki, S.; Nakasa, T.; Otsuki, S.; Grogan, S.P.; Higashiyama, R.; Inoue, A.; Kato, Y.; Sato, T.; Lotz, M.K.; Asahara, H. MicroRNA-140 is expressed in differentiated human articular chondrocytes and modulates interleukin-1 responses. Arthritis Rheum. 2009, 60, 2723–2730. [Google Scholar] [CrossRef] [Green Version]
- Jeon, O.H.; Wilson, D.R.; Clement, C.C.; Rathod, S.; Cherry, C.; Powell, B.; Lee, Z.; Khalil, A.M.; Green, J.J.; Campisi, J.; et al. Senescence cell-associated extracellular vesicles serve as osteoarthritis disease and therapeutic markers. JCI Insight 2019, 4, e125019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Gibson, G.; Kim, J.S.; Kroin, J.; Xu, S.; van Wijnen, A.J.; Im, H.J. MicroRNA-146a is linked to pain-related pathophysiology of osteoarthritis. Gene 2011, 480, 34–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, W.S.; Ko, J.Y.; Wu, R.W.; Sun, Y.C.; Chen, Y.S.; Wu, S.L.; Weng, L.H.; Jahr, H.; Wang, F.S. MicroRNA-128a represses chondrocyte autophagy and exacerbates knee osteoarthritis by disrupting Atg12. Cell Death Dis. 2018, 9, 919. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, N.; Rasheed, Z.; Ramamurthy, S.; Anbazhagan, A.N.; Voss, F.R.; Haqqi, T.M. MicroRNA-27b regulates the expression of matrix metalloproteinase 13 in human osteoarthritis chondrocytes. Arthritis Rheum. 2010, 62, 1361–1371. [Google Scholar] [CrossRef]
- Wang, X.-b.; Zhao, F.-c.; Yi, L.-h.; Tang, J.-l.; Zhu, Z.-y.; Pang, Y.; Chen, Y.-s.; Li, D.-y.; Guo, K.-j.; Zheng, X. MicroRNA-21-5p as a novel therapeutic target for osteoarthritis. Rheumatology 2019, 58, 1485–1497. [Google Scholar] [CrossRef]
- Santini, P.; Politi, L.; Dalla Vedova, P.; Scandurra, R.; d’Abusco, A.S. The inflammatory circuitry of miR-149 as a pathological mechanism in osteoarthritis. Rheumatol. Int. 2014, 34, 711–716. [Google Scholar] [CrossRef]
- Evans, C.H.; Ghivizzani, S.C.; Robbins, P.D. Arthritis gene therapy and its tortuous path into the clinic. Transl. Res. 2013, 161, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef]
- Schmitz, S.U.; Grote, P.; Herrmann, B.G. Mechanisms of long noncoding RNA function in development and disease. Cell. Mol. Life Sci. 2016, 73, 2491–2509. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.-H.; Kim, J.; Gorospe, M. Long noncoding RNA turnover. Biochimie 2015, 117, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-Z.; Liu, H.; Chen, S.-R.J.C. Mechanisms of long non-coding RNAs in cancers and their dynamic regulations. Cancers 2020, 12, 1245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sun, X.; Chen, S.; Yang, C.; Shi, B.; Zhou, L.; Zhao, J. Long noncoding RNA DANCR regulates miR-1305-Smad 4 axis to promote chondrogenic differentiation of human synovium-derived mesenchymal stem cells. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, P.; Sun, X.; Zhou, L.; Zhao, J. Long non-coding RNA DANCR regulates proliferation and apoptosis of chondrocytes in osteoarthritis via miR-216a-5p-JAK2-STAT3 axis. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, F.; Su, K.; Sun, J.; Liao, W.; Yao, Y.; Zheng, Y.; Zhang, Z. The LncRNA ZBED3-AS1 induces chondrogenesis of human synovial fluid mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2017, 487, 457–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, H.L.; Quinn, J.J.; Yang, Y.W.; Thornburg, C.K.; Chang, H.Y.; Stadler, H.S. LncRNA-HIT functions as an epigenetic regulator of chondrogenesis through its recruitment of p100/CBP complexes. PLoS Genet. 2015, 11, e1005680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Peng, G.; Ning, X.; Wang, J.; Yang, H.; Deng, J. Emerging roles of long noncoding RNA in chondrogenesis, osteogenesis, and osteoarthritis. Am. J. Transl. Res. 2019, 11, 16–30. [Google Scholar]
