
Virtual and In Vitro Screening of Natural Products Identifies Indole and Benzene Derivatives as Inhibitors of SARS-CoV-2 Main Protease (Mpro)
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Ligand-Based Virtual Screening
2.3. Performance Measures
2.4. Molecular Docking
2.5. Visual Validation of Drug-Target Interactions
2.6. In Vitro Protease Inhibition Assay
3. Results
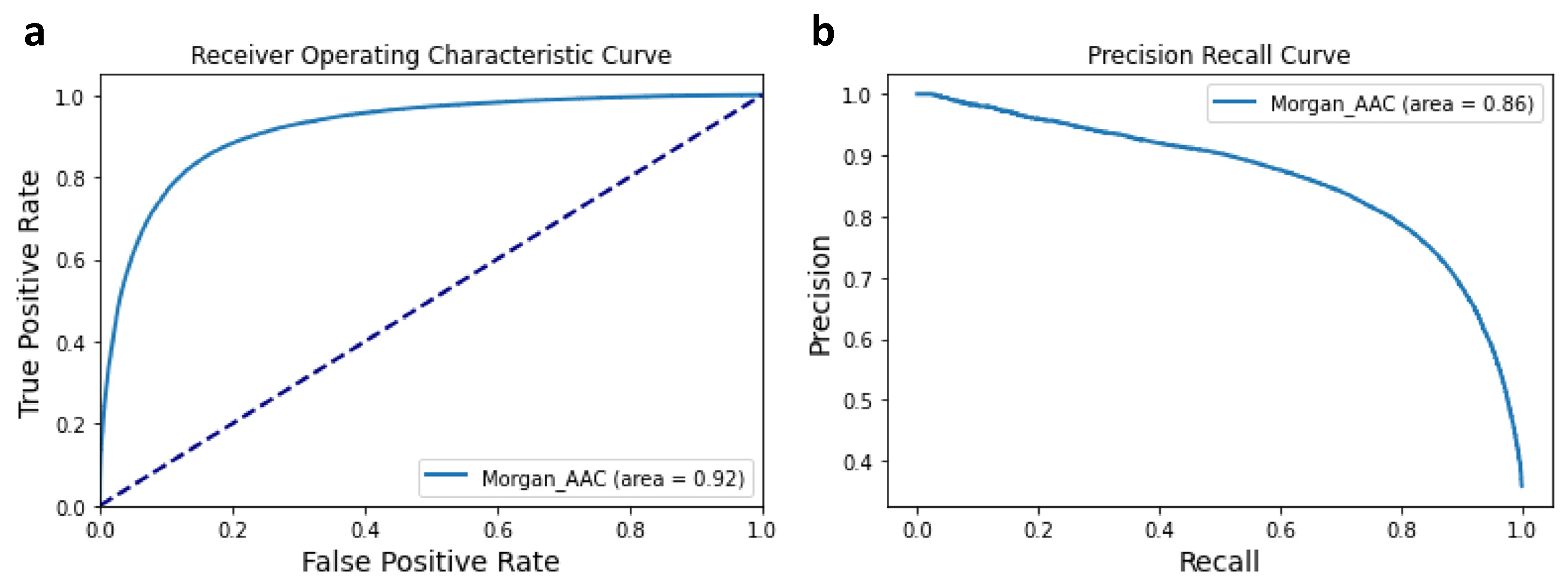
3.1. Performance of the Machine Learning Classifier
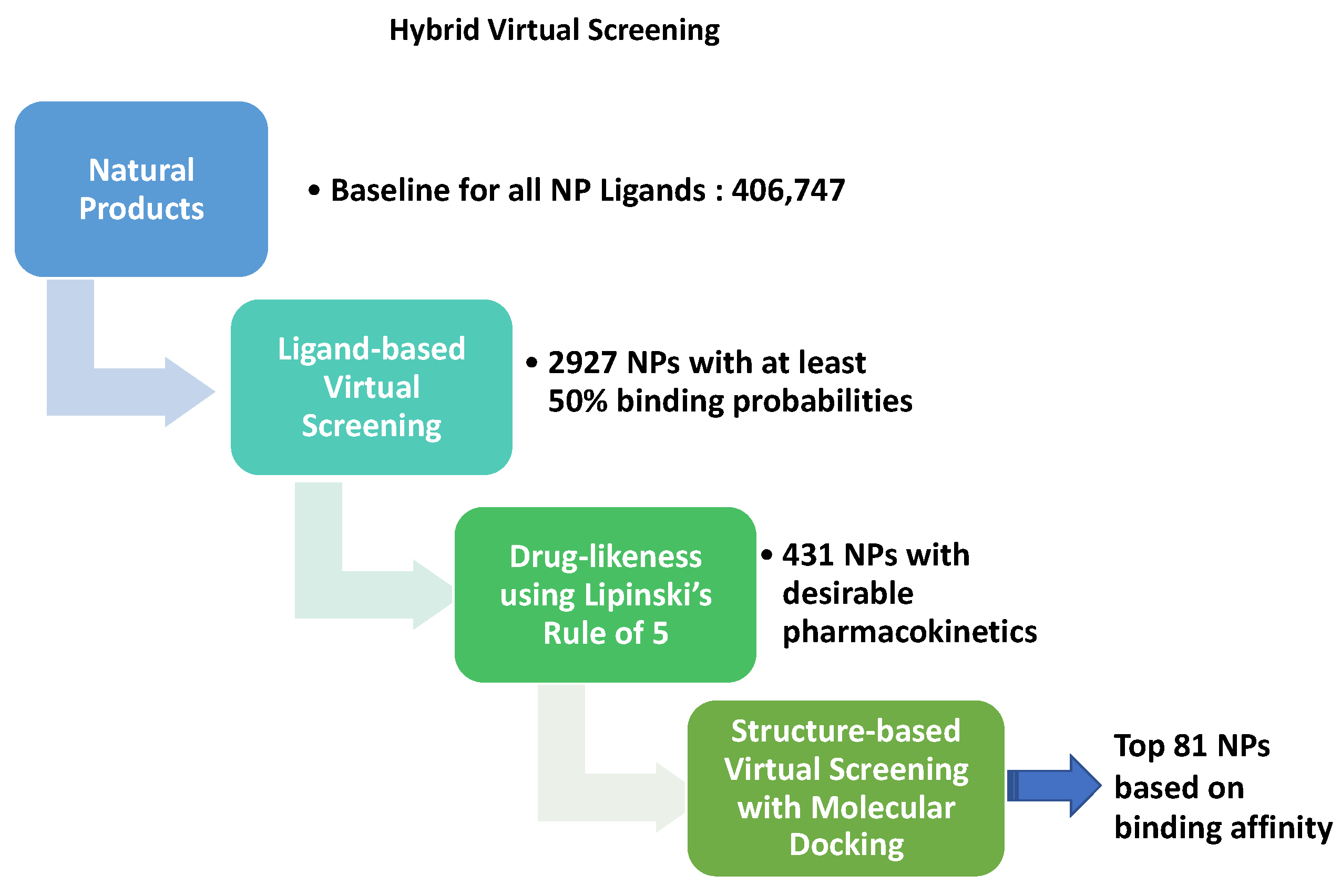
3.2. Virtual Screening for NP inhibitors of Mpro
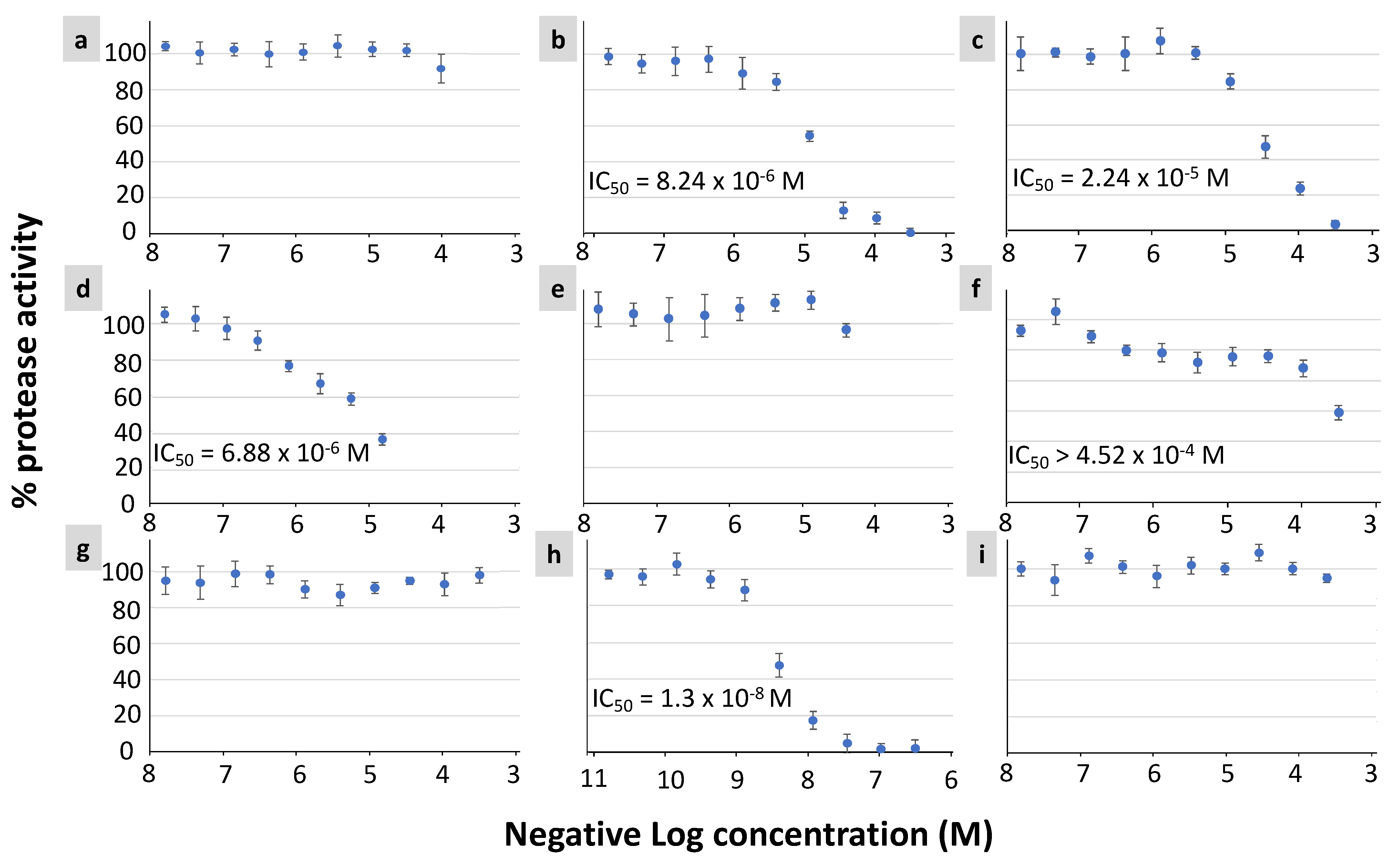
3.3. In Vitro Protease Inhibition Assay of Selected NP Candidates
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tang, J.W.; Tambyah, P.A.; Hui, D.S. Emergence of a new SARS-CoV-2 variant in the UK. J. Infect. 2021, 82, e27–e28. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.D.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Bernal, J.L.; Andrews, N.; Gower, C.; Gallagher, E.; Simmons, R.; Thelwall, S.; Stowe, J.; Tessier, E.; Groves, N.; Dabrera, G.; et al. Effectiveness of Covid-19 vaccines against the B.1.617.2 (Delta) variant. N. Engl. J. Med. 2021, 385, 585–594. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Hong, W.; Pan, X.; Lu, G.; Wei, X. SARS-CoV-2 Omicron variant: Characteristics and prevention. Med. Comm. 2021, 2, 838–845. [Google Scholar] [CrossRef]
- Moghadas, S.M.; Vilches, T.N.; Zhang, K.; Wells, C.R.; Shoukat, A.; Singer, B.H.; Meyers, L.A.; Neuzil, K.M.; Langley, J.M.; Fitzpatrick, M.C.; et al. The impact of vaccination on COVID-19 outbreaks in the United States. Clin. Infect. Dis. 2021, 73, 2257–2264. [Google Scholar] [CrossRef]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome composition and divergence of the novel Coronavirus (2019-nCoV) originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [Green Version]
- Kandeel, M.; Al-Nazawi, M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020, 251, 117627. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar]
- Parks, J.M.; Smith, J.C. How to discover antiviral drugs quickly. N. Engl. J. Med. 2020, 382, 2261–2264. [Google Scholar] [CrossRef]
- Sharma, V.; Wakode, S.; Kumar, H. Structure- and ligand-based drug design: Concepts, approaches, and challenges. In Chemoinformatics and Bioinformatics in the Pharmaceutical Sciences; Sharma, N., Ojha, H., Raghav, P.K., Goyal, R.K., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 27–53. [Google Scholar]
