

Dynamic Regulation Genes at Microtubule Plus Ends: A Novel Class of Glioma Biomarkers
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition
2.2. Genetic Alteration and Methylation Analyses
2.3. Differential Expression Analyses
2.4. Protein–Protein Interaction Analyses
2.5. Unsupervised Consensus Clustering
2.6. Functional Enrichment Analyses
2.7. Development and Validation of Risk Signature and Nomogram Model
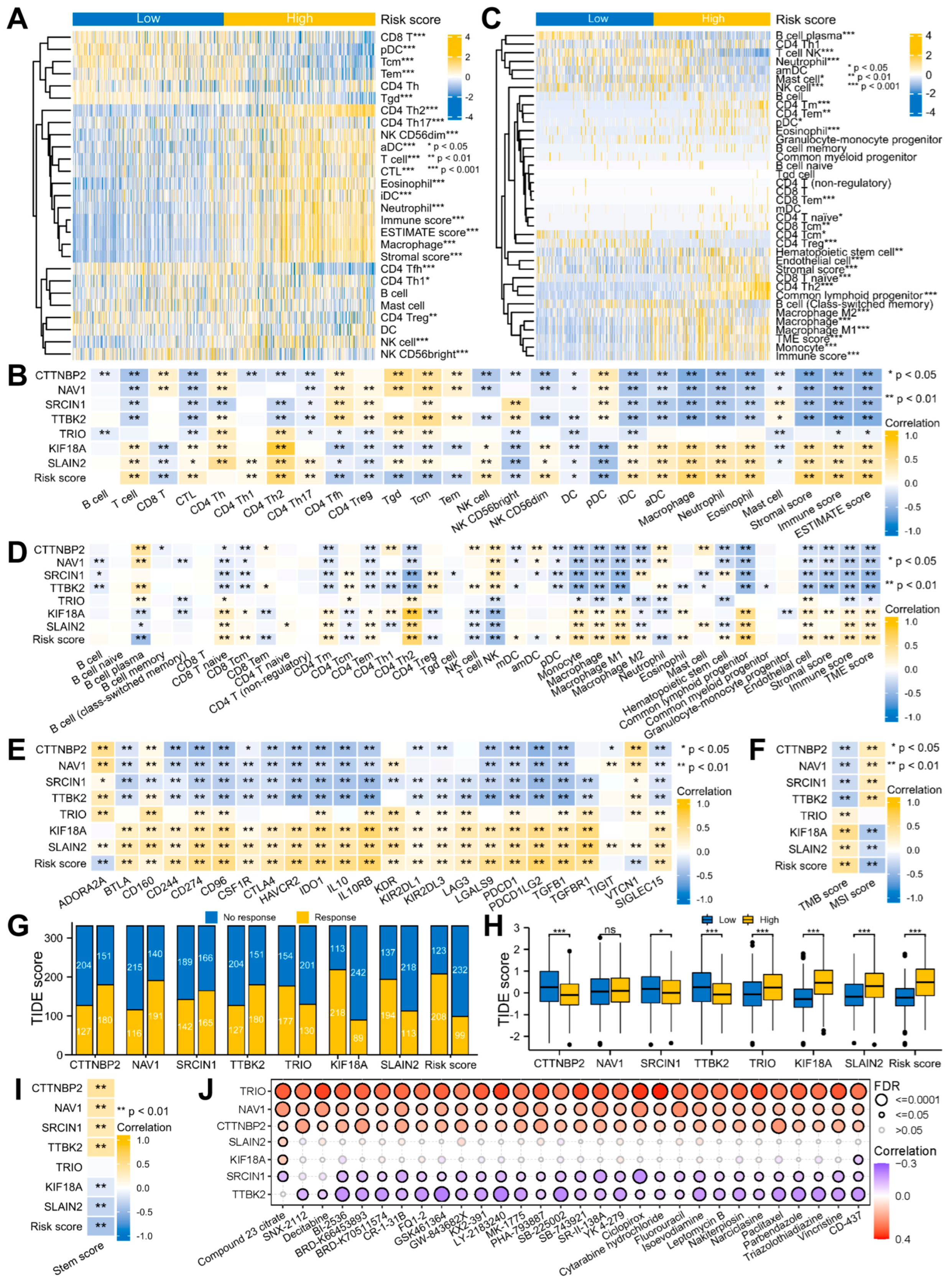
2.8. TME Infiltration Analyses
2.9. Immunotherapy and Drug Sensitivity Analyses
3. Results
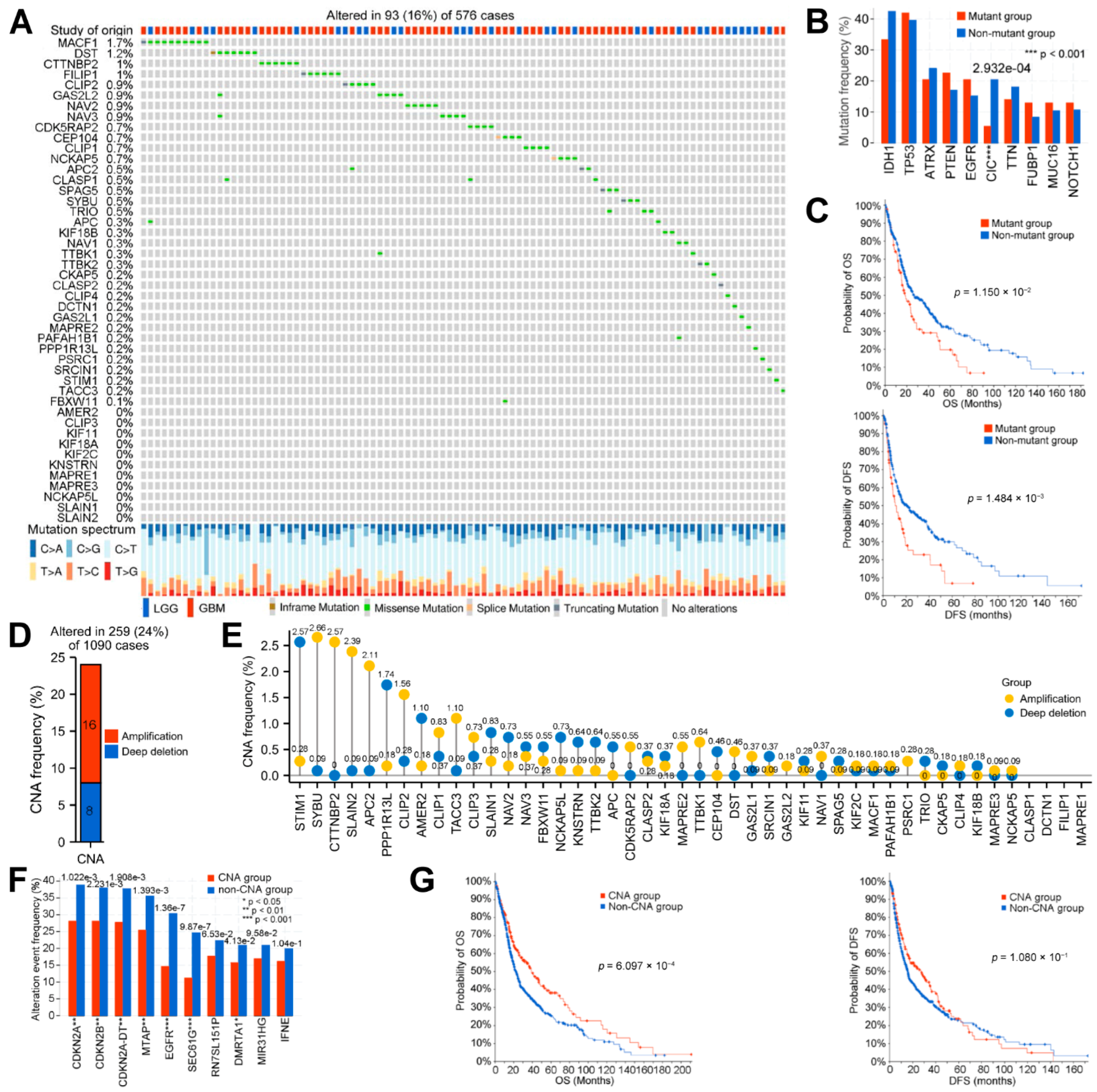
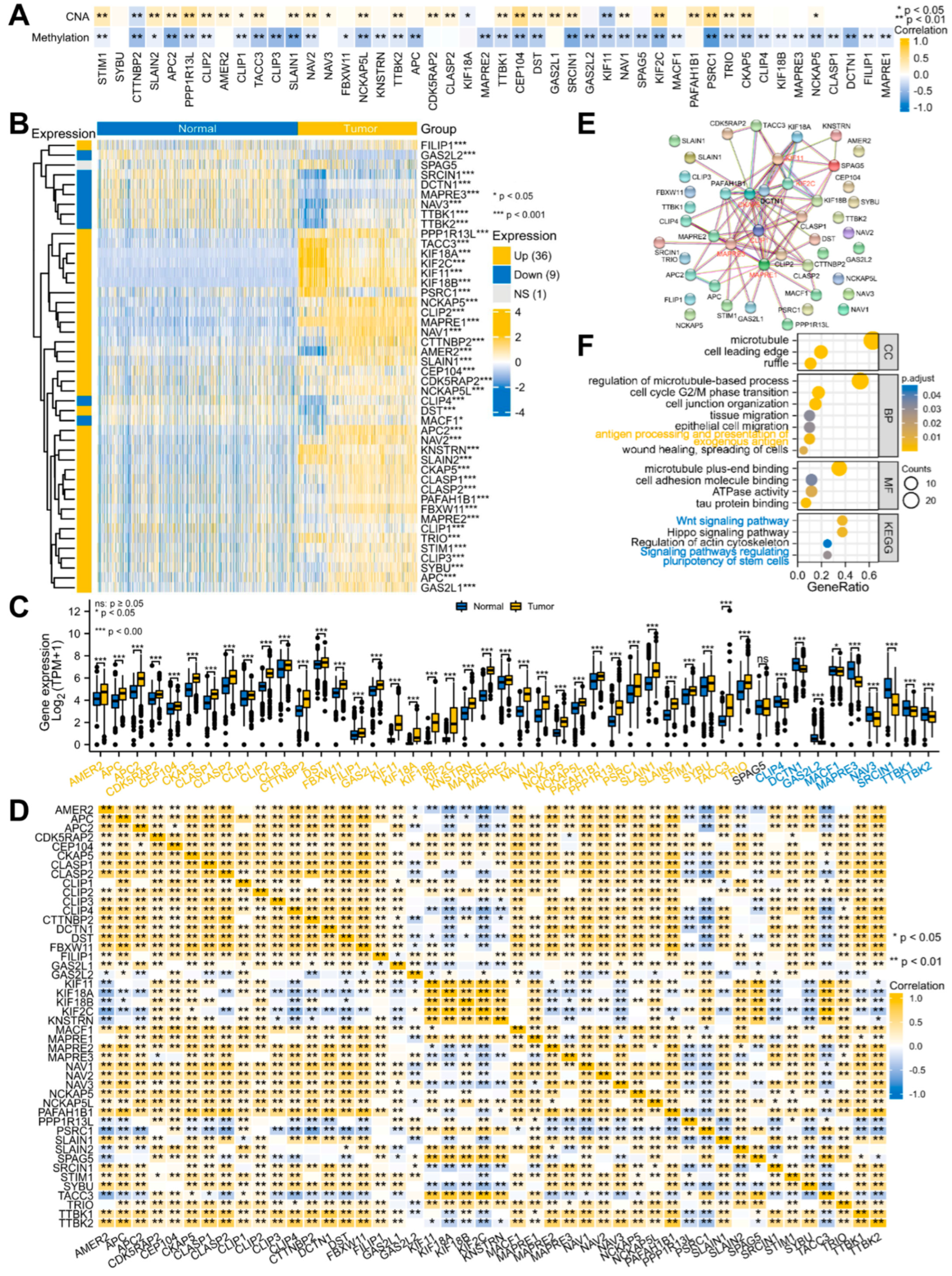
3.1. Systematic Analyses of MPERG Expression, Genetic Alteration, Correlation, and Interaction in Glioma
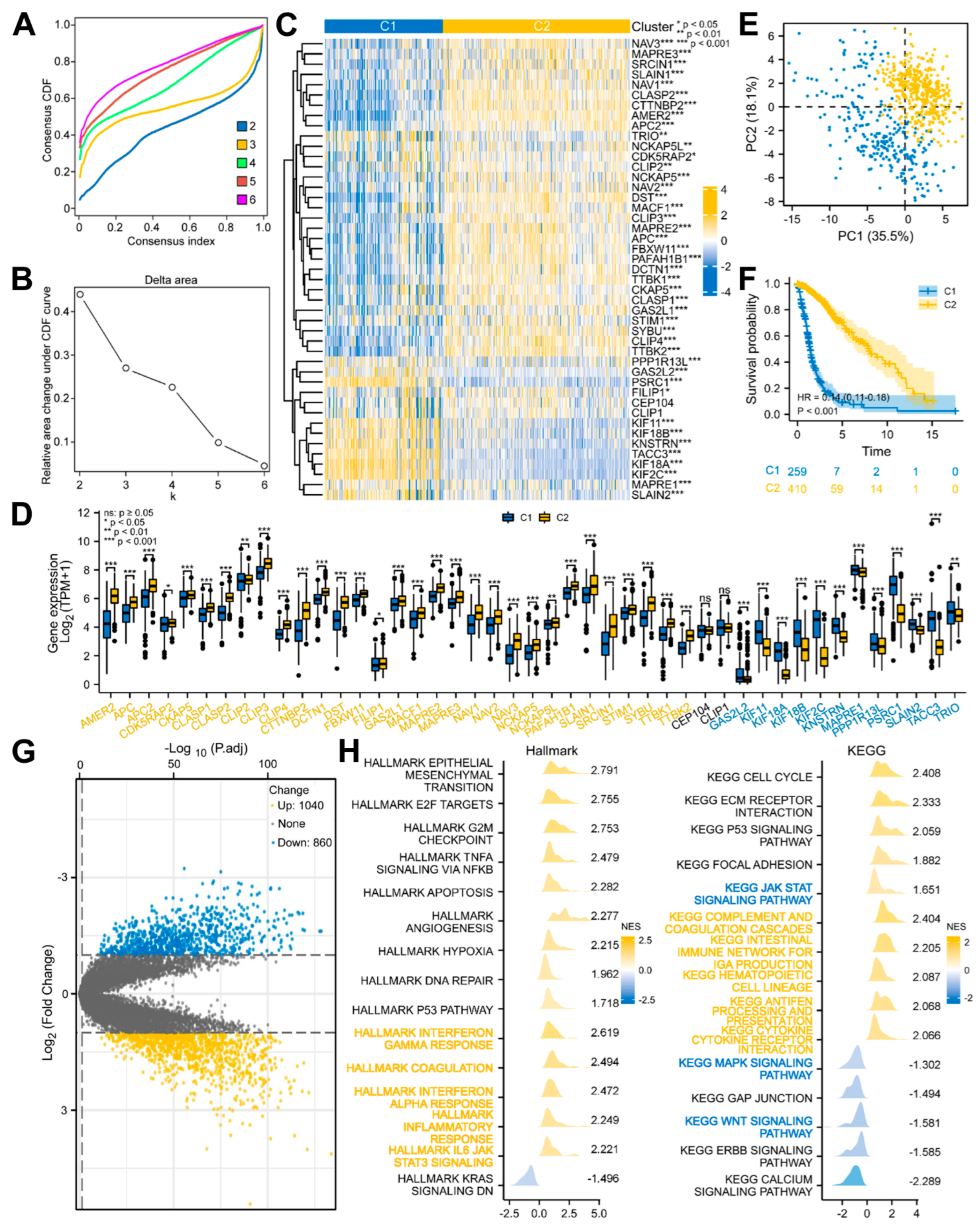
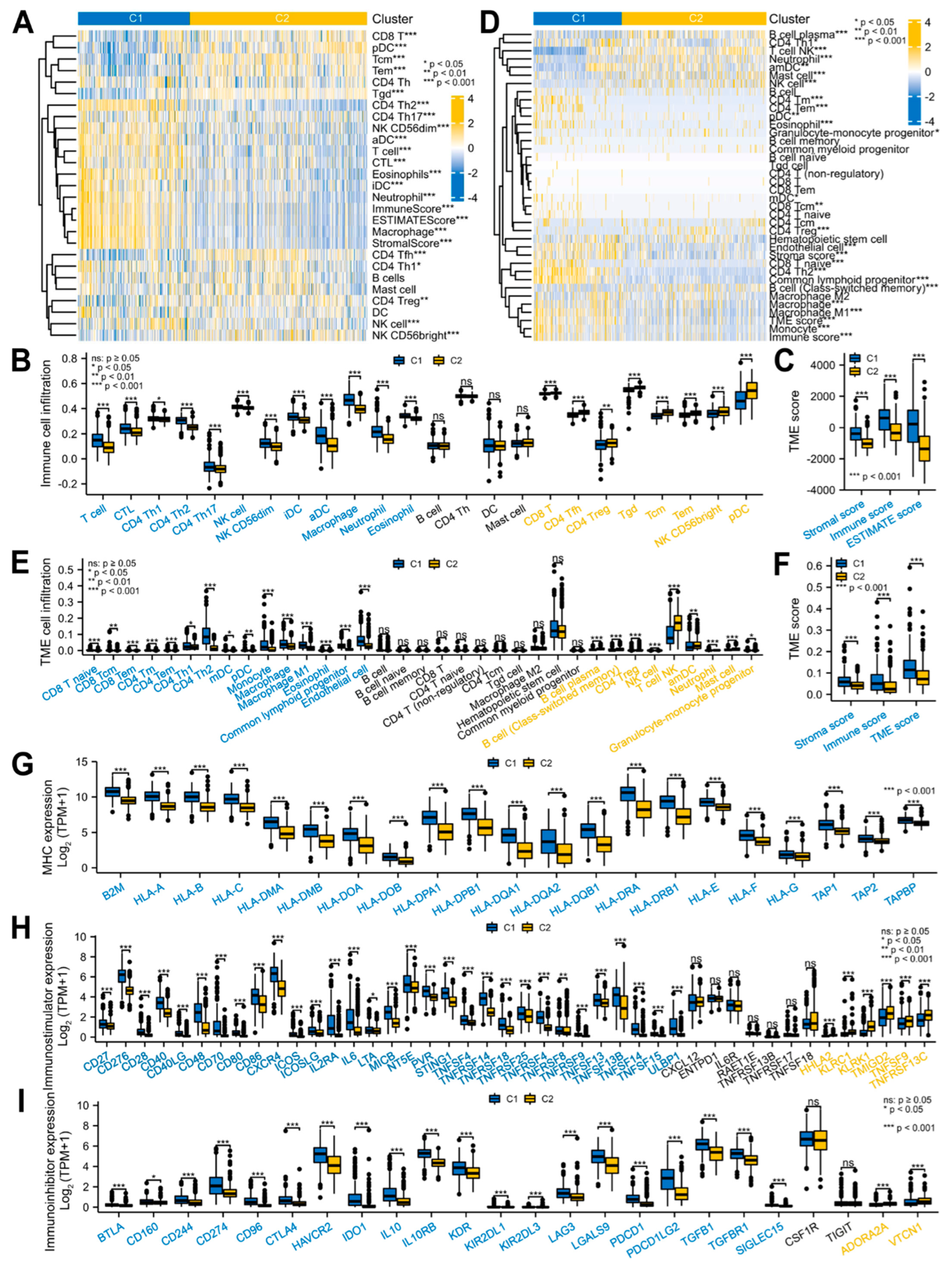
3.2. Glioma Patients with DEMPERGs Were Well Distinguished into Two Subgroups with Survival and TME Infiltration Differences
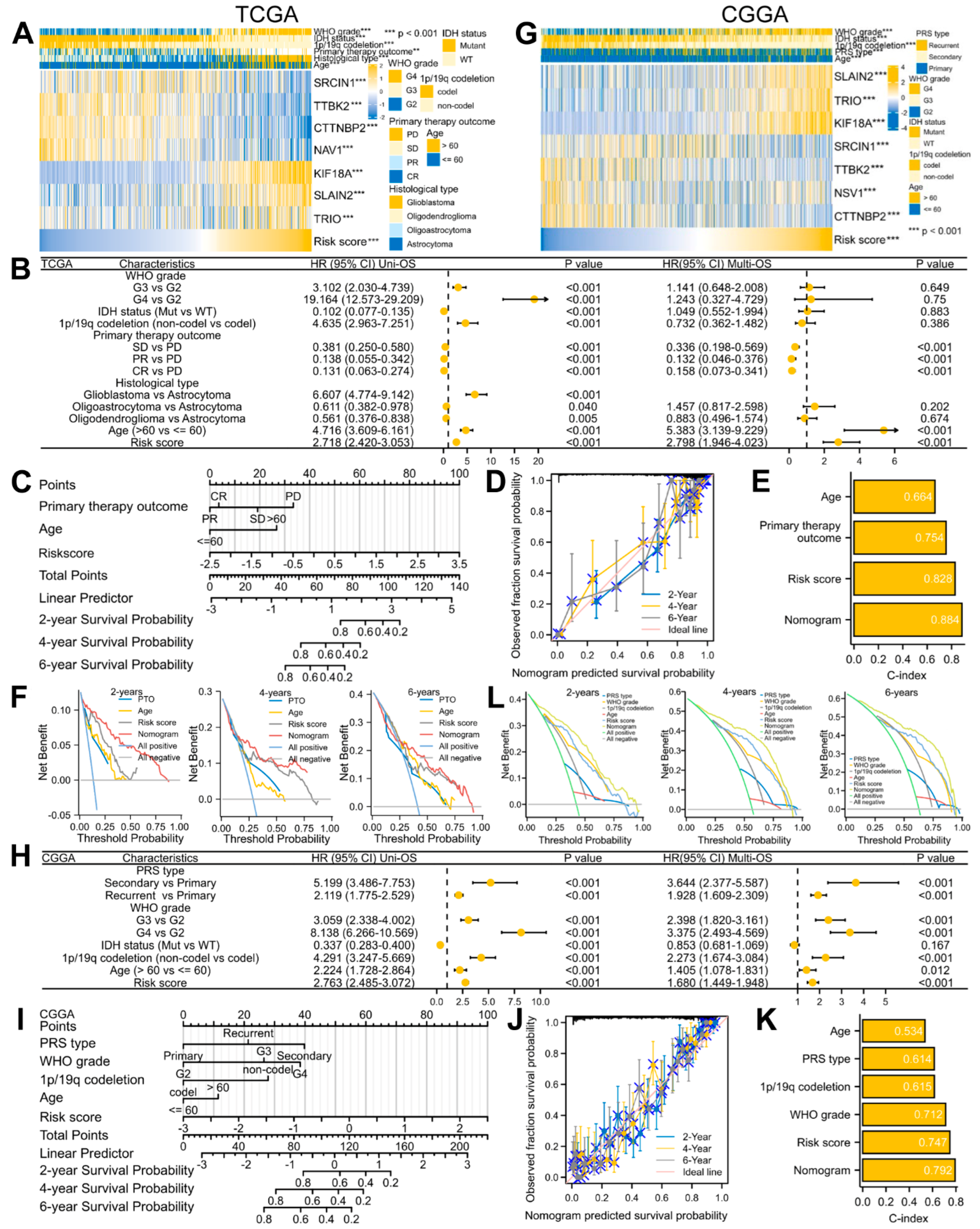
3.3. A Seven-Gene Prognostic Signature Was Constructed and Validated in Glioma
3.4. A Predictive Nomogram Model Was Established and Verified to Facilitate the Clinical Application of the Risk Signature in Glioma
3.5. Signature-Associated Glioma Patients Were Predicted to Benefit from Immunotherapy and Chemotherapy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Chen, F.; Wendl, M.C.; Wyczalkowski, M.A.; Bailey, M.H.; Li, Y.; Ding, L. Moving pan-cancer studies from basic research toward the clinic. Nat. Cancer 2021, 2, 879–890. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.L.; Sun, X.S.; Chen, Q.Y.; Liu, Z.X.; Bian, L.J.; Yuan, L.; Xiao, B.B.; Lu, Z.J.; Li, X.Y.; Yan, J.J.; et al. Development and validation of a transcriptomics-based gene signature to predict distant metastasis and guide induction chemotherapy in locoregionally advanced nasopharyngeal carcinoma. Eur. J. Cancer 2022, 163, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef]
- Kelly, W.J.; Giles, A.J.; Gilbert, M. T lymphocyte-targeted immune checkpoint modulation in glioma. J. Immunother. Cancer 2020, 8, e000379. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Tang, L.; Li, X.; Fan, F.; Liu, Z. Immunotherapy for glioma: Current management and future application. Cancer Lett. 2020, 476, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Karahalil, B.; Yardim-Akaydin, S.; Nacak Baytas, S. An overview of microtubule targeting agents for cancer therapy. Arh. Hig. Rada Toksikol. 2019, 70, 160–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meiring, J.C.M.; Shneyer, B.I.; Akhmanova, A. Generation and regulation of microtubule network asymmetry to drive cell polarity. Curr. Opin. Cell Biol. 2020, 62, 86–95. [Google Scholar] [CrossRef]