- Xing, D.; Liang, J.Q.; Li, Y.; Lu, J.; Jia, H.B.; Xu, L.Y.; Ma, X.L. Identification of long noncoding RNA associated with osteoarthritis in humans. Orthop. Surg. 2014, 6, 288–293. [Google Scholar] [CrossRef]
- Li, X.; Ren, W.; Xiao, Z.Y.; Wu, L.F.; Wang, H.; Guo, P.Y. GACAT3 promoted proliferation of osteoarthritis synoviocytes by IL-6/STAT3 signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 5114–5120. [Google Scholar] [CrossRef]
- Xiao, Y.; Yan, X.; Yang, Y.; Ma, X. Downregulation of long noncoding RNA HOTAIRM1 variant 1 contributes to osteoarthritis via regulating miR-125b/BMPR2 axis and activating JNK/MAPK/ERK pathway. Biomed. Pharmacother. 2019, 109, 1569–1577. [Google Scholar] [CrossRef]
- Sun, J.; Song, X.; Su, L.; Cao, S. Long non-coding RNA LncHIFCAR promotes osteoarthritis development via positively regulating HIF-1α and activating the PI3K/AKT/mTOR pathway. Int. J. Exp. Pathol. 2018, 11, 3000–3009. [Google Scholar]
- Ye, D.; Jian, W.; Feng, J.; Liao, X. Role of long noncoding RNA ZFAS1 in proliferation, apoptosis and migration of chondrocytes in osteoarthritis. Biomed. Pharmacother. 2018, 104, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.-D.; Lu, J.; Deng, Z.-H.; Li, Y.-S.; Lei, G.-H. Long noncoding RNAs in osteoarthritis. Joint Bone Spine 2017, 84, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Perrin, S. Preclinical research: Make mouse studies work. Nature 2014, 507, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmi, G.; Faust, R. Suitability of the C57 black mouse as an experimental animal for the study of skeletal changes due to ageing, with special reference to osteo-arthrosis and its response to tribenoside. Pharmacology 1976, 14, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Walton, M. Degenerative joint disease in the mouse knee; histological observations. J. Pathol. 1977, 123, 109–122. [Google Scholar] [CrossRef]
- Poulet, B.; Ulici, V.; Stone, T.C.; Pead, M.; Gburcik, V.; Constantinou, E.; Palmer, D.B.; Beier, F.; Timmons, J.A.; Pitsillides, A.A. Time-series transcriptional profiling yields new perspectives on susceptibility to murine osteoarthritis. Arthritis Rheum. 2012, 64, 3256–3266. [Google Scholar] [CrossRef]
- Jimenez, P.A.; Glasson, S.S.; Trubetskoy, O.V.; Haimes, H.B. Spontaneous osteoarthritis in Dunkin Hartley guinea pigs: Histologic, radiologic, and biochemical changes. Lab. Anim. Sci. 1997, 47, 598–601. [Google Scholar]
- Simmons, H.A. Age-associated pathology in rhesus macaques (Macaca mulatta). Vet. Pathol. 2016, 53, 399–416. [Google Scholar] [CrossRef] [Green Version]
- Macfadyen, M.A.; Daniel, Z.; Kelly, S.; Parr, T.; Brameld, J.M.; Murton, A.J.; Jones, S.W. The commercial pig as a model of spontaneously-occurring osteoarthritis. BMC Musculoskel. Disord. 2019, 20, 70. [Google Scholar] [CrossRef]
- Glasson, S.S.; Blanchet, T.J.; Morris, E.A. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthr. Cartil. 2007, 15, 1061–1069. [Google Scholar] [CrossRef] [Green Version]
- Hayami, T.; Pickarski, M.; Wesolowski, G.A.; McLane, J.; Bone, A.; Destefano, J.; Rodan, G.A.; Duong, L.T. The role of subchondral bone remodeling in osteoarthritis: Reduction of cartilage degeneration and prevention of osteophyte formation by alendronate in the rat anterior cruciate ligament transection model. Arthritis Rheum. 2004, 50, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Knights, C.B.; Gentry, C.; Bevan, S. Partial medial meniscectomy produces osteoarthritis pain-related behaviour in female C57BL/6 mice. Pain 2012, 153, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Teeple, E.; Jay, G.D.; Elsaid, K.A.; Fleming, B.C. Animal models of osteoarthritis: Challenges of model selection and analysis. AAPS J. 2013, 15, 438–446. [Google Scholar] [CrossRef] [Green Version]