- Swamy, M.K. Plant-Derived Bioactives, 1st ed.; Springer: Singapore, 2020; Volume 1, p. 589. [Google Scholar]
- Edwards, A. What are the odds of finding a COVID-19 drug from a lab repurposing screen? J. Chem. Inf. Model. 2020, 60, 5727–5729. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- de Wilde, A.H.; Jochmans, D.; Posthuma, C.C.; Zevenhoven-Dobbe, J.C.; van Nieuwkoop, S.; Bestebroer, T.M.; van den Hoogen, B.G.; Neyts, J.; Snijder, E.J. Screening of an FDA-approved compound library identifies four small-molecule inhibitors of Middle East respiratory syndrome coronavirus replication in cell culture. Antimicrob. Agents Chemother. 2014, 58, 4875–4884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A trial of lopinavir-ritonavir in adults hospitalized with severe COVID-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Geleris, J.; Sun, Y.; Platt, J.; Zucker, J.; Baldwin, M.; Hripcsak, G.; Labella, A.; Manson, D.K.; Kubin, C.; Barr, R.G.; et al. Observational study of hydroxychloroquine in hospitalized patients with COVID-19. N. Engl. J. Med. 2020, 382, 2411–2418. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kiso, M.; Sakai-Tagawa, Y.; Iwatsuki-Horimoto, K.; Imai, M.; Takeda, M.; Kinoshita, N.; Ohmagari, N.; Gohda, J.; Semba, K.; et al. The anticoagulant nafamostat potently inhibits SARS-CoV-2 S protein-mediated fusion in a cell fusion assay system and viral infection in vitro in a cell-type-dependent manner. Viruses 2020, 12, 629. [Google Scholar] [CrossRef]
- Yamaya, M.; Nishimura, H.; Deng, X.; Kikuchi, A.; Nagatomi, R. Protease inhibitors: Candidate drugs to inhibit severe acute respiratory syndrome coronavirus 2 replication. Tohoku J. Exp. Med. 2020, 251, 27–30. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.; Mu, C.; Kwok, H.F.; Xu, J.; Wu, Y.; Liu, W.; Xie, Y. Capivasertib restricts SARS-CoV-2 cellular entry: A potential clinical application for COVID-19. Int. J. Biol. Sci. 2021, 17, 2348. [Google Scholar] [CrossRef]
