
Bacterial Subcellular Architecture, Structural Epistasis, and Antibiotic Resistance

 , ,
, , {kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
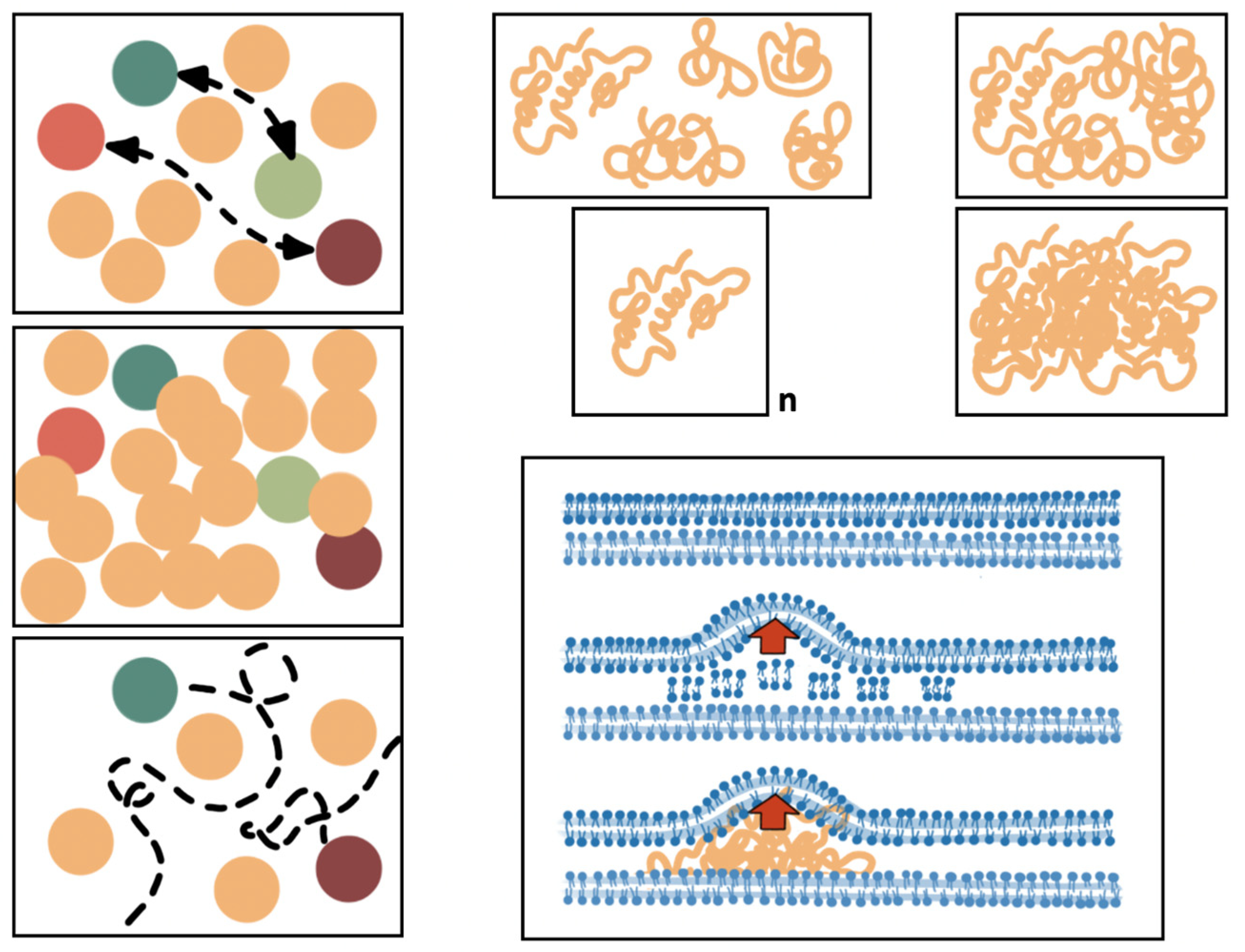
2. The Molecular Components Involved in Structural Epistasis
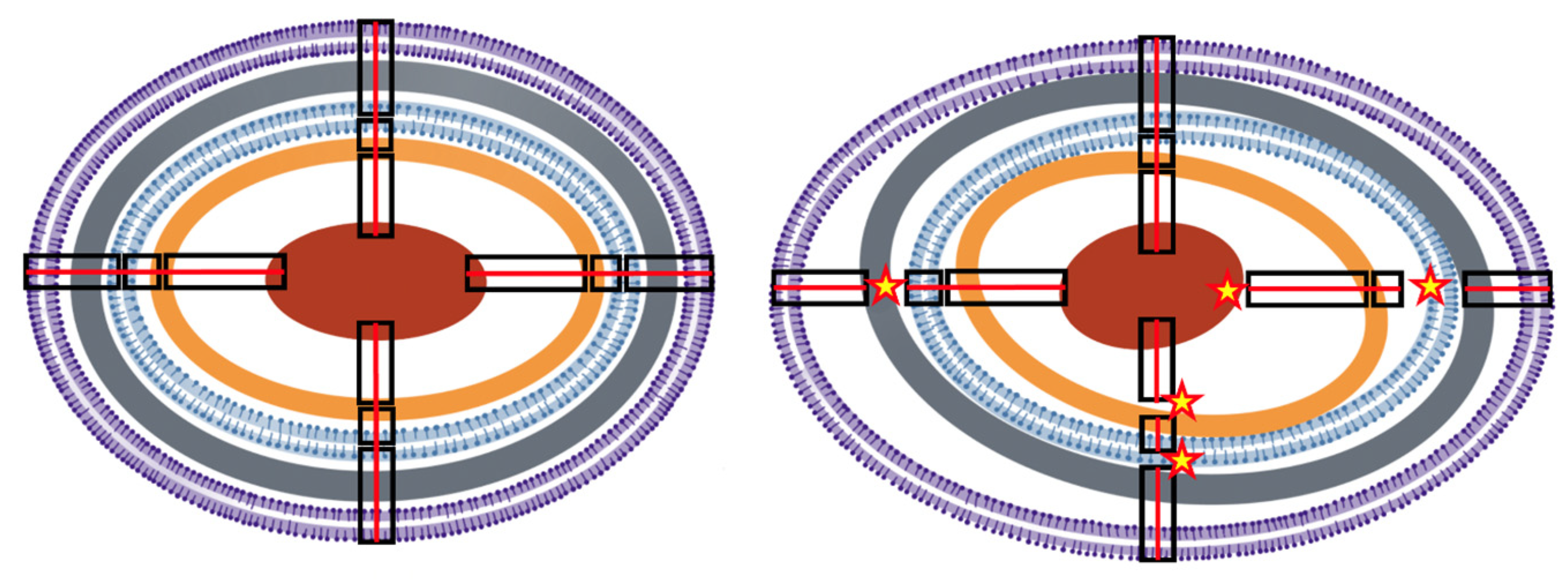
3. The Bacterial Cell Molecular Architecture and Shape Is Altered by Antibiotic Exposure
4. The Bacterial Cell Architecture and Shape Is Altered by Antibiotic Resistance
5. Cell Architecture and Cell Size Influences Antibiotic Effects
6. Spatial Cell Biology, Molecular Interactome, and Antibiotic Actions
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Starr, T.N.; Thornton, J.W. Epistasis in Protein Evolution. Protein Sci. 2016, 25, 1204–1218. [Google Scholar] [CrossRef]
- Phillips, P.C. Epistasis—The Essential Role of Gene Interactions in the Structure and Evolution of Genetic Systems. Nat. Rev. Gen. 2008, 9, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Baquero, F. Epigenetics, Epistasis and Epidemics. Evol. Med. Public Health 2013, 1, 86–88. [Google Scholar] [CrossRef]
- Olivares, J.; Alvarez-Ortega, C.; Linares, J.F.; Rojo, F.; Köhler, T.; Martínez, J.L. Overproduction of the Multidrug Efflux Pump MexEF-OprN Does Not Impair Pseudomonas aeruginosa Fitness in Competition Tests, but Produces Specific Changes in Bacterial Regulatory Networks. Environ. Microbiol. 2012, 14, 1968–1981. [Google Scholar] [CrossRef]
- Olivares Pacheco, J.; Alvarez-Ortega, C.; Alcalde Rico, M.; Martínez, J.L. Metabolic Compensation of Fitness Costs is a General Outcome for Antibiotic-Resistant Pseudomonas aeruginosa Mutants Overexpressing Efflux Pumps. MBio 2017, 8, e00500-17. [Google Scholar] [CrossRef] [PubMed]
- Hernando-Amado, S.; Laborda, P.; Valverde, J.R.; Martínez, J.L. Mutational Background Influences, P. aeruginosa Ciprofloxacin Resistance Evolution but Preserves Collateral Sensitivity Robustness. Proc. Natl. Acad. Sci. USA 2022, 119, e2109370119. [Google Scholar] [CrossRef]
- Zimmerman, S.B.; Trach, S.O. Estimation of Macromolecule Concentrations and Excluded Volume Effects for the Cytoplasm of Escherichia coli. J. Mol. Biol. 1991, 222, 599–620. [Google Scholar] [CrossRef] [PubMed]
- Speer, S.L.; Zheng, W.; Jiang, X.; Chu, I.T.; Guseman, A.J.; Liu, M.; Pielak, J.; Li, C. The Intracellular Environment Affects Protein–Protein Interactions. Proc. Natl. Acad. Sci. USA 2021, 118, e2019918118. [Google Scholar] [CrossRef]
- Wolf, J.B.; Brodie, E.D.; Wade, M.J.; Wade, M.J. (Eds.) Epistasis and the Evolutionary Process; Oxford University Press: Cary, NC, USA, 2000. [Google Scholar]
- Li, Y.; Naveed, H.; Kachalo, S.; Xu, L.X.; Liang, J. Mechanisms of Regulating Cell Topology in Proliferating Epithelia: Impact of Division Plane, Mechanical Forces, and Cell Memory. PLoS ONE 2012, 7, e43108. [Google Scholar] [CrossRef]
- Baum, M.; Erdel, F.; Wachsmuth, M.; Rippe, K. Retrieving the Intracellular Topology from Multi-Scale Protein Mobility Mapping in Living Cells. Nat. Comm. 2014, 5, 4494. [Google Scholar] [CrossRef]
- Weber, S.C.; Spakowitz, A.J.; Theriot, J.A. Bacterial Chromosomal Loci Move Subdiffusively through a Viscoelastic Cytoplasm. Phys. Rev. Lett. 2010, 104, 238102. [Google Scholar] [CrossRef]