- Gregory, M.H.; Capito, N.; Kuroki, K.; Stoker, A.M.; Cook, J.L.; Sherman, S.L. A review of translational animal models for knee osteoarthritis. Arthritis 2012, 2012, 764621. [Google Scholar] [CrossRef] [PubMed]
- Cake, M.A.; Read, R.A.; Corfield, G.; Daniel, A.; Burkhardt, D.; Smith, M.M.; Little, C.B. Comparison of gait and pathology outcomes of three meniscal procedures for induction of knee osteoarthritis in sheep. Osteoarthr. Cartil. 2013, 21, 226–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christiansen, B.A.; Guilak, F.; Lockwood, K.A.; Olson, S.A.; Pitsillides, A.A.; Sandell, L.J.; Silva, M.J.; van der Meulen, M.C.H.; Haudenschild, D.R. Non-invasive mouse models of post-traumatic osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1627–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furman, B.D.; Strand, J.; Hembree, W.C.; Ward, B.D.; Guilak, F.; Olson, S.A. Joint degeneration following closed intraarticular fracture in the mouse knee: A model of posttraumatic arthritis. J. Orthop. Res. 2007, 25, 578–592. [Google Scholar] [CrossRef]
- De Souza, R.L.; Matsuura, M.; Eckstein, F.; Rawlinson, S.C.; Lanyon, L.E.; Pitsillides, A.A. Non-invasive axial loading of mouse tibiae increases cortical bone formation and modifies trabecular organization: A new model to study cortical and cancellous compartments in a single loaded element. Bone 2005, 37, 810–818. [Google Scholar] [CrossRef]
- Poulet, B.; Hamilton, R.W.; Shefelbine, S.; Pitsillides, A.A. Characterizing a novel and adjustable noninvasive murine joint loading model. Arthritis Rheum. 2011, 63, 137–147. [Google Scholar] [CrossRef]
- Christiansen, B.A.; Anderson, M.J.; Lee, C.A.; Williams, J.C.; Yik, J.H.; Haudenschild, D.R. Musculoskeletal changes following non-invasive knee injury using a novel mouse model of post-traumatic osteoarthritis. Osteoarthr. Cartil. 2012, 20, 773–782. [Google Scholar] [CrossRef] [Green Version]
- Alexander, P.G.; Song, Y.; Taboas, J.M.; Chen, F.H.; Melvin, G.M.; Manner, P.A.; Tuan, R.S. Development of a spring-loaded impact device to deliver injurious mechanical impacts to the articular cartilage surface. Cartilage 2013, 4, 52–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnevie, E.D.; Delco, M.L.; Fortier, L.A.; Alexander, P.G.; Tuan, R.S.; Bonassar, L.J. Characterization of tissue response to impact loads delivered using a hand-held instrument for studying articular cartilage injury. Cartilage 2015, 6, 226–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thijssen, E.; van Caam, A.; van der Kraan, P.M. Obesity and osteoarthritis, more than just wear and tear: Pivotal roles for inflamed adipose tissue and dyslipidaemia in obesity-induced osteoarthritis. Rheumatology 2015, 54, 588–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azamar-Llamas, D.; Hernández-Molina, G.; Ramos-Ávalos, B.; Furuzawa-Carballeda, J. Adipokine contribution to the pathogenesis of osteoarthritis. Mediators Inflamm. 2017, 2017, 5468023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansone, V.; Applefield, R.C.; De Luca, P.; Pecoraro, V.; Gianola, S.; Pascale, W.; Pascale, V. Does a high-fat diet affect the development and progression of osteoarthritis in mice?: A systematic review. Bone Jt. Res. 2019, 8, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Griffin, T.M.; Fermor, B.; Huebner, J.L.; Kraus, V.B.; Rodriguiz, R.M.; Wetsel, W.C.; Cao, L.; Setton, L.A.; Guilak, F. Diet-induced obesity differentially regulates behavioral, biomechanical, and molecular risk factors for osteoarthritis in mice. Arthritis Res. Ther. 2010, 12, R130. [Google Scholar] [CrossRef] [Green Version]