- Kolinsky, M.P.; Rescigno, P.; Bianchini, D.; Zafeiriou, Z.; Mehra, N.; Mateo, J.; de Bono, J.S. A phase I dose-escalation study of enzalutamide in combination with the AKT inhibitor AZD5363 (capivasertib) in patients with metastatic castration-resistant prostate cancer. Ann. Oncol. 2020, 31, 619–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Xie, W.; Xue, X.; Yang, K.; Ma, J.; Liang, W.; Zhao, Q.; Zhou, Z.; Pei, D.; Ziebuhr, J.; et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005, 3, e324. [Google Scholar]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Achdout, H.; Aimon, A.; Bar-David, E.; Morris, G.M. COVID moonshot: Open science discovery of SARS-CoV-2 main protease inhibitors by combining crowdsourcing, high-throughput experiments, computational simulations, and machine learning. bioRxiv 2020. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Zhu, Y. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Bjarnason, I.; Hayllar, J.; MacPherson, A.J.; Russell, A.S. Side effects of nonsteroidal anti-inflammatory drugs on the small and large intestine in humans. Gastroenterology 1993, 104, 1832–1847. [Google Scholar] [CrossRef]
- Bjarnason, I.; Thjodleifsson, B. Gastrointestinal toxicity of non-steroidal anti-inflammatory drugs: The effect of nimesulide compared with naproxen on the human gastrointestinal tract. Rheumatology 1999, 38 (Suppl. S1), 24–32. [Google Scholar] [CrossRef] [Green Version]
- Jóźwiak-Bebenista, M.; Nowak, J.Z. Paracetamol: Mechanism of action, applications and safety concern. Acta Pol. Pharm. 2014, 71, 11–23. [Google Scholar]
- Karimi, A.; Majlesi, M.; Rafieian-Kopaei, M. Herbal versus synthetic drugs; beliefs and facts. J. Nephropharmacology 2015, 4, 27–30. [Google Scholar]
- Su, H.; Yao, S.; Zhao, W.; Li, M.; Liu, J.; Shang, W.; Xie, H.; Ke, C.; Gao, M.; Yu, K.; et al. Discovery of baicalin and baicalein as novel, natural product inhibitors of SARS-CoV-2 3CL protease in vitro. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Bordeleau, M.E.; Robert, F.; Gerard, B.; Lindqvist, L.; Chen, S.M.; Wendel, H.G.; Brem, B.; Greger, H.; Lowe, S.W.; Porco, J.A., Jr.; et al. Therapeutic suppression of translation initiation modulates chemosensitivity in a mouse lymphoma model. J. Clin. Investig. 2008, 118, 2651–2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biedenkopf, N.; Lange-Grunweller, K.; Schulte, F.W.; Weisser, A.; Muller, C.; Becker, D.; Becker, S.; Hartmann, R.K.; Grunweller, A. The natural compound silvestrol is a potent inhibitor of Ebola virus replication. Antivir. Res. 2017, 137, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Elgner, F.; Sabino, C.; Basic, M.; Ploen, D.; Grunweller, A.; Hildt, E. Inhibition