- Lu, J.; Deutsch, C. Electrostatics in the Ribosomal Tunnel Modulate Chain Elongation Rates. J. Mol. Biol. 2008, 384, 73–86. [Google Scholar] [CrossRef]
- Scholz, S.A.; Diao, R.; Wolfe, M.B.; Fivenson, E.M.; Lin, X.N.; Freddolino, P.L. High-Resolution Mapping of the Escherichia coli Chromosome Reveals Positions of High and Low transcription. Cell Syst. 2019, 8, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Lioy, V.S.; Cournac, A.; Marbouty, M.; Duigou, S.; Mozziconacci, J.; Espéli, O.; Broccard, F.; Koszul, R. Multiscale Structuring of the E. coli Chromosome by Nucleoid-Associated and Condensing Proteins. Cell 2018, 172, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Dorman, C.J. H-NS: A Universal Regulator for a Dynamic Genome. Nat. Rev. Microbiol. 2004, 2, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Vora, T.; Hottes, A.K.; Tavazoie, S. Protein Occupancy Landscape of a Bacterial Genome. Mol. Cell 2009, 35, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Merrikh, H.; Zhang, Y.; Grossman, A.D.; Wang, J.D. Replication–transcription conflicts in bacteria. Nat. Rev. Microbiol. 2012, 10, 449–458. [Google Scholar] [CrossRef]
- Sadoon, A.A.; Wang, Y. Anomalous, non-Gaussian, Viscoelastic, and Age-Dependent Dynamics of Histone-Like Nucleoid-Structuring Proteins in Live Escherichia coli. Phys. Rev. 2018, E98, 042411. [Google Scholar]
- Lewis, P.J. Bacterial Subcellular Architecture: Recent Advances and Future Prospects. Mol. Microbiol. 2004, 54, 1135–1150. [Google Scholar] [CrossRef]
- Karig, D.; Martini, K.M.; Lu, T.; De Lateur, N.A.; Goldenfeld, N.; Weiss, R. Stochastic Turing Patterns in a Synthetic Bacterial Population. Proc. Natl. Acad. Sci. USA 2018, 115, 6572–6577. [Google Scholar] [CrossRef]
- Silhavy, T.J.; Kahne, D.; Walker, S. The Bacterial Cell Envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef] [PubMed]
- Denich, T.J.; Beaudette, L.A.; Lee, H.; Trevors, J.T. Effect of Selected Environmental and Physico-Chemical Factors on Bacterial Cytoplasmic Membranes. J. Microb. Meth. 2003, 52, 149–182. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.A. The Role of Membrane Lipid Remodelling in the Antimicrobial Resistance Arsenal of Pseudomonas aeruginosa. Ph.D. Thesis, University of Warwick, Warwick, UK, 2020. [Google Scholar]
- Asmar, A.T.; Collet, J.F. Lpp, The Braun Lipoprotein, Turns 50—Major Achievements and Remaining Issues. FEMS Microbiol. Lett. 2018, 365, fny199. [Google Scholar] [CrossRef]
- Kleanthous, C.; Rassam, P.; Baumann, C.G. Protein–Protein Interactions and the Spatiotemporal Dynamics of Bacterial Outer Membrane Proteins. Curr. Opin. Struct. Biol. 2015, 35, 109–115. [Google Scholar] [CrossRef]
- Randich, A.M.; Brun, Y.V. Molecular Mechanisms for the Evolution of Bacterial Morphologies and Growth Modes. Front. Microbiol. 2015, 6, 580. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, W.; Blanot, D.; de Pedro, M.A. Peptidoglycan Structure And Architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [Google Scholar] [CrossRef]