- Griffin, T.M.; Huebner, J.L.; Kraus, V.B.; Yan, Z.; Guilak, F. Induction of osteoarthritis and metabolic inflammation by a very high-fat diet in mice: Effects of short-term exercise. Arthritis Rheum. 2012, 64, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Schott, E.M.; Farnsworth, C.W.; Grier, A.; Lillis, J.A.; Soniwala, S.; Dadourian, G.H.; Bell, R.D.; Doolittle, M.L.; Villani, D.A.; Awad, H.; et al. Targeting the gut microbiome to treat the osteoarthritis of obesity. JCI Insight 2018, 3, e95997. [Google Scholar] [CrossRef]
- Louer, C.R.; Furman, B.D.; Huebner, J.L.; Kraus, V.B.; Olson, S.A.; Guilak, F. Diet-induced obesity significantly increases the severity of posttraumatic arthritis in mice. Arthritis Rheum. 2012, 64, 3220–3230. [Google Scholar] [CrossRef]
- Datta, P.; Zhang, Y.; Parousis, A.; Sharma, A.; Rossomacha, E.; Endisha, H.; Wu, B.; Kacprzak, I.; Mahomed, N.N.; Gandhi, R.; et al. High-fat diet-induced acceleration of osteoarthritis is associated with a distinct and sustained plasma metabolite signature. Sci. Rep. 2017, 7, 8205. [Google Scholar] [CrossRef] [Green Version]
- Barve, R.A.; Minnerly, J.C.; Weiss, D.J.; Meyer, D.M.; Aguiar, D.J.; Sullivan, P.M.; Weinrich, S.L.; Head, R.D. Transcriptional profiling and pathway analysis of monosodium iodoacetate-induced experimental osteoarthritis in rats: Relevance to human disease. Osteoarthr. Cartil. 2007, 15, 1190–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guingamp, C.; Gegout-Pottie, P.; Philippe, L.; Terlain, B.; Netter, P.; Gillet, P. Mono-iodoacetate-induced experimental osteoarthritis: A dose-response study of loss of mobility, morphology, and biochemistry. Arthritis Rheum. 1997, 40, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Adães, S.; Mendonça, M.; Santos, T.N.; Castro-Lopes, J.M.; Ferreira-Gomes, J.; Neto, F.L. Intra-articular injection of collagenase in the knee of rats as an alternative model to study nociception associated with osteoarthritis. Arthritis Res. Ther. 2014, 16, R10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sendzik, J.; Lode, H.; Stahlmann, R. Quinolone-induced arthropathy: An update focusing on new mechanistic and clinical data. Int. J. Antimicrob. Agents 2009, 33, 194–200. [Google Scholar] [CrossRef]
- Guzman, R.E.; Evans, M.G.; Bove, S.; Morenko, B.; Kilgore, K. Mono-iodoacetate-induced histologic changes in subchondral bone and articular cartilage of rat femorotibial joints: An animal model of osteoarthritis. Toxicol. Pathol. 2003, 31, 619–624. [Google Scholar] [CrossRef]
- Cifuentes, D.J.; Rocha, L.G.; Silva, L.A.; Brito, A.C.; Rueff-Barroso, C.R.; Porto, L.C.; Pinho, R.A. Decrease in oxidative stress and histological changes induced by physical exercise calibrated in rats with osteoarthritis induced by monosodium iodoacetate. Osteoarthr. Cartil. 2010, 18, 1088–1095. [Google Scholar] [CrossRef] [Green Version]
- Ogbonna, A.C.; Clark, A.K.; Gentry, C.; Hobbs, C.; Malcangio, M. Pain-like behaviour and spinal changes in the monosodium iodoacetate model of osteoarthritis in C57Bl/6 mice. Eur. J. Pain 2013, 17, 514–526. [Google Scholar] [CrossRef]
- Justice, M.J.; Dhillon, P. Using the mouse to model human disease: Increasing validity and reproducibility. Dis. Models Mech. 2016, 9, 101. [Google Scholar] [CrossRef] [Green Version]
- Gurumurthy, C.B.; Lloyd, K.C.K. Generating mouse models for biomedical research: Technological advances. Dis. Models Mech. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Malfait, A.M.; Ritchie, J.; Gil, A.S.; Austin, J.S.; Hartke, J.; Qin, W.; Tortorella, M.D.; Mogil, J.S. ADAMTS-5 deficient mice do not develop mechanical allodynia associated with osteoarthritis following medial meniscal destabilization. Osteoarthr. Cartil. 2010, 18, 572–580. [Google Scholar] [CrossRef] [Green Version]