of Zika virus replication by silvestrol. Viruses 2018, 10, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, C.; Schulte, F.W.; Lange-Grunweller, K.; Obermann, W.; Madhugiri, R.; Pleschka, S.; Ziebuhr, J.; Hartmann, R.K.; Grunweller, A. Broad-spectrum antiviral activity of the eIF4A inhibitor silvestrol against corona- and picornaviruses. Antivir. Res. 2018, 150, 123–129. [Google Scholar] [CrossRef]
- Todt, D.; Moeller, N.; Praditya, D.; Kinast, V.; Friesland, M.; Engelmann, M.; Verhoye, L.; Sayed, I.M.; Behrendt, P.; Dao Thi, V.L.; et al. The natural compound silvestrol inhibits hepatitis E virus (HEV) replication in vitro and in vivo. Antivir. Res. 2018, 157, 151–158. [Google Scholar] [CrossRef]
- Gentile, F.; Agrawal, V.; Hsing, M.; Ton, A.T.; Ban, F.; Norinder, U.; Gleave, M.E.; Cherkasov, A. Deep docking: A deep learning platform for augmentation of structure based drug discovery. ACS Cent. Sci. 2020, 6, 939–949. [Google Scholar] [CrossRef]
- Andrade, B.S.; Ghosh, P.; Barh, D.; Tiwari, S.; Silva, R.J.S.; de Assis Soares, W.R.; Melo, T.S.; Freitas, A.S.; Gonzalez-Grande, P.; Palmeira, L.S.; et al. Computational screening for potential drug candidates against the SARS-CoV-2 main protease. F1000Research 2020. [Google Scholar] [CrossRef]
- Sorokina, M.; Steinbeck, C. Review on natural products databases: Where to find data in 2020. J. Cheminformatics 2020, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef]
- Huang, K.; Fu, T.; Glass, L.M.; Zitnik, M.; Xiao, C.; Sun, J. DeepPurpose: A deep learning library for drug-target interaction prediction. Bioinformatics 2021, 36, 5545–5547. [Google Scholar] [CrossRef]
- Vazquez, J.; Lopez, M.; Gibert, E.; Herrero, E.; Luque, F.J. Merging ligand-based and structure-based methods in drug discovery: An overview of combined virtual screening approaches. Molecules 2020, 25, 4723. [Google Scholar] [CrossRef] [PubMed]
- Alhossary, A.; Handoko, S.D.; Mu, Y.; Kwoh, C.K. Fast, accurate, and reliable molecular docking with QuickVina 2. Bioinformatics 2015, 31, 2214–2216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fawcett, T. An introduction to ROC analysis. Pattern Recognit. Lett. 2006, 27, 861–874. [Google Scholar] [CrossRef]
- Narkhede, S. Understanding AUC-ROC Curve, 2018. Available online: https://towardsdatascience.com/understanding-auc-roc-curve-68b2303cc9c5 (accessed on 20 September 2021).