- Howard, J.; Hyman, A.A. Dynamics and mechanics of the microtubule plus end. Nature 2003, 422, 753–758. [Google Scholar] [CrossRef]
- van de Willige, D.; Hoogenraad, C.C.; Akhmanova, A. Microtubule plus-end tracking proteins in neuronal development. Cell Mol. Life Sci. 2016, 73, 2053–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aher, A.; Akhmanova, A. Tipping microtubule dynamics, one protofilament at a time. Curr. Opin. Cell. Biol. 2018, 50, 86–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borys, F.; Joachimiak, E.; Krawczyk, H.; Fabczak, H. Intrinsic and Extrinsic Factors Affecting Microtubule Dynamics in Normal and Cancer Cells. Molecules 2020, 25, 3705. [Google Scholar] [CrossRef]
- Wattanathamsan, O.; Pongrakhananon, V. Emerging role of microtubule-associated proteins on cancer metastasis. Front. Pharmacol. 2022, 13, 935493. [Google Scholar] [CrossRef] [PubMed]
- Vivian, J.; Rao, A.A.; Nothaft, F.A.; Ketchum, C.; Armstrong, J.; Novak, A.; Pfeil, J.; Narkizian, J.; Deran, A.D.; Musselman-Brown, A.; et al. Toil enables reproducible, open source, big biomedical data analyses. Nat. Biotechnol. 2017, 35, 314–316. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Zhang, K.N.; Wang, Q.; Li, G.; Zeng, F.; Zhang, Y.; Wu, F.; Chai, R.; Wang, Z.; Zhang, C.; et al. Chinese Glioma Genome Atlas (CGGA): A Comprehensive Resource with Functional Genomic Data from Chinese Glioma Patients. Genom. Proteom. Bioinform. 2021, 19, 1–12. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, L.; Wu, G.; Guo, L.; Zou, X.; Huang, P. Comprehensive Analysis of the PD-L1 and Immune Infiltrates of m(6)A RNA Methylation Regulators in Head and Neck Squamous Cell Carcinoma. Mol. Ther. Nucleic Acids 2020, 21, 299–314. [Google Scholar] [CrossRef]
- Li, Y.; Xiao, J.; Bai, J.; Tian, Y.; Qu, Y.; Chen, X.; Wang, Q.; Li, X.; Zhang, Y.; Xu, J. Molecular characterization and clinical relevance of m(6)A regulators across 33 cancer types. Mol. Cancer 2019, 18, 137. [Google Scholar] [CrossRef]
- Zhang, Z.; Lin, E.; Zhuang, H.; Xie, L.; Feng, X.; Liu, J.; Yu, Y. Construction of a novel gene-based model for prognosis prediction of clear cell renal cell carcinoma. Cancer Cell. Int. 2020, 20, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Vickers, A.J.; Elkin, E.B. Decision curve analysis: A novel method for evaluating prediction models. Med. Decis. Mak. 2006, 26, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [Green Version]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Bonneville, R.; Krook, M.A.; Kautto, E.A.; Miya, J.; Wing, M.R.; Chen, H.Z.; Reeser, J.W.; Yu, L.; Roychowdhury, S. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precis. Oncol. 2017, 2017, PO.17.00073. [Google Scholar] [CrossRef] [PubMed]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kaminska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354.e15. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Wang, Q.; Li, M.; Yang, M.; Yang, Y.; Song, F.; Zhang, W.; Li, X.; Chen, K. Analysis of immune-related signatures of lung adenocarcinoma identified two distinct subtypes: Implications for immune checkpoint blockade therapy. Aging 2020, 12, 3312–3339. [Google Scholar] [CrossRef]
- Liu, C.J.; Hu, F.F.; Xia, M.X.; Han, L.; Zhang, Q.; Guo, A.Y. GSCALite: A web server for gene set cancer analysis. Bioinformatics 2018, 34, 3771–3772. [Google Scholar] [CrossRef] [PubMed]
- Lansbergen, G.; Akhmanova, A. Microtubule plus end: A hub of cellular activities. Traffic 2006, 7, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Mendiratta, G.; Ke, E.; Aziz, M.; Liarakos, D.; Tong, M.; Stites, E.C. Cancer gene mutation frequencies for the U.S. population. Nat. Commun. 2021, 12, 5961. [Google Scholar] [CrossRef]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Z.; Wang, S.; Wu, C.; Wu, T.; Zhao, X.; Ning, W.; Wang, G.; Wang, J.; Chen, J.; Diao, K.; et al. The repertoire of copy number alteration signatures in human cancer. Brief. Bioinform. 2023, bbad053. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, P.Y.; Lee, S.P.; Chen, Y.K.; Hsueh, Y.P. Cortactin-binding protein 2 increases microtubule stability and regulates dendritic arborization. J. Cell Sci. 2014, 127, 3521–3534. [Google Scholar] [CrossRef] [Green Version]
- Si, L.; Chen, J.; Yang, S.; Liu, Z.; Chen, Y.; Peng, M.; Jia, Y. lncRNA HEIH accelerates cell proliferation and inhibits cell senescence by targeting miR-3619-5p/CTTNBP2 axis in ovarian cancer. Menopause 2020, 27, 1302–1314. [Google Scholar] [CrossRef]
- Marquis, C.; Fonseca, C.L.; Queen, K.A.; Wood, L.; Vandal, S.E.; Malaby, H.L.H.; Clayton, J.E.; Stumpff, J. Chromosomally unstable tumor cells specifically require KIF18A for proliferation. Nat. Commun. 2021, 12, 1213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhu, C.; Chen, H.; Li, L.; Guo, L.; Jiang, W.; Lu, S.H. Kif18A is involved in human breast carcinogenesis. Carcinogenesis 2010, 31, 1676–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, W.; Huang, G.; Liao, Y.; Yang, J.; Chen, Q.; Xiao, S.; Jin, J.; He, S.; Wang, C. High KIF18A expression correlates with unfavorable prognosis in primary hepatocellular carcinoma. Oncotarget 2014, 5, 10271–10279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.I.; Cao, B.; Nan, N.; Wang, Y.U.; Zhai, X.U.; Li, Y.; Chong, T. Elevated expression of KIF18A enhances cell proliferation and predicts poor survival in human clear cell renal carcinoma. Exp. Ther. Med. 2016, 12, 377–383. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Liao, M.; Liao, Y.; Chen, X.; Huang, C.; Fan, J.; Liao, W. The role of kinesin KIF18A in the invasion and metastasis of hepatocellular carcinoma. World J. Surg. Oncol. 2018, 16, 36. [Google Scholar] [CrossRef] [Green Version]
- Alfarsi, L.H.; Elansari, R.; Toss, M.S.; Diez-Rodriguez, M.; Nolan, C.C.; Ellis, I.O.; Rakha, E.A.; Green, A.R. Kinesin family member-18A (KIF18A) is a predictive biomarker of poor benefit from endocrine therapy in early ER+ breast cancer. Breast Cancer Res. Treat. 2019, 173, 93–102. [Google Scholar] [CrossRef]
- Chen, F.T.; Zhong, F.K. Kinesin Family Member 18A (KIF18A) Contributes to the Proliferation, Migration, and Invasion of Lung Adenocarcinoma Cells In Vitro and In Vivo. Dis. Markers 2019, 2019, 6383685. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Shen, T.; Zhang, Z.; Li, Y.; Pan, Z. Expression of KIF18A Is Associated with Increased Tumor Stage and Cell Proliferation in Prostate Cancer. Med. Sci. Monit. 2019, 25, 6418–6428. [Google Scholar] [CrossRef]
- Zhong, Y.; Jiang, L.; Lin, H.; Li, X.; Long, X.; Zhou, Y.; Li, B.; Li, Z. Overexpression of KIF18A promotes cell proliferation, inhibits apoptosis, and independently predicts unfavorable prognosis in lung adenocarcinoma. IUBMB Life 2019, 71, 942–955. [Google Scholar] [CrossRef]
- Qian, L.X.; Cao, X.; Du, M.Y.; Ma, C.X.; Zhu, H.M.; Peng, Y.; Hu, X.Y.; He, X.; Yin, L. KIF18A knockdown reduces proliferation, migration, invasion and enhances radiosensitivity of esophageal cancer. Biochem. Biophys. Res. Commun. 2021, 557, 192–198. [Google Scholar] [CrossRef]
- Tao, B.Y.; Liu, Y.Y.; Liu, H.Y.; Zhang, Z.H.; Guan, Y.Q.; Wang, H.; Shi, Y.; Zhang, J. Prognostic Biomarker KIF18A and Its Correlations With Immune Infiltrates and Mitosis in Glioma. Front. Genet. 2022, 13, 852049. [Google Scholar] [CrossRef]
- van der Vaart, B.; Manatschal, C.; Grigoriev, I.; Olieric, V.; Gouveia, S.M.; Bjelic, S.; Demmers, J.; Vorobjev, I.; Hoogenraad, C.C.; Steinmetz, M.O.; et al. SLAIN2 links microtubule plus end-tracking proteins and controls microtubule growth in interphase. J. Cell Biol. 2011, 193, 1083–1099. [Google Scholar] [CrossRef] [Green Version]