- Ferrell, W.R.; Kelso, E.B.; Lockhart, J.C.; Plevin, R.; McInnes, I.B. Protease-activated receptor 2: A novel pathogenic pathway in a murine model of osteoarthritis. Ann. Rheum. Dis. 2010, 69, 2051–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Rozelle, A.L.; Lepus, C.M.; Scanzello, C.R.; Song, J.J.; Larsen, D.M.; Crish, J.F.; Bebek, G.; Ritter, S.Y.; Lindstrom, T.M.; et al. Identification of a central role for complement in osteoarthritis. Nat. Med. 2011, 17, 1674–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, J.H.; Shin, Y.; Huh, Y.H.; Yang, S.; Chun, C.H.; Chun, J.S. Hypoxia-inducible factor-2α regulates Fas-mediated chondrocyte apoptosis during osteoarthritic cartilage destruction. Cell Death Differ. 2012, 19, 440–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Little, C.B.; Barai, A.; Burkhardt, D.; Smith, S.M.; Fosang, A.J.; Werb, Z.; Shah, M.; Thompson, E.W. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009, 60, 3723–3733. [Google Scholar] [CrossRef] [PubMed]
- Neuhold, L.A.; Killar, L.; Zhao, W.; Sung, M.L.; Warner, L.; Kulik, J.; Turner, J.; Wu, W.; Billinghurst, C.; Meijers, T.; et al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J. Clin. Investig. 2001, 107, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Säämänen, A.K.; Salminen, H.J.; Dean, P.B.; De Crombrugghe, B.; Vuorio, E.I.; Metsäranta, M.P. Osteoarthritis-like lesions in transgenic mice harboring a small deletion mutation in type II collagen gene. Osteoarthr. Cartil. 2000, 8, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Hu, K.; Xu, L.; Cao, L.; Flahiff, C.M.; Brussiau, J.; Ho, K.; Setton, L.A.; Youn, I.; Guilak, F.; Olsen, B.R.; et al. Pathogenesis of osteoarthritis-like changes in the joints of mice deficient in type IX collagen. Arthritis Rheum. 2006, 54, 2891–2900. [Google Scholar] [CrossRef]
- Tortorella, M.D.; Malfait, A.M.; Deccico, C.; Arner, E. The role of ADAM-TS4 (aggrecanase-1) and ADAM-TS5 (aggrecanase-2) in a model of cartilage degradation. Osteoarthr. Cartil. 2001, 9, 539–552. [Google Scholar] [CrossRef] [Green Version]
- Glasson, S.S.; Askew, R.; Sheppard, B.; Carito, B.A.; Blanchet, T.; Ma, H.L.; Flannery, C.R.; Kanki, K.; Wang, E.; Peluso, D.; et al. Characterization of and osteoarthritis susceptibility in ADAMTS-4-knockout mice. Arthritis Rheum. 2004, 50, 2547–2558. [Google Scholar] [CrossRef]
- Glasson, S.S.; Askew, R.; Sheppard, B.; Carito, B.; Blanchet, T.; Ma, H.L.; Flannery, C.R.; Peluso, D.; Kanki, K.; Yang, Z.; et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 2005, 434, 644–648. [Google Scholar] [CrossRef]
- Little, C.B.; Meeker, C.T.; Golub, S.B.; Lawlor, K.E.; Farmer, P.J.; Smith, S.M.; Fosang, A.J. Blocking aggrecanase cleavage in the aggrecan interglobular domain abrogates cartilage erosion and promotes cartilage repair. J. Clin. Investig. 2007, 117, 1627–1636. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Li, J.; Wang, B.; Jin, H.; Wang, M.; Zhang, Y.; Yang, Y.; Im, H.J.; O’Keefe, R.; Chen, D. Deletion of the transforming growth factor β receptor type II gene in articular chondrocytes leads to a progressive osteoarthritis-like phenotype in mice. Arthritis Rheum. 2013, 65, 3107–3119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Chen, Q.; Lanske, B.; Fleming, B.C.; Terek, R.; Wei, X.; Zhang, G.; Wang, S.; Li, K.; Wei, L. Disrupting the Indian hedgehog signaling pathway in vivo attenuates surgically induced osteoarthritis progression in Col2a1-CreERT2; Ihhfl/fl mice. Arthritis Res. Ther. 2014, 16, R11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corr, M. Wnt–β-catenin signaling in the pathogenesis of osteoarthritis. Nat. Clin. Pract. Rheumatol. 2008, 4, 550–556. [Google Scholar] [CrossRef]