- Word, J.M.; Lovell, S.C.; Richardson, J.S.; Richardson, D.C. Asparagine and glutamine: Using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 1999, 285, 1735–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Iketani, S.; Zask, A.; Khanizeman, N.; Bednarova, E.; Forouhar, F.; Stockwell, B.R. Development of optimized drug-like small molecule inhibitors of the SARS-CoV-2 3CL protease for treatment of COVID-19. Nat. Commun. 2022, 13, 1891. [Google Scholar] [CrossRef] [PubMed]
- Hosmer, D.W.; Lemeshow, S. Applied Logistic Regression, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2000. [Google Scholar]
- Vardhan, S.; Sahoo, S.K. In silico ADMET and molecular docking study on searching potential inhibitors from limonoids and triterpenoids for COVID-19. Comput. Biol. Med. 2020, 124, 103936. [Google Scholar] [CrossRef]
- Mansour, M.A.; AboulMagd, A.M.; Abdel-Rahman, H.M. Quinazoline-Schiff base conjugates: In silico study and ADMET predictions as multi-target inhibitors of coronavirus (SARS-CoV-2) proteins. RSC Adv. 2020, 10, 34033–34045. [Google Scholar] [CrossRef]
- Kaushik, N.K.; Kaushik, N.; Attri, P.; Kumar, N.; Kim, C.H.; Verma, A.K.; Choi, E.H. Biomedical importance of indoles. Molecules 2013, 18, 6620–6662. [Google Scholar] [CrossRef]
- Hamid, H.A.; Ramli, A.N.; Yusoff, M.M. Indole alkaloids from plants as potential leads for antidepressant drugs: A mini review. Front. Pharmacol. 2017, 8, 96. [Google Scholar] [CrossRef] [Green Version]
- Takayama, H. Chemistry and pharmacology of analgesic indole alkaloids from the rubiaceous plant, Mitragyna speciosa. Chem. Pharm. Bull. 2004, 52, 916–928. [Google Scholar] [CrossRef] [Green Version]
- Netz, N.; Opatz, T. Marine indole alkaloids. Mar. Drugs 2015, 13, 4814–4914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piechowska, P.; Zawirska-Wojtasiak, R.; Mildner-Szkudlarz, S. Bioactive β-carbolines in food: A review. Nutrients 2019, 11, 814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waidha, K.; Saxena, A.; Kumar, P.; Sharma, S.; Ray, D.; Saha, B. Design and identification of novel annomontine analogues against SARS-CoV-2: An in-silico approach. Heliyon 2021, 7, e06657. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Xin, H.; Tao, Y.; Mei, L.; Wang, Z. Arenaria kansuensis attenuates pulmonary fibrosis in mice via the activation of Nrf2 pathway and the inhibition of NF-kB/TGF-beta1/Smad2/3 pathway. Phytother. Res. 2021, 35, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.J.; Chen, P.N.; Li, H.J.; Wu, F.; Zhang, G.Y.; Liu, X.P.; Shao, Z.Z. β-Carboline alkaloids fom the deep-sea fungus Trichoderma sp. MCCC 3A01244 as a new type of anti-pulmonary fibrosis agent that inhibits TGF-β/Smad signaling pathway. Front. Microbiol. 2022, 13, 947226. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ma, H.; Li, L.; Chen, Y.; Sun, X.; Dong, Z.; Liu, J.H.; Zhang, J.T. Novel synthetic bisindolylmaleimide alkaloids inhibit STAT3 activation by binding to the SH2 domain and suppress breast xenograft tumor growth. Oncogene 2018, 37, 2469–2480. [Google Scholar] [CrossRef]