- Mullineaux, C.W. Classic Spotlight: To the Periplasm and Beyond—Protein Secretion in Escherichia coli. J. Bacteriol. 2016, 198, 2017. [Google Scholar] [CrossRef]
- Zgurskaya, H.I.; Nikaido, H. Multidrug Resistance Mechanisms: Drug Efflux Across Two Membranes. Mol. Microbiol. 2000, 37, 219–225. [Google Scholar] [CrossRef]
- De Geyter, J.; Tsirigotaki, A.; Orfanoudaki, G.; Zorzini, V.; Economou, A.; Karamanou, S. Protein Folding in the Cell Envelope of Escherichia coli. Nat. Microbiol. 2016, 1, 16107. [Google Scholar] [CrossRef]
- Papanastasiou, M.; Orfanoudaki, G.; Koukaki, M.; Kountourakis, N.; Sardis, M.F.; Aivaliotis, M.; Karamanou, S.; Economou, A. The Escherichia coli Peripheral Inner Membrane Proteome. Mol. Cell Proteom. 2013, 2, 599–610. [Google Scholar] [CrossRef]
- Matsumoto, K.; Hara, H.; Fishov, I.; Mileykovskaya, E.; Norris, V. The Membrane: Transertion as an Organizing Principle in Membrane Heterogeneity. Front. Microbiol. 2015, 12, 572. [Google Scholar] [CrossRef]
- Mayer, B.; Schwan, M.; Oviedo-Bocanegra, L.M.; Bange, G.; Thormann, K.M.; Graumann, P.L. Dynamics of Bacterial Signal Recognition Particle at a Single Molecule Level. Front. Microbiol. 2021, 12, 663747. [Google Scholar] [CrossRef]
- Herskovits, A.A.; Bibi, E. Association of Escherichia coli Ribosomes with the Inner Membrane Requires the Signal Recognition Particle Receptor but is Independent of the Signal Recognition Particle. Proc. Natl. Acad. Sci. USA 2000, 97, 4621–4626. [Google Scholar] [CrossRef]
- Bakshi, S.; Siryaporn, A.; Goulian, M.; Weisshaar, J.C. Superresolution Imaging of Ribosomes and Rna Polymerase in Live Escherichia coli Cells. Mol. Microbiol. 2012, 85, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Robinow, C.; Kellenberger, E. The Bacterial Nucleoid Revisited. Microbiol. Rev. 1994, 58, 211–232. [Google Scholar] [CrossRef]
- Gohara, D.W.; Yap, M.N.F. Survival of The Drowsiest: The Hibernating 100S Ribosome in Bacterial Stress Management. Curr. Genet. 2018, 64, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Saberi, S.; Emberly, E. Chromosome driven spatial patterning of proteins in bacteria. PLoS Computat. Biol. 2010, 6, e1000986. [Google Scholar] [CrossRef]
- Thanbichler, M.; Wang, S.C.; Shapiro, L. The Bacterial Nucleoid: A Highly Organized and Dynamic Structure. J. Cell. Biochem. 2005, 96, 506–521. [Google Scholar] [CrossRef] [PubMed]
- Badrinarayanan, A.; Reyes-Lamothe, R.; Uphoff, S.; Leake, M.C.; Sherratt, D.J. In Vivo Architecture and Action of Bacterial Structural Maintenance of Chromosome Proteins. Science 2012, 338, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Irastortza-Olaziregi, M.; Amster-Choder, O. Coupled Transcription-Translation in Prokaryotes: An old Couple with New Surprises. Front. Microbiol. 2021, 11, 624830. [Google Scholar] [CrossRef]
- Firshein, W.; Kim, P. Plasmid Replication and Partition in Escherichia coli: Is the Cell Membrane the Key? Mol. Microbiol. 1997, 23, 1–10. [Google Scholar] [CrossRef]
- Helinski, D.R. A Brief History of Plasmids. EcoSal Plus 2022, 10, eESP-0028. [Google Scholar] [CrossRef]