- Luyten, F.P.; Tylzanowski, P.; Lories, R.J. Wnt signaling and osteoarthritis. Bone 2009, 44, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Little, C.B.; Hunter, D.J. Post-traumatic osteoarthritis: From mouse models to clinical trials. Nat. Rev. Rheumatol. 2013, 9, 485–497. [Google Scholar] [CrossRef]
- Fujisawa, T.; Hattori, T.; Takahashi, K.; Kuboki, T.; Yamashita, A.; Takigawa, M. Cyclic mechanical stress induces extracellular matrix degradation in cultured chondrocytes via gene expression of matrix metalloproteinases and interleukin-11. J. Biochem. 1999, 125, 966–975. [Google Scholar] [CrossRef]
- Honda, K.; Ohno, S.; Tanimoto, K.; Ijuin, C.; Tanaka, N.; Doi, T.; Kato, Y.; Tanne, K. The effects of high magnitude cyclic tensile load on cartilage matrix metabolism in cultured chondrocytes. Eur. J. Cell Biol. 2000, 79, 601–609. [Google Scholar] [CrossRef]
- Huang, J.; Ballou, L.R.; Hasty, K.A. Cyclic equibiaxial tensile strain induces both anabolic and catabolic responses in articular chondrocytes. Gene 2007, 404, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.; Kanda, V.; Bush-Joseph, C.; Verma, N.; Chubinskaya, S.; Mikecz, K.; Glant, T.T.; Malfait, A.M.; Crow, M.K.; Spear, G.T.; et al. Synovial fluid from patients with early osteoarthritis modulates fibroblast-like synoviocyte responses to toll-like receptor 4 and toll-like receptor 2 ligands via soluble CD14. Arthritis Rheum. 2012, 64, 2268–2277. [Google Scholar] [CrossRef] [Green Version]
- Ribel-Madsen, S.; Bartels, E.M.; Stockmarr, A.; Borgwardt, A.; Cornett, C.; Danneskiold-Samsøe, B.; Bliddal, H. A synoviocyte model for osteoarthritis and rheumatoid arthritis: Response to Ibuprofen, betamethasone, and ginger extract-a cross-sectional in vitro study. Arthritis 2012, 2012, 505842. [Google Scholar] [CrossRef]
- Pearson, M.J.; Herndler-Brandstetter, D.; Tariq, M.A.; Nicholson, T.A.; Philp, A.M.; Smith, H.L.; Davis, E.T.; Jones, S.W.; Lord, J.M. IL-6 secretion in osteoarthritis patients is mediated by chondrocyte-synovial fibroblast cross-talk and is enhanced by obesity. Sci. Rep. 2017, 7, 3451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, M.C.; Saunders, K.M.; Burton-Wurster, N.; Macleod, J.N. Phenotypic stability of articular chondrocytes in vitro: The effects of culture models, bone morphogenetic protein 2, and serum supplementation. J. Bone Miner. Res. 2000, 15, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Caron, M.M.J.; Emans, P.J.; Coolsen, M.M.E.; Voss, L.; Surtel, D.A.M.; Cremers, A.; van Rhijn, L.W.; Welting, T.J.M. Redifferentiation of dedifferentiated human articular chondrocytes: Comparison of 2D and 3D cultures. Osteoarthr. Cartil. 2012, 20, 1170–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, P.; Cheng, K.H.; Yan, C.H.; Chan, B.P. Collagen microsphere based 3D culture system for human osteoarthritis chondrocytes (hOACs). Sci. Rep. 2019, 9, 12453. [Google Scholar] [CrossRef]
- Samavedi, S.; Diaz-Rodriguez, P.; Erndt-Marino, J.D.; Hahn, M.S. A three-dimensional chondrocyte-macrophage coculture system to probe inflammation in experimental osteoarthritis. Tissue Eng. Part A 2016, 23, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wang, X.; Kaplan, D.L. A 3D cartilage-inflammatory cell culture system for the modeling of human osteoarthritis. Biomaterials 2011, 32, 5581–5589. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.H.; Lo, W.C.; Hsu, W.C.; Wei, H.J.; Liu, H.Y.; Lee, C.H.; Tina Chen, S.Y.; Shieh, Y.H.; Williams, D.F.; Deng, W.P. Synergistic anabolic actions of hyaluronic acid and platelet-rich plasma on cartilage regeneration in osteoarthritis therapy. Biomaterials 2014, 35, 9599–9607. [Google Scholar] [CrossRef]