- Sun, X.; Li, L.; Ma, H.G.; Sun, P.; Wang, Q.L.; Zhang, T.T.; Li, X. Bisindolylmaleimide alkaloid BMA-155Cl induces autophagy and apoptosis in human hepatocarcinoma HepG-2 cells through the NF-κB p65 pathway. Acta Pharmacol. Sin. 2017, 38, 524–538. [Google Scholar] [CrossRef] [Green Version]
- Toullec, D.; Pianetti, P.; Coste, H.; Bellevergue, P.; Grand-Perret, T.; Ajakane, M.; Loriolle, F. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J. Biol. Chem. 1991, 266, 15771–15781. [Google Scholar] [CrossRef]
- Shaikh, T.M.; Debebe, H. Synthesis and evaluation of antimicrobial activities of novel N-substituted indole derivatives. J. Chem. 2020, 20, 1–9. [Google Scholar] [CrossRef]
- Wildermuth, M.C. Variations on a theme: Synthesis and modification of plant benzoic acids. Curr. Opin. Plant Biol. 2006, 93, 288–296. [Google Scholar] [CrossRef]
- Widhalm, J.R.; Dudareva, N. A familiar ring to it: Biosynthesis of plant benzoic acids. Mol. Plant 2015, 8, 83–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Olmo, A.; Calzada, J.; Nuñez, M. Benzoic acid and its derivatives as naturally occurring compounds in foods and as additives: Uses, exposure, and controversy. Crit. Rev. Food Sci. Nutr. 2017, 57, 3084–3103. [Google Scholar] [CrossRef] [PubMed]
- Armani, E.; Amari, G.; Rizzi, A.; Fanti, R.D.; Ghidini, E.; Capaldi, C.; Carzaniga, L.; Villetti, G. Novel class of benzoic acid ester derivatives as potent PDE4 inhibitors for inhaled administration in the treatment of respiratory diseases. J. Med. Chem. 2014, 57, 793–816. [Google Scholar] [CrossRef] [PubMed]
- Pais, J.P.; Magalhães, M.; Antoniuk, O.; Barbosa, I.; Freire, R.; Pires, D.; Valente, E.; Constantino, L. Benzoic acid derivatives as prodrugs for the treatment of tuberculosis. Pharmaceuticals 2022, 15, 1118. [Google Scholar] [CrossRef]
- Tejera, E.; Pérez-Castillo, Y.; Toscano, G.; Noboa, A.L.; Ochoa-Herrera, V.; Giampieri, F.; Álvarez-Suarez, J.M. Computational modeling predicts potential effects of the herbal infusion “horchata” against COVID-19. Food Chem. 2022, 366, 130589. [Google Scholar] [CrossRef]
- Stefaniu, A.; Pirvu, L.; Albu, B.; Pintilie, L. Molecular docking study on several benzoic acid derivatives against SARS-CoV-2. Molecules 2020, 25, 5828. [Google Scholar] [CrossRef]
- Patowary, L.; Borthakur, M.S. Computational studies of Bridelia retusa phytochemicals for the identification of promising molecules with inhibitory potential against the spike protein and papain-like protease of SARS-CoV-2. Sci. Phytochem. 2022, 1, 29–41. [Google Scholar] [CrossRef]