- Hiraga, S. Dynamic Localization of Bacterial and Plasmid Chromosomes. Annu. Rev. Genet. 2000, 34, 21–59. [Google Scholar] [CrossRef]
- Ramos-León, F.; Ramamurthi, K.S. Cytoskeletal Proteins: Lessons Learned from Bacteria. Phys. Biol. 2022, 19, 021005. [Google Scholar] [CrossRef] [PubMed]
- Yeong, V.; Werth, E.G.; Brown, L.M.; Obermeyer, A.C. Formation Of Biomolecular Condensates in Bacteria by Tuning Protein Electrostatics. ACS Cent. Sci. 2020, 6, 2301–2310. [Google Scholar] [CrossRef]
- Wei, S.P.; Qian, Z.G.; Hu, C.F.; Pan, F.; Chen, M.T.; Lee, S.Y.; Xia, X.X. Formation and Functionalization of Membraneless Compartments in Escherichia coli. Nat. Chem. Biol. 2020, 16, 1143–1148. [Google Scholar] [CrossRef]
- Khanna, K.; Villa, E. Revealing Bacterial Cell Biology Using Cryo-Electron Tomography. Curr. Opin. Struct. Biol. 2022, 75, 102419. [Google Scholar] [CrossRef]
- Leiva, L.E.; Zegarra, V.; Bange, G.; Ibba, M. At the Crossroad of Nucleotide Dynamics and Protein Synthesis in Bacteria. Microbiol. Mol. Biol. Rev. 2023, 87, e00044-22. [Google Scholar] [CrossRef]
- Ezraty, B.; Vergnes, A.; Banzhaf, M.; Duverger, Y.; Huguenot, A.; Brochado, A.R.; Su, S.Y.; Espinosa Loiseau, L.; Py, B.; Typas, A.; et al. Fe-S cluster biosynthesis controls uptake of aminoglycosides in a ROS-less death pathway. Science 2013, 340, 1583–1587. [Google Scholar] [CrossRef] [PubMed]
- Khondker, A.; Bider, R.C.; Passos-Gastaldo, I.; Wright, G.D.; Rheinstädter, M.C. Membrane interactions of non-membrane targeting antibiotics: The case of aminoglycosides, macrolides, and fluoroquinolones. Biochim. Biophys. Acta (BBA)-Biomembr. 2021, 1863, 183448. [Google Scholar] [CrossRef] [PubMed]
- Castellana, M.; Wilson, M.Z.; Xu, Y.; Joshi, P.; Cristea, I.M.; Rabinowitz, J.D.; Gitai, Z.; Wingreen, N.S. Enzyme Clustering Accelerates Processing of Intermediates Through Metabolic Channeling. Nat. Biotechnol. 2014, 32, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.L.; Beveridge, T.J. Gentamicin Interaction with Pseudomonas aeruginosa Cell Envelope. Antimicrob. Agents Chemother. 1986, 29, 1079–1087. [Google Scholar] [CrossRef]
- Wong, F.; Stokes, J.M.; Cervantes, B.; Penkov, S.; Friedrichs, J.; Renner, L.D.; Collins, J.J. Cytoplasmic Condensation Induced by Membrane Damage is Associated with Antibiotic Lethality. Nat. Commun. 2021, 12, 2321. [Google Scholar] [CrossRef]
- Bakshi, S.; Choi, H.; Mondal, J.; Weisshaar, J.C. Time-Dependent Effects of Transcription-and Translation-Halting Drugs on the Spatial Distributions of the Escherichia coli Chromosome and Ribosomes. Mol. Microbiol. 2014, 94, 871–887. [Google Scholar] [CrossRef]
- Wlodarski, M.; Mancini, L.; Raciti, B.; Sclavi, B.; Lagomarsino, M.C.; Cicuta, P. Cytosolic Crowding Drives The Dynamics of Both Genome and Cytosol in Escherichia coli Challenged with Sub-Lethal Antibiotic Treatments. IScience 2020, 23, 101560. [Google Scholar] [CrossRef] [PubMed]
- Hasnain, S.; McClendon, C.L.; Hsu, M.T.; Jacobson, M.P.; Bandyopadhyay, P. A New Coarse-Grained Model for E. coli Cytoplasm: Accurate Calculation of the Diffusion Coefficient of Proteins and Observation of Anomalous Diffusion. PLoS ONE 2014, 9, e106466. [Google Scholar] [CrossRef]