- Deng, Y.; Lei, G.; Lin, Z.; Yang, Y.; Lin, H.; Tuan, R.S. Engineering hyaline cartilage from mesenchymal stem cells with low hypertrophy potential via modulation of culture conditions and Wnt/β-catenin pathway. Biomaterials 2019, 192, 569–578. [Google Scholar] [CrossRef]
- Shen, H.; Lin, H.; Sun, A.X.; Song, S.; Wang, B.; Yang, Y.; Dai, J.; Tuan, R.S. Acceleration of chondrogenic differentiation of human mesenchymal stem cells by sustained growth factor release in 3D graphene oxide incorporated hydrogels. Acta Biomater. 2020, 105, 44–55. [Google Scholar] [CrossRef]
- Delise, A.M.; Tuan, R.S. Analysis of N-cadherin function in limb mesenchymal chondrogenesis in vitro. Dev. Dyn. 2002, 225, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kuang, B.; Rothrauff, B.B.; Tuan, R.S.; Lin, H. Robust bone regeneration through endochondral ossification of human mesenchymal stem cells within their own extracellular matrix. Biomaterials 2019, 218, 119336. [Google Scholar] [CrossRef] [PubMed]
- Grenier, S.; Bhargava, M.M.; Torzilli, P.A. An in vitro model for the pathological degradation of articular cartilage in osteoarthritis. J. Biomech. 2014, 47, 645–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Jiang, Y.; Alexander, P.G.; Ulici, V.; Zhu, Y.; Wu, S.; Tuan, R.S. Infrapatellar fat pad aggravates degeneration of acute traumatized cartilage: A possible role for interleukin-6. Osteoarthr. Cartil. 2017, 25, 138–145. [Google Scholar] [CrossRef]
- Nishimuta, J.F.; Bendernagel, M.F.; Levenston, M.E. Co-culture with infrapatellar fat pad differentially stimulates proteoglycan synthesis and accumulation in cartilage and meniscus tissues. Connect. Tissue Res. 2017, 58, 447–455. [Google Scholar] [CrossRef] [Green Version]
- Nishimuta, J.F.; Levenston, M.E. Adipokines induce catabolism of newly synthesized matrix in cartilage and meniscus tissues. Connect. Tissue Res. 2017, 58, 246–258. [Google Scholar] [CrossRef]
- Beekhuizen, M.; Bastiaansen-Jenniskens, Y.M.; Koevoet, W.; Saris, D.B.F.; Dhert, W.J.A.; Creemers, L.B.; van Osch, G.J.V.M. Osteoarthritic synovial tissue inhibition of proteoglycan production in human osteoarthritic knee cartilage: Establishment and characterization of a long-term cartilage–synovium coculture. Arthritis Rheum. 2011, 63, 1918–1927. [Google Scholar] [CrossRef]
- Greenberg, D.D.; Stoker, A.; Kane, S.; Cockrell, M.; Cook, J.L. Biochemical effects of two different hyaluronic acid products in a co-culture model of osteoarthritis. Osteoarthr. Cartil. 2006, 14, 814–822. [Google Scholar] [CrossRef] [Green Version]
- Occhetta, P.; Mainardi, A.; Votta, E.; Vallmajo-Martin, Q.; Ehrbar, M.; Martin, I.; Barbero, A.; Rasponi, M. Hyperphysiological compression of articular cartilage induces an osteoarthritic phenotype in a cartilage-on-a-chip model. Nat. Biomed. Eng. 2019, 3, 1. [Google Scholar] [CrossRef]
- Rosser, J.; Bachmann, B.; Jordan, C.; Ribitsch, I.; Haltmayer, E.; Gueltekin, S.; Junttila, S.; Galik, B.; Gyenesei, A.; Haddadi, B.; et al. Microfluidic nutrient gradient–based three-dimensional chondrocyte culture-on-a-chip as an in vitro equine arthritis model. Mater. Today 2019, 4, 100023. [Google Scholar] [CrossRef]
- Lin, H.; Lozito, T.P.; Alexander, P.G.; Gottardi, R.; Tuan, R.S. Stem cell-based microphysiological osteochondral system to model tissue response to interleukin-1β. Mol. Pharm. 2014, 11, 2203–2212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Li, Z.; Li, E.N.; Li, X.; Del Duke, C.J.; Shen, H.; Hao, T.; O’Donnell, B.; Bunnell, B.A.; Goodman, S.B.; et al. Osteochondral tissue chip derived from iPSCs: Modeling OA pathologies and testing drugs. Front. Bioeng. Biotechnol. 2019, 7, 411. [Google Scholar] [CrossRef] [PubMed]