- Refaat, H.; Mady, F.M.; Sarhan, H.A.; Rateb, H.S.; Alaaeldin, E. Optimization and evaluation of propolis liposomes as a promising therapeutic approach for COVID-19. Int. J. Pharm. 2021, 592, 120028. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Structure | Center | Size | ||||
|---|---|---|---|---|---|---|
| X | Y | Z | X | Y | Z | |
| 6LU7 | −14.607 | 19.162 | 64.101 | 25 | 25 | 25 |
| Enzyme | SARS-CoV-2 Mpro |
|---|---|
| Enzyme in rxn (nM) | 12 |
| Substrate | NH2-C(EDANS)VNSTQSGLRK(DABCYL)M-CO2H |
| Substrate in rxn (µM) | 5 |
| Excitation/Emission | 340/490 |
| Mpro buffer | 50 mM Tris pH 7.3, 1 mM EDTA, 1 mM DTT, 0.005% Triton X-100 |
| Binding Affinity (kcal/mol) | |||||
|---|---|---|---|---|---|
| COCONUT Id | Name | Direct Parent/Class | Autodoc Vina | SwissDock | CB Dock |
| CNP0403473 | N-[(2H-1,3-benzodioxol-5-yl)methyl]-2-(2,5-dioxo-2,3,4,5-tetrahydro-1H-1,4-benzodiazepin-3-yl)acetamide | 1,4-benzodiazepines | −8.1 | −7.65 | −8.6 |
| CNP0391183 | 5-bromo-N-(5-chloro-2-methoxyphenyl)-5′-(1-hydroxyethyl)-2-oxo-1,2-dihydrospiro[indole-3,2′-pyrrolidine]-3′-carboxamide | Indoles | −8.7 | −7.94 | −7.8 |
| CNP0064681 | 4′-(methoxycarbonyl)-1′-methyl-2-oxo-1,2,4′a,5′,5′a,7′,8′,9′,10′,10′a-decahydro-1′H-spiro[indole-3,6′-pyrano [3,4-f]indolizin]-9′-ium | Indolizidines | −8.0 | −7.97 | −7.5 |
| CNP0381522 | (8R)-6-[(E)-[(4-nitrophenyl)methylidene]amino]-3,6,17-triazatetracyclo [8.7.0.03,8.011,16]heptadeca-1(10),11,13,15-tetraene-4,7-dione | NA | −8.9 | NA | −9 |
| CNP0161104 | 3-[(2-hydroxy-2,2-diphenylacetyl)oxy]-1,1-dimethylpiperidin-1-ium | Diphenylmethanes | −8.1 | NA | −7.9 |
| CNP0333632 | 11-(1-hydroxy-4-methylpentyl)-4-(3-methoxy-4-methyl-5-oxo-2,5-dihydrofuran-2-ylidene)-3-methyl-5-oxa-10-azatetracyclo [6.6.2.01,10.02,6]hexadeca-6,15-dien-10-ium | Azaspirodecane | −8.0 | NA | −7.7 |
| CNP0043743 | N-[13-(4-chlorophenyl)-2,8-dioxo-3,9-diazatricyclo [8.4.0.03,7]tetradeca-1(14),10,12-trien-5-yl]methanesulfonamide | Benzodiazepines | −8.1 | −8.12 | −8 |
| CNP0047370 | 4-[bis(4-fluorophenyl)methylidene]-1-(2-{7-methyl-5-oxo-5H-[1,3]thiazolo [3,2-a]pyrimidin-6-yl}ethyl)piperidin-1-ium | Diphenylmethanes | −8.3 | −8.03 | −8.3 |
| CNP0336034 | 4-chloro-2-[4-(2,3-dihydro-1,4-benzodioxin-6-yl)-1,2-oxazol-5-yl]phenol | Benzodioxanes | −8.2 | −7.81 | −7.8 |
| CNP0375828 | (12aS)-2-{[(E)-(4-nitrophenyl)methylidene]amino}-2,3,6,7,12,12a-hexahydropyrazino [1′,2′:1,6]pyrido [3,4-b]indole-1,4-dione | NA | −9.3 | NA | −8.2 |
| CNP0403000 | 3-(2,5-dioxo-2,3,4,5-tetrahydro-1H-1,4-benzodiazepin-3-yl)-N-[(1-methyl-1H-1,3-benzodiazol-2-yl)methyl]propanamide | Benzodiazepines | −8.7 | −8.84 | −8.7 |
| CNP0061237 | 2-[(5-methylfuran-2-yl)methylidene]-3-oxo-2,3-dihydro-1-benzofuran-6-yl 4-methylbenzoate | Benzene and derivatives | −8.2 | −8.33 | −8 |
| CNP0372136 | 17-[(4-nitrophenyl)methyl]-9-oxo-12-oxa-8,17-diazaheptacyclo [15.5.2.01,18.02,7.08,22.011,21.015,20]tetracosa-2,4,6,14-tetraen-17-ium | Strychnos alkaloids | −8.4 | NA | −8.9 |