- Bouchet, B.P.; Noordstra, I.; van Amersfoort, M.; Katrukha, E.A.; Ammon, Y.C.; Ter Hoeve, N.D.; Hodgson, L.; Dogterom, M.; Derksen, P.W.B.; Akhmanova, A. Mesenchymal Cell Invasion Requires Cooperative Regulation of Persistent Microtubule Growth by SLAIN2 and CLASP1. Dev. Cell 2016, 39, 708–723. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, M.; Zhao, S.; Jiang, Z.; Wang, S.; Sun, P.; Quan, J.; Yan, D.; Wang, X. MALAT1 sponges miR-106b-5p to promote the invasion and metastasis of colorectal cancer via SLAIN2 enhanced microtubules mobility. eBioMedicine 2019, 41, 286–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasso, S.; Cangelosi, D.; Chapelle, J.; Alzona, M.; Centonze, G.; Lamolinara, A.; Salemme, V.; Angelini, C.; Morellato, A.; Saglietto, A.; et al. The SRCIN1/p140Cap adaptor protein negatively regulates the aggressiveness of neuroblastoma. Cell. Death Differ. 2020, 27, 790–807. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yuan, J.; Guo, M.; Xiang, R.; Wang, X.; Xie, T.; Zhuang, X.; Li, Q.; Lai, Q. miR-657 Targets SRCIN1 via the Slug Pathway to Promote NSCLC Tumor Growth and EMT Induction. Dis. Markers 2022, 2022, 4842454. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.S.; Hou, P.; Kong, Y. Hepatitis B virus promotes proliferation and metastasis in male Chinese hepatocellular carcinoma patients through the LEF-1/miR-371a-5p/SRCIN1/pleiotrophin/Slug pathway. Exp. Cell Res. 2018, 370, 174–188. [Google Scholar] [CrossRef]
- Xu, X.; Wang, W.; Su, N.; Zhu, X.; Yao, J.; Gao, W.; Hu, Z.; Sun, Y. miR-374a promotes cell proliferation, migration and invasion by targeting SRCIN1 in gastric cancer. FEBS Lett. 2015, 589, 407–413. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Luo, L.J.; Zhang, L.; Wang, D.D.; Yang, S.J.; Ding, L.; Li, J.; Chen, D.; Ma, R.; Wu, J.Z.; et al. MiR-346 promotes the biological function of breast cancer cells by targeting SRCIN1 and reduces chemosensitivity to docetaxel. Gene 2017, 600, 21–28. [Google Scholar] [CrossRef]
- Nguyen, A.; Goetz, S.C. TTBK2 controls cilium stability by regulating distinct modules of centrosomal proteins. Mol. Biol. Cell. 2022, 34, mbcE22080373. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kakeno, M.; Matsui, T.; Sugiyama, I.; Arimura, N.; Matsuzawa, K.; Shirahige, A.; Ishidate, F.; Nishioka, T.; Taya, S.; et al. TTBK2 with EB1/3 regulates microtubule dynamics in migrating cells through KIF2A phosphorylation. J. Cell Biol. 2015, 210, 737–751. [Google Scholar] [CrossRef] [Green Version]
- Bender, C.; Ullrich, A. PRKX, TTBK2 and RSK4 expression causes Sunitinib resistance in kidney carcinoma- and melanoma-cell lines. Int. J. Cancer 2012, 131, E45–E55. [Google Scholar] [CrossRef]