- Macchi, V.; Stocco, E.; Stecco, C.; Belluzzi, E.; Favero, M.; Porzionato, A.; De Caro, R. The infrapatellar fat pad and the synovial membrane: An anatomo-functional unit. J. Anat. 2018, 233, 146–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eymard, F.; Pigenet, A.; Citadelle, D.; Tordjman, J.; Foucher, L.; Rose, C.; Flouzat Lachaniette, C.H.; Rouault, C.; Clément, K.; Berenbaum, F.; et al. Knee and hip intra-articular adipose tissues (IAATs) compared with autologous subcutaneous adipose tissue: A specific phenotype for a central player in osteoarthritis. Ann. Rheum. Dis. 2017, 76, 1142–1148. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.S.; Ellman, M.B.; Yan, D.; Kroin, J.S.; Cole, B.J.; van Wijnen, A.J.; Im, H.J. A current review of molecular mechanisms regarding osteoarthritis and pain. Gene 2013, 527, 440–447. [Google Scholar] [CrossRef] [Green Version]
- Deshmukh, V.; O’Green, A.L.; Bossard, C.; Seo, T.; Lamangan, L.; Ibanez, M.; Ghias, A.; Lai, C.; Do, L.; Cho, S.; et al. Modulation of the Wnt pathway through inhibition of CLK2 and DYRK1A by lorecivivint as a novel, potentially disease-modifying approach for knee osteoarthritis treatment. Osteoarthr. Cartil. 2019, 27, 1347–1360. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chen, L.; Yin, Q.; Gao, H.; Dong, P.; Zhang, X.; Kang, J. Reciprocal interferences of TNF-α and Wnt1/β-catenin signaling axes shift bone marrow-derived stem cells towards osteoblast lineage after ethanol exposure. Cell. Physiol. Biochem. 2013, 32, 755–765. [Google Scholar] [CrossRef]
- Oo, W.M.; Yu, S.P.-C.; Daniel, M.S.; Hunter, D.J. Disease-modifying drugs in osteoarthritis: Current understanding and future therapeutics. Expert Opin. Emerg. Drugs 2018, 23, 331–347. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Cells Studied | Effects | Ref. |
|---|---|---|---|
| Wnt/β-catenin |
|
| [90,91,92,93,94] |
|
| [97,98] | |
| PI3K/Akt/mTOR |
|
| [105,106] |
| Notch |
|
| [110,118,119] |
| [120,121] | ||
| SIRT1/AMPK |
|
| [123,124,125,126] |
| Hippo/YAP/TAZ |
|
| [129,130] |
|
| [132,133,134,135] | |
| DOT1L |
|
| [139,140,141,142] |
| In Vivo Models | Advantages | Limitations | Ref. |
|---|---|---|---|
| Aging-induced spontaneous OA models |
|
| [176,177] |
| Trauma-induced OA models |
|
| [186,226] |
| Obesity-induced OA models |
|
| [197,200] |
| Chemically induced OA models |
|
| [204,206] |
| OA models involving genetic manipulations |
|
| [212,214] |
| Current In Vitro Models | Advantages | Limitations | Ref. |
|---|---|---|---|
| Monolayer culture |
|
| [227,232] |
| 3D engineered cartilage tissues |
|
| [235,236] |
| Tissue explant models |
|
| [245,247] |
| Microphysiological systems |
|
| [249,252] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; Li, Z.; Alexander, P.G.; Ocasio-Nieves, B.D.; Yocum, L.; Lin, H.; Tuan, R.S. Pathogenesis of Osteoarthritis: Risk Factors, Regulatory Pathways in Chondrocytes, and Experimental Models. Biology 2020, 9, 194. https://doi.org/10.3390/biology9080194
He Y, Li Z, Alexander PG, Ocasio-Nieves BD, Yocum L, Lin H, Tuan RS. Pathogenesis of Osteoarthritis: Risk Factors, Regulatory Pathways in Chondrocytes, and Experimental Models. Biology. 2020; 9(8):194. https://doi.org/10.3390/biology9080194
Chicago/Turabian StyleHe, Yuchen, Zhong Li, Peter G. Alexander, Brian D. Ocasio-Nieves, Lauren Yocum, Hang Lin, and Rocky S. Tuan. 2020. "Pathogenesis of Osteoarthritis: Risk Factors, Regulatory Pathways in Chondrocytes, and Experimental Models" Biology 9, no. 8: 194. https://doi.org/10.3390/biology9080194