- Bellotto, N.; Agudo-Canalejo, J.; Colin, R.; Golestanian, R.; Malengo, G.; Sourjik, V. Dependence of Diffusion in Escherichia coli Cytoplasm on Protein Size, Environmental Conditions, and Cell Growth. eLife 2022, 11, e82654. [Google Scholar] [CrossRef] [PubMed]
- Van Laar, T.A.; Chen, T.; You, T.; Leung, K.P. Sublethal Concentrations of Carbapenems Alter Cell Morphology and Genomic Expression of Klebsiella pneumoniae Biofilms. Antimicrob. Agents Chemother. 2015, 59, 1707–1717. [Google Scholar] [CrossRef]
- Popham, D.L.; Young, K.D. Role of penicillin-binding proteins in bacterial cell morphogenesis. Curr. Opin. Microbiol. 2003, 6, 594–599. [Google Scholar] [CrossRef]
- Cohen, S.E.; Lewis, C.A.; Mooney, R.A.; Kohanski, M.A.; Collins, J.J.; Landick, R.; Walker, G.C. Roles for the Transcription Elongation Factor NusA in both DNA Repair and Damage Tolerance Pathways in Escherichia coli. Proc. Natl. Acad. Sci. USA 2010, 107, 15517–15522. [Google Scholar] [CrossRef]
- Raivio, T.L.; Leblanc, S.K.; Price, N.L. The Escherichia coli Cpx Envelope Stress Response Regulates Genes of Diverse Function that Impact Antibiotic Resistance and Membrane Integrity. J. Bacteriol. 2013, 195, 2755–2767. [Google Scholar] [CrossRef] [PubMed]
- Phillips, I.; Culebras, E.; Moreno, F.; Baquero, F. Induction of the SOS Response By new 4-Quinolones. J. Antimicrob. Chemother. 1987, 20, 631–638. [Google Scholar] [CrossRef]
- Ojkic, N.; Serbanescu, D.; Banerjee, S. Antibiotic Resistance Via Bacterial Cell Shape-Shifting. Mbio 2022, 13, e00659-22. [Google Scholar] [CrossRef] [PubMed]
- Lopatkin, A.J.; Bening, S.C.; Manson, A.L.; Stokes, J.M.; Kohanski, M.A.; Badran, A.H.; Earl, A.M.; Cheney, N.J.; Yang, J.H.; Collins, J.J. Clinically Relevant Mutations in Core Metabolic Genes Confer Antibiotic Resistance. Science 2021, 371, eaba0862. [Google Scholar] [CrossRef]
- Honda, T.; Cremer, J.; Mancini, L.; Zhang, Z.; Pilizota, T.; Hwa, T. Coordination of Gene Expression with Cell Size Enables Escherichia coli to Efficiently Maintain Motility Across Conditions. Proc. Natl. Acad. Sci. USA 2022, 119, e2110342119. [Google Scholar] [CrossRef] [PubMed]
- Rowlett, V.W.; Mallampalli, V.K.P.S.; Karlstaedt, A.; Dowhan, W.; Taegtmeyer, H.; Margolin, W.; Vitrac, H. Impact of Membrane Phospholipid Alterations in Escherichia coli on Cellular Function And Bacterial Stress Adaptation. J. Bacteriol. 2017, 199, e00849-16. [Google Scholar] [CrossRef]
- Tanaka, M.; Ueno, Y.; Miyake, T.; Sakuma, T.; Okochi, M. Enrichment of Membrane Curvature-Sensing Proteins from Escherichia coli Using Spherical Supported Lipid Bilayers. J. Biosci. Bioeng. 2022, 133, 98–104. [Google Scholar] [CrossRef]
- Villaseñor, C.G.; Dimitrova, V.; Karagiaridi, A.; Rivera, K.; Pinkett, H.; Kamat, N.P. The Role of Lipid Bilayer Composition in Cell-Free Expressed ABC Transporter Folding. Biophys. J. 2023, 122, 342a–343a. [Google Scholar] [CrossRef]
- Pospíšil, J.; Vítovská, D.; Kofroňová, O.; Muchová, K.; Šanderová, H.; Hubálek, M.; Šiková, M.; Modrák, M.; Benada, O.; Barák, I.; et al. Bacterial Nanotubes as a Manifestation of Cell Death. Nat. Commun. 2020, 11, 4963. [Google Scholar] [CrossRef]