- Powers, R.M.; Daza, R.; Koehler, A.E.; Courchet, J.; Calabrese, B.; Hevner, R.F.; Halpain, S. Growth cone macropinocytosis of neurotrophin receptor and neuritogenesis are regulated by neuron navigator 1. Mol. Biol. Cell 2022, 33, ar64. [Google Scholar] [CrossRef] [PubMed]
- van Haren, J.; Boudeau, J.; Schmidt, S.; Basu, S.; Liu, Z.; Lammers, D.; Demmers, J.; Benhari, J.; Grosveld, F.; Debant, A.; et al. Dynamic microtubules catalyze formation of navigator-TRIO complexes to regulate neurite extension. Curr. Biol. 2014, 24, 1778–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seipel, K.; Medley, Q.G.; Kedersha, N.L.; Zhang, X.A.; O’Brien, S.P.; Serra-Pages, C.; Hemler, M.E.; Streuli, M. Trio amino-terminal guanine nucleotide exchange factor domain expression promotes actin cytoskeleton reorganization, cell migration and anchorage-independent cell growth. J. Cell Sci. 1999, 112 Pt 12, 1825–1834. [Google Scholar] [CrossRef] [PubMed]
- Deinhardt, K.; Kim, T.; Spellman, D.S.; Mains, R.E.; Eipper, B.A.; Neubert, T.A.; Chao, M.V.; Hempstead, B.L. Neuronal growth cone retraction relies on proneurotrophin receptor signaling through Rac. Sci. Signal. 2011, 4, ra82. [Google Scholar] [CrossRef] [Green Version]
- Hirahara, K.; Nakayama, T. CD4+ T-cell subsets in inflammatory diseases: Beyond the Th1/Th2 paradigm. Int. Immunol. 2016, 28, 163–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, E.N.; Gocher, A.M.; Workman, C.J.; Vignali, D.A.A. Regulatory T Cells: Barriers of Immune Infiltration Into the Tumor Microenvironment. Front. Immunol. 2021, 12, 702726. [Google Scholar] [CrossRef]
- Liu, X.; Liu, Y.; Qi, Y.; Huang, Y.; Hu, F.; Dong, F.; Shu, K.; Lei, T. Signal Pathways Involved in the Interaction Between Tumor-Associated Macrophages/TAMs and Glioblastoma Cells. Front. Oncol. 2022, 12, 822085. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Protein Name | Binding Mode |
|---|---|---|
| AMER2 (FAM123A) | APC membrane recruitment protein 2 | Via MAPRE1/2/3 |
| APC (DP2.5) | Adenomatous polyposis coli protein | Autonomous or via MAPRE1/2/3 |
| APC2 (APCL) | Adenomatous polyposis coli protein 2 | Via MAPRE1/2/3 |
| CDK5RAP2 (CEP215) | CDK5 regulatory subunit-associated protein 2 | Via MAPRE1/2/3 |
| CEP104 (KIAA0562) | Centrosomal protein of 104 kDa | Via MAPRE1/2/3 |
| CKAP5 (ch-TOG) | Cytoskeleton-associated protein 5 | Autonomous |
| CLASP1 (MAST1) | CLIP-associating protein 1 | Via MAPRE1/2/3 |
| CLASP2 (KIAA0627) | CLIP-associating protein 2 | Via MAPRE1/2/3 |
| CLIP1 (CYLN1, CLIP-170) | CAP-Gly domain-containing linker protein 1 | Via MAPRE1/2/3 |
| CLIP2 (CYLN2, CLIP-115) | CAP-Gly domain-containing linker protein 2 | Via MAPRE1/2/3 |
| CLIP3 (CLIPR59) | CAP-Gly domain-containing linker protein 3 | Via MAPRE1/2/3 |