| CNP0038881 | 3-[(2-hydroxy-2,2-diphenylacetyl)oxy]-8-methyl-8-azabicyclo [3.2.1]octan-8-ium | Diphenylmethanes | −8.0 | −7.53 | −7.6 |
| CNP0402005 | 3-(2,5-dioxo-2,3,4,5-tetrahydro-1H-1,4-benzodiazepin-3-yl)-N-(4-oxo-3,4-dihydroquinazolin-6-yl)propanamide | Diazanaphthalenes | −8.4 | −8.58 | −8.9 |
| CNP0366487 | 1-({2-[(1-ethyl-5-methoxy-1H-indol-3-yl)methylidene]-6-oxido-3-oxo-2,3-dihydro-1-benzofuran-7-yl}methyl)-2-methylpiperidin-1-ium | Indoles and derivatives | −8.2 | NA | −8.2 |
| CNP0105187 | 2-[(4-fluorophenyl)methylidene]-3-oxo-2,3-dihydro-1-benzofuran-6-yl morpholine-4-carboxylate | Aurone flavonoids | −8 | −8.56 | −8.2 |
| CNP0028523 | NA | NA | −8 | −9.51 | −7.4 |
| CNP0259483 | 6-cyclopentyl-2-(3-nitrophenyl)-3,6,17-triazatetracyclo [8.7.0.03,8.011,16]heptadeca-1(10),11,13,15-tetraene-4,7-dione | NA | −9.9 | NA | −10.1 |
| CNP0331537 | Capsimycin B | Macrolactams | −8.3 | −7.02 | −8.9 |
| COCONUT Id | Chemical SuperClass | Chemical Class | Chemical SubClass | DirectParent Classification |
|---|---|---|---|---|
| CNP0403473 | Organoheterocyclic compounds | Benzodiazepines | 1,4-benzodiazepines | 1,4-benzodiazepines |
| CNP0381522 * | Organoheterocyclic compounds | Indoles and derivatives | Pyridoindoles | Beta carbolines |
| CNP0375828 * | Organoheterocyclic compounds | Indoles and derivatives | Pyridoindoles | Beta carbolines |
| CNP0061237 * | Benzenoids | Benzene and derivatives | Benzoic acids and derivatives | Benzoic acid esters |
| CNP0402005 | Organoheterocyclic compounds | Diazanaphthalenes | Benzodiazines | Quinazolinamines |
| CNP0366487 * | Organoheterocyclic compounds | Indoles and derivatives | N-alkylindoles | N-alkylindoles |
| CNP0105187 | Phenylpropanoids and polyketides | Aurone flavonoids | Unknown | Aurone flavonoids |
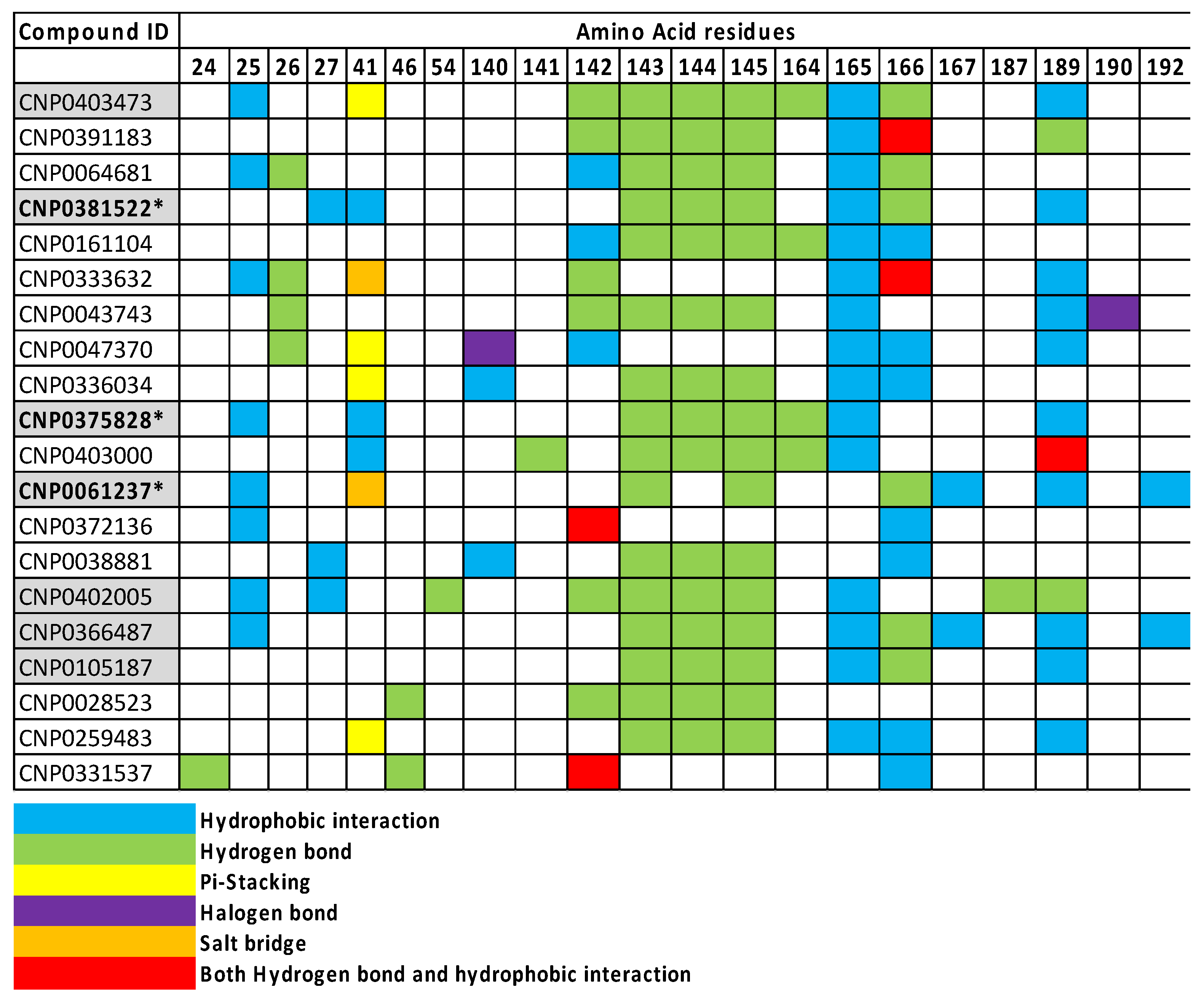
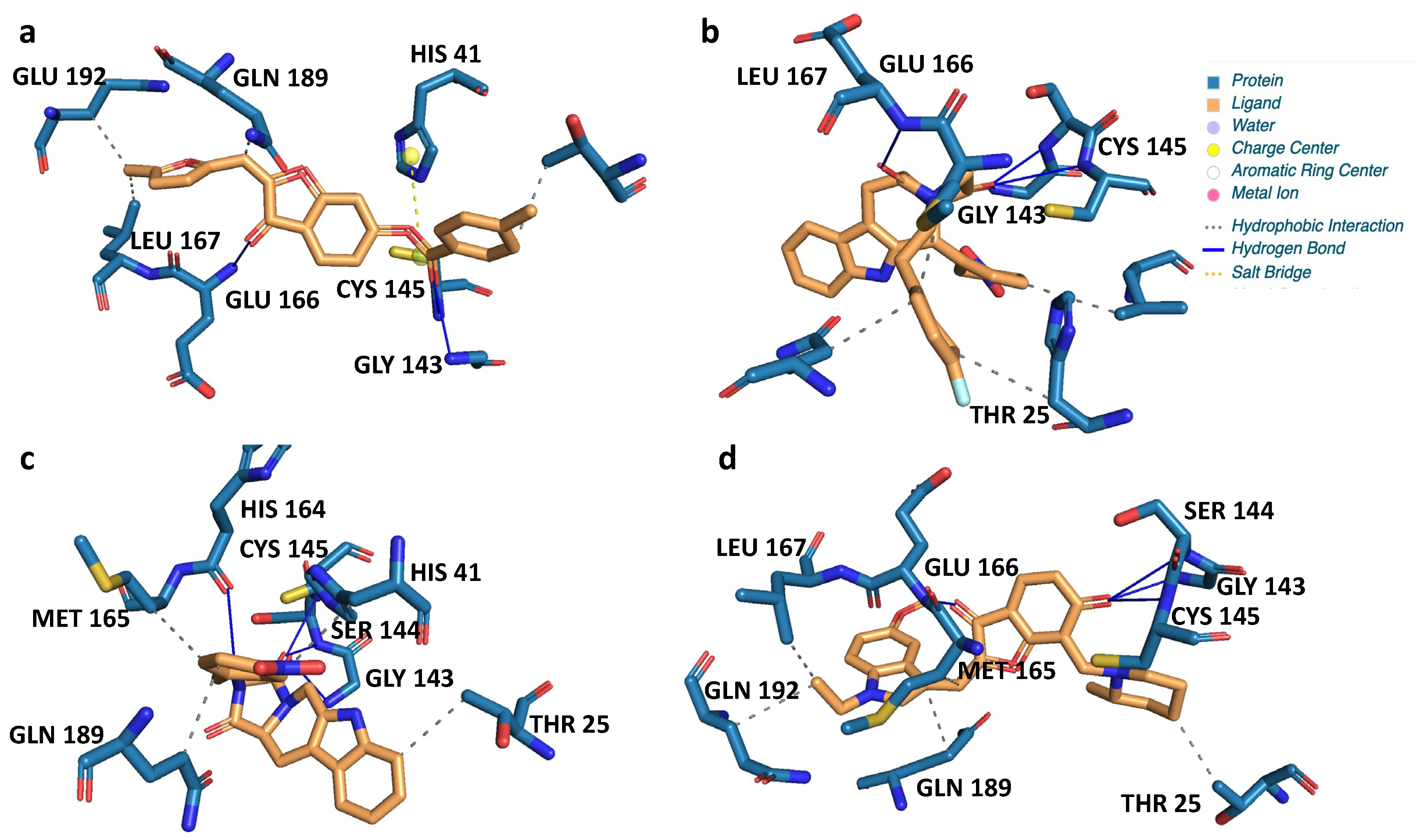
| Name | 2D Structure | 2D Interaction with Mpro Residues |
|---|---|---|
| CNP0381522 |  |  |
| CNP0375828 |  |  |
| CNP0061237 |  |  |
| CNP0366487 |  |  |
| Key for 2D Interaction Maps |  | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ang, D.; Kendall, R.; Atamian, H.S. Virtual and In Vitro Screening of Natural Products Identifies Indole and Benzene Derivatives as Inhibitors of SARS-CoV-2 Main Protease (Mpro). Biology 2023, 12, 519. https://doi.org/10.3390/biology12040519
Ang D, Kendall R, Atamian HS. Virtual and In Vitro Screening of Natural Products Identifies Indole and Benzene Derivatives as Inhibitors of SARS-CoV-2 Main Protease (Mpro). Biology. 2023; 12(4):519. https://doi.org/10.3390/biology12040519
Chicago/Turabian StyleAng, Dony, Riley Kendall, and Hagop S. Atamian. 2023. "Virtual and In Vitro Screening of Natural Products Identifies Indole and Benzene Derivatives as Inhibitors of SARS-CoV-2 Main Protease (Mpro)" Biology 12, no. 4: 519. https://doi.org/10.3390/biology12040519