- Uzoechi, C.S.; Abu-Lail, N.I. Variations in the Morphology, Mechanics and Adhesion of Persister and Resister, E. coli Cells in Response to Ampicillin: AFM Study. Antibiotics 2020, 9, 235. [Google Scholar] [CrossRef]
- Johns, B.E.; Purdy, K.J.; Tucker, N.P.; Maddocks, S.E. Phenotypic and Genotypic Characteristics of Small Colony Variants and their Role in Chronic Infection. Microbiol. Insights. 2015, 8, 15–23. [Google Scholar] [CrossRef]
- Berryhill, B.; Gil-Gil, T.; Manuel, J.; Smith, A.; McCall, I.C.; Baquero, F.; Levin, B.R. The Evolution of Heteroresistance and Small Colony Variants in Escherichia coli Following Long Term Bacteriostatic Drug Exposure; Development Biology, Emory University: Atlanta, GE, USA, 2023; manuscript in preparation. [Google Scholar]
- Munder, M.C.; Midtvedt, D.; Franzmann, T.; Nuske, E.; Otto, O.; Herbig, M.; Ulbricht, E.; Müller, P.; Taubenberger, A.; Maharana, S.; et al. A pH-Driven Transition of The Cytoplasm from a Fluid-To a Solid-Like State Promotes Entry into Dormancy. eLife 2016, 5, e09347. [Google Scholar] [CrossRef]
- Parry, B.R.; Surovtsev, I.V.; Cabeen, M.T.; O’Hern, C.S.; Dufresne, E.R.; Jacobs-Wagner, C. The Bacterial Cytoplasm has Glass-Like Properties and is Fluidized by Metabolic Activity. Cell 2014, 156, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Mullineaux, C.W.; Nenninger, A.; Ray, N.; Robinson, C. Diffusion of Green Fluorescent Protein In Three Cell Environments in Escherichia coli. J. Bacteriol. 2006, 188, 3442–3448. [Google Scholar] [CrossRef] [PubMed]
- Braet, J.; Catteeuw, D.; Van Damme, P. Recent Advancements in Tracking Bacterial Effector Protein Translocation. Microorganisms 2022, 10, 260. [Google Scholar] [CrossRef]
- Jiang, X.R.; Chen, G.Q. Morphology Engineering of Bacteria for Bio-Production. Biotechnol. Adv. 2016, 34, 435–440. [Google Scholar] [CrossRef]
- Bowden, G.A.; Georgiou, G. Folding And Aggregation of Beta-Lactamase in The Periplasmic Space of Escherichia coli. J. Biol. Chem. 1990, 265, 16760–16766. [Google Scholar] [CrossRef] [PubMed]
- Morosini, M.I.; Ayala, J.A.; Baquero, F.; Martinez, J.L.; Blazquez, J. Biological Cost of AmpC Production for Salmonella enterica Serotype Typhimurium. Antimicrob. Agents Chemother. 2000, 44, 3137–3143. [Google Scholar] [CrossRef]
- Moya, B.; Juan, C.; Alberti, S.; Perez, J.L.; Oliver, O. Benefit of Having Multiple AmpD Genes for Acquiring Β-Lactam Resistance Without Losing Fitness and Virulence of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2008, 52, 3694–3700. [Google Scholar] [CrossRef]
- López, C.; Prunotto, A.; Bahr, G.; Bonomo, R.A.; González, L.J.; Dal Peraro, M.; Vila, A.J. Specific Protein-Membrane Interactions Promote Packaging of Metallo-Β-Lactamases into Outer Membrane Vesicles. Antimicrob. Agents Chemother. 2021, 65, e00507-21. [Google Scholar] [CrossRef]
- Alonso-Santos, A.M. Molecular Basis of Multiple Antibiotic Resistance in Stenotrophomonas maltophilia; Development Molecular Biology, Autonomous University: Madrid, Spain, 2000. [Google Scholar]