| CLIP4 (RSNL2) | CAP-Gly domain-containing linker protein 4 | Via MAPRE1/2/3 |
| CTTNBP2 (CORTBP2) | Cortactin-binding protein 2 | Via MAPRE1/2/3 |
| DCTN1 (p150glued) | Dynactin subunit 1 | Via MAPRE1/2/3 |
| DST | Dystonin | Via MAPRE1/2/3 |
| FBXW11 | F-box/WD repeat-containing protein 11 | Via MAPRE1/2/3 |
| FILIP1 (KIAA1275) | Filamin-A-interacting protein 1 | Via MAPRE1/2/3 |
| GAS2L1 (GAR22) | GAS2-like protein 1 | Via MAPRE1/2/3 |
| GAS2L2 (GAR17) | GAS2-like protein 2 | Via MAPRE1/2/3 |
| KIF11 (Eg5) | Kinesin-like protein KIF11 | Via MAPRE1/2/3 |
| KIF18A | Kinesin-like protein KIF18A | Via MAPRE1/2/3 |
| KIF18B | Kinesin-like protein KIF18B | Via MAPRE1/2/3 |
| KIF2C (MCAK) | Kinesin-like protein KIF2C | Via MAPRE1/2/3 |
| KNSTRN (SKAP) | Small kinetochore-associated protein | Via MAPRE1/2/3 |
| MACF1 (ACF7) | Microtubule-actin cross-linking factor1 | Via MAPRE1/2/3 |
| MAPRE1 (EB1) | Microtubule-associated protein RP/EB family member 1 | Autonomous |
| MAPRE2 (EB2) | Microtubule-associated protein RP/EB family member 2 | Via MAPRE1/2/3 |
| MAPRE3 (EB3) | Microtubule-associated protein RP/EB family member 3 | Via MAPRE1/2/3 |
| NAV1 (POMFIL3) | Neuron navigator 1 | Via MAPRE1/2/3 |
| NAV2 (POMFIL2) | Neuron navigator 2 | Via MAPRE1/2/3 |
| NAV3 (POMFIL1) | Neuron navigator 3 | Via MAPRE1/2/3 |
| NCKAP5 (ERIH, NAP5) | Nck-associated protein 5 | Via MAPRE1/2/3 |
| NCKAP5L (CEP169) | Nck-associated protein 5-like | Via MAPRE1/2/3 |
| PAFAH1B1 (LIS1) | Lissencephaly-1 homolog | Via CLIP-1 |
| PPP1R13L (iASPP) | RelA-associated inhibitor | Via MAPRE1/2/3 |
| PSRC1 (DDA3) | Proline/serine-rich coiled-coil protein 1 | Via MAPRE1/2/3 |
| SLAIN1 (C13orf32) | SLAIN motif-containing protein 1 | Via MAPRE1/2/3 |
| SLAIN2 (KIAA1458) | SLAIN motif-containing protein 2 | Via MAPRE1/2/3 |
| SPAG5 (Astrin) | Sperm-associated antigen 5 | Via MAPRE1/2/3 |
| SRCIN1 (P140) | SRC kinase signaling inhibitor 1 | Via MAPRE1/2/3 |
| STIM1 (GOK) | Stromal interaction molecule 1 | Via MAPRE1/2/3 |
| SYBU (GOLSYN) | Syntabulin | Via MAPRE1/2/3 |
| TACC3 (ERIC1) | Transforming acidic coiled-coil-containing protein 3 | Unclear |
| TRIO (ARHGEF23) | Triple functional domain protein | Via MAPRE1/2/3 |
| TTBK1 (BDTK) | Tau-tubulin kinase 1 | Via MAPRE1/2/3 |
| TTBK2 (KIAA0847) | Tau-tubulin kinase 2 | Via MAPRE1/2/3 |
| Characteristic | Low Risk (334) | High Risk (335) | p |
|---|---|---|---|
| WHO grade, n (%) | <0.001 *** | ||
| G2 | 169 (27.6%) | 46 (7.2%) | |
| G3 | 127 (20.8%) | 110 (18%) | |
| G4 | 1 (0.2%) | 159 (26%) | |
| IDH status, n (%) | <0.001 *** | ||
| WT | 10 (1.5%) | 227 (34.4%) | |
| Mut | 322 (48.8%) | 101 (15.3%) | |
| 1p/19q codeletion, n (%) | <0.001 *** | ||
| Codel | 158 (23.8%) | 9 (1.4%) | |
| Non-codel | 176 (26.5%) | 320 (48.3%) | |
| Histological type, n (%) | <0.001 *** | ||
| Astrocytoma | 88 (13.2%) | 104 (15.5%) | |
| Glioblastoma | 1 (0.1%) | 159 (23.8%) | |
| Oligoastrocytoma | 86 (12.9%) | 42 (6.3%) | |
| Oligodendroglioma | 159 (23.8%) | 30 (4.5%) | |
| Primary therapy outcome (PTO), n (%) | 0.001 ** | ||
| Progressive disease (PD) | 53 (12%) | 50 (11.3%) | |
| Stable disease (SD) | 102 (23%) | 42 (9.5%) | |
| Partial response (PR) | 42 (9.5%) | 20 (4.5%) | |
| Complete response (CR) | 100 (22.6%) | 34 (7.7%) | |
| Gender, n (%) | 0.331 | ||
| Female | 148 (22.1%) | 135 (20.2%) | |
| Male | 186 (27.8%) | 200 (29.9%) | |
| Race, n (%) | 0.200 | ||
| Asian | 6 (0.9%) | 7 (1.1%) | |
| Black or African American | 11 (1.7%) | 21 (3.2%) | |
| White | 309 (47%) | 303 (46.1%) | |
| Age, n (%) | <0.001 *** | ||
| ≤60 | 305 (45.6%) | 225 (33.6%) | |
| >60 | 29 (4.3%) | 110 (16.4%) |
| Characteristic | Low Risk (485) | High Risk (485) | p |
|---|---|---|---|
| WHO grade, n (%) | <0.001 *** | ||
| G2 | 39 (4%) | 231 (23.9%) | |
| G3 | 131 (13.6%) | 191 (19.8%) | |
| G4 | 311 (32.2%) | 63 (6.5%) | |
| IDH status, n (%) | <0.001 *** | ||
| WT | 346 (37.6%) | 75 (8.1%) | |
| Mutant | 132 (14.3%) | 368 (40%) | |
| 1p/19q codeletion, n (%) | <0.001 *** | ||
| Codel | 14 (1.6%) | 185 (20.6%) | |
| Non-codel | 439 (49%) | 258 (28.8%) | |
| Primary-recurrent-secondary (PRS) type, n (%) | <0.001 *** | ||
| Primary | 273 (28.3%) | 353 (36.5%) | |
| Secondary | 23 (2.4%) | 6 (0.6%) | |
| Recurrent | 185 (19.2%) | 126 (13%) | |
| Age, n (%) | <0.001 *** | ||
| ≤60 | 414 (42.7%) | 463 (47.8%) | |
| >60 | 70 (7.2%) | 22 (2.3%) | |
| Gender, n (%) | 0.117 | ||
| Female | 187 (19.3%) | 212 (21.9%) | |
| Male | 298 (30.7%) | 273 (28.1%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Li, W.; Pan, L.; Li, L.; Xu, Y.; Wang, Y.; Zhang, X.; Zhang, S. Dynamic Regulation Genes at Microtubule Plus Ends: A Novel Class of Glioma Biomarkers. Biology 2023, 12, 488. https://doi.org/10.3390/biology12030488
Wang W, Li W, Pan L, Li L, Xu Y, Wang Y, Zhang X, Zhang S. Dynamic Regulation Genes at Microtubule Plus Ends: A Novel Class of Glioma Biomarkers. Biology. 2023; 12(3):488. https://doi.org/10.3390/biology12030488
Chicago/Turabian StyleWang, Wenwen, Weilong Li, Lifang Pan, Lingjie Li, Yasi Xu, Yuqing Wang, Xiaochen Zhang, and Shirong Zhang. 2023. "Dynamic Regulation Genes at Microtubule Plus Ends: A Novel Class of Glioma Biomarkers" Biology 12, no. 3: 488. https://doi.org/10.3390/biology12030488