- Olivares, J.; Álvarez-Ortega, C.; Martinez, J.L. Metabolic Compensation of Fitness Costs Associated with Overexpression of the Multidrug Efflux Pump MexEF-OprN in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2014, 58, 3904–3913. [Google Scholar] [CrossRef] [PubMed]
- Linares, J.F.; López, J.A.; Camafeita, E.; Albar, J.P.; Rojo, F.; Martínez, J.L. Overexpression of the Multidrug Efflux Pumps MexCD-OprJ and MexEF-OprN is Associated With a Reduction of Type III Secretion in Pseudomonas aeruginosa. J. Bacteriol. 2005, 187, 1384–1391. [Google Scholar] [CrossRef]
- Ohgita, T.; Saito, H. Biophysical Mechanism of Protein Export by Bacterial Type III Secretion System. Chem. Pharm. Bull 2019, 67, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Grahn, A.M.; Haase, J.; Bamford, D.H.; Lanka, E. Components of the RP4 Conjugative Transfer Apparatus Form an Envelope Structure Bridging Inner and Outer Membranes of Donor Cells: Implications For Related Macromolecule Transport Systems. J. Bacteriol. 2000, 182, 1564–1574. [Google Scholar] [CrossRef]
- Couturier, A.; Virolle, C.; Goldlust, K.; Berne-Dedieu, A.; Reuter, A.; Nolivos, S.; Yamaichi, T.; Bigot, S.; Lesterlin, C. Real-Time Visualisation of the Intracellular Dynamics of Conjugative Plasmid Transfer. Nat. Commun. 2023, 14, 294. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Bershtein, S.; Yan, J.; Argun, T.; Gilson, A.I.; Trauger, S.A.; Shakhnovich, E.I. Transient Protein-Protein Interactions Perturb, E. Coli Metabolome and Cause Gene Dosage Toxicity. eLife 2016, 5, e20309. [Google Scholar] [CrossRef]
- Serbanescu, D.; Ojkic, N.; Banerjee, S. Nutrient-Dependent Trade-Offs Between Ribosomes and Division Protein Synthesis Control Bacterial Cell Size and Growth. Cell Rep. 2020, 32, 108183. [Google Scholar] [CrossRef] [PubMed]
- García-Fernández, E.; Koch, G.; Wagner, R.M.; Fekete, A.; Stengel, S.T.; Schneider, J.; Mielich-Süss, B.; Geibel, S.; Markert, S.M.; Stigloher, C.; et al. Membrane Microdomain Disassembly Inhibits MRSA Antibiotic Resistance. Cell 2017, 17, 1354–1367.e20. [Google Scholar] [CrossRef]
- Iosifidis, G.; Duggin, I.G. Distinct Morphological Fates of Uropathogenic Escherichia coli Intracellular Bacterial Communities: Dependency on Urine Composition and pH. Infect. Immun. 2020, 88, e00884-19. [Google Scholar] [CrossRef]
- Horvath, D.J., Jr.; Li, B.; Casper, T.; Partida-Sanchez, S.; Hunstad, D.A.; Hultgren, S.J.; Justice, S.S. Morphological Plasticity Promotes Resistance To Phagocyte Killing Of Uropathogenic Escherichia coli. Microbes Infect. 2011, 13, 426–437. [Google Scholar] [CrossRef]
- Justice, S.S.; Hunstad, D.A.; Seed, P.C.; Hultgren, S.J. Filamentation by Escherichia coli Subverts Innate Defenses During Urinary Tract Infection. Proc. Natl. Acad. Sci. USA 2006, 103, 19884–19889. [Google Scholar] [CrossRef]
- Andersen, T.E.; Khandige, S.; Madelung, M.; Brewer, J.; Kolmos, H.J.; Moller-Jensen, J. Escherichia coli Uropathogenesis In Vitro: Invasion, Cellular Escape, and Secondary Infection Analyzed in a Human Bladder Cell Infection Model. Infect. Immun. 2012, 80, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.D.; Ali, M.A.; Lee, D.; Félix, M.A.; Luallen, R.J. Bacterial Filamentation As A Mechanism for Cell-to-Cell Spread within an Animal Host. Nat. Commun. 2022, 13, 693. [Google Scholar] [CrossRef]
- Bos, J.; Zhang, Q.; Vyawahare, S.; Rogers, E.; Rosenberg, S.M.; Austin, R.H. Emergence of Antibiotic Resistance from Multinucleated Bacterial Filaments. Proc. Natl. Acad. Sci. USA 2015, 112, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Asensio, C. Molecular Ecology. In Reflections in Biochemistry; Kornberg, A., Horecker, B.L., Cornudella, L., Oro, J., Eds.; Pergamon Press: Oxford, UK, 1976; pp. 235–240. [Google Scholar]
- Ishihama, Y.; Schmidt, T.; Rappsilber, J.; Mann, M.; Hartl, F.; Kerner, M.J.; Frishman, D. Protein Abundance Profiling of the Escherichia coli Cytosol. BMC Genom. 2008, 9, 102. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, L.; McAdams, H.H.; Losick, R. Why and how bacteria localize proteins. Science 2009, 326, 1225–1228. [Google Scholar] [CrossRef]
- Rajagopala, S.V.; Sikorski, P.; Kumar, A.; Mosca, R.; Vlasblom, J.; Arnold, R.; Franca-Koh, J.; Pakala, S.B.; Phanse, S.; Ceol, A.; et al. The Binary Protein-Protein Interaction Landscape of Escherichia coli. Nat. Biotechnol. 2014, 32, 285–290. [Google Scholar] [CrossRef]
- Cohen, R.D.; Pielak, G.J. A Cell is More than The Sum of its (Dilute) Parts: A Brief History of Quinary Structure. Protein Sci. 2017, 26, 403–413. [Google Scholar] [CrossRef]
- Soleymani, F.; Paquet, E.; Viktor, H.L.; Michalowski, W.; Spinello, D. ProtInteract: A deep Learning Framework for Predicting Protein—Protein Interactions. Comp. Struct. Biotech. J. 2023, 21, 1324–1348. [Google Scholar] [CrossRef]
- Yu, B.; Wang, X.; Zhang, Y.; Gao, H.; Wang, Y.; Liu, Y.; Gao, X. RPI-MDLStack: Predicting RNA–Protein Interactions Through Deep Learning with Stacking Strategy and LASSO. Appl. Soft. Comp. 2022, 120, 108676. [Google Scholar] [CrossRef]
- Baquero, F.; Levin, B.R. Proximate And Ultimate Causes of the Bactericidal Action of Antibiotics. Nat. Rev. Microbiol. 2021, 19, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Łapińska, U.; Voliotis, M.; Lee, K.K.; Campey, A.; Stone, M.R.L.; Tuck, B.; Phetsang, W.; Zhang, B.; Tsaneva-Atanasova, K.; Blaskovich, M.A.T.; et al. Fast Bacterial Growth Reduces Antibiotic Accumulation And Efficacy. eLife 2022, 11, e74062. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baquero, F.; Martínez, J.-L.; Sánchez, A.; Fernández-de-Bobadilla, M.D.; San-Millán, A.; Rodríguez-Beltrán, J. Bacterial Subcellular Architecture, Structural Epistasis, and Antibiotic Resistance. Biology 2023, 12, 640. https://doi.org/10.3390/biology12050640
Baquero F, Martínez J-L, Sánchez A, Fernández-de-Bobadilla MD, San-Millán A, Rodríguez-Beltrán J. Bacterial Subcellular Architecture, Structural Epistasis, and Antibiotic Resistance. Biology. 2023; 12(5):640. https://doi.org/10.3390/biology12050640
Chicago/Turabian StyleBaquero, Fernando, José-Luis Martínez, Alvaro Sánchez, Miguel D. Fernández-de-Bobadilla, Alvaro San-Millán, and Jerónimo Rodríguez-Beltrán. 2023. "Bacterial Subcellular Architecture, Structural Epistasis, and Antibiotic Resistance" Biology 12, no. 5: 640. https://doi.org/10.3390/biology12050640