
Exploiting Multi-Omics Profiling and Systems Biology to Investigate Functions of TOMM34
 , , , , , , , and
, , , , , , , and 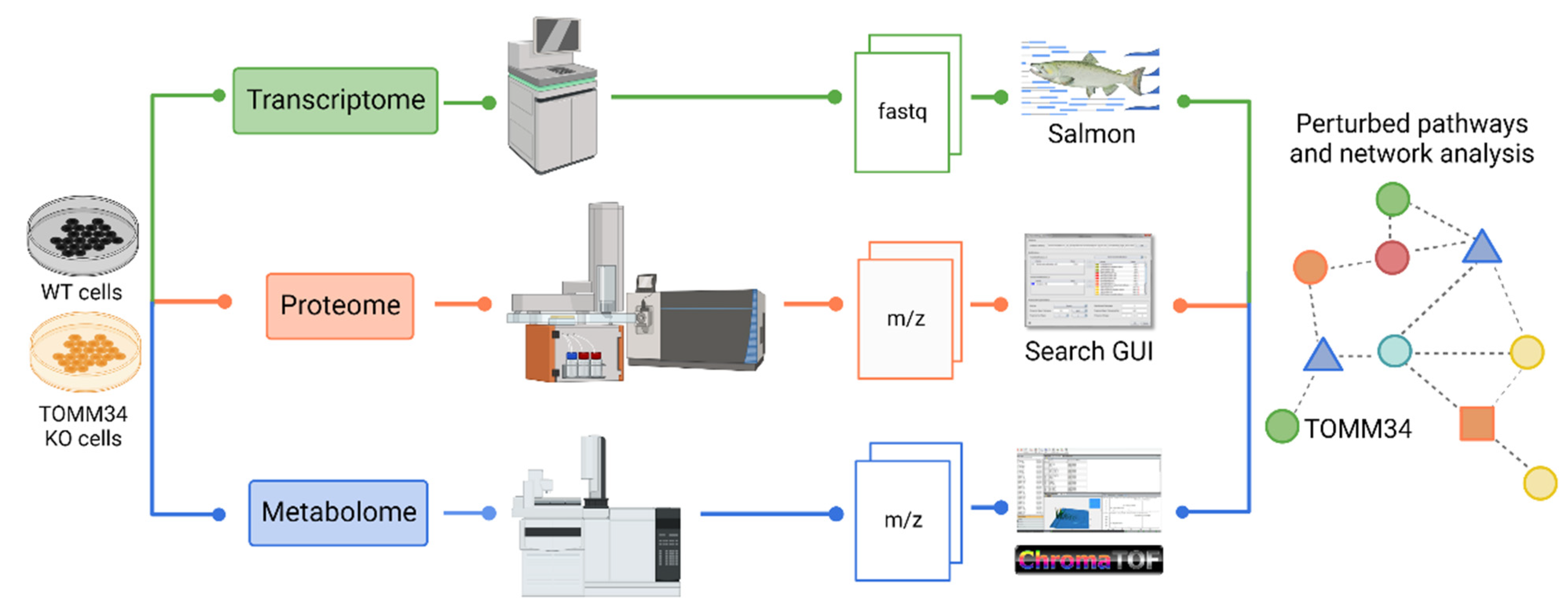{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells Preparation
2.2. CRISPR-CAS9
2.3. Western Blotting
2.4. Transcriptome Profiling
2.5. Proteome Profiling
2.6. Metabolome Profiling
2.7. Bioinformatics and Statistical Data Analysis
3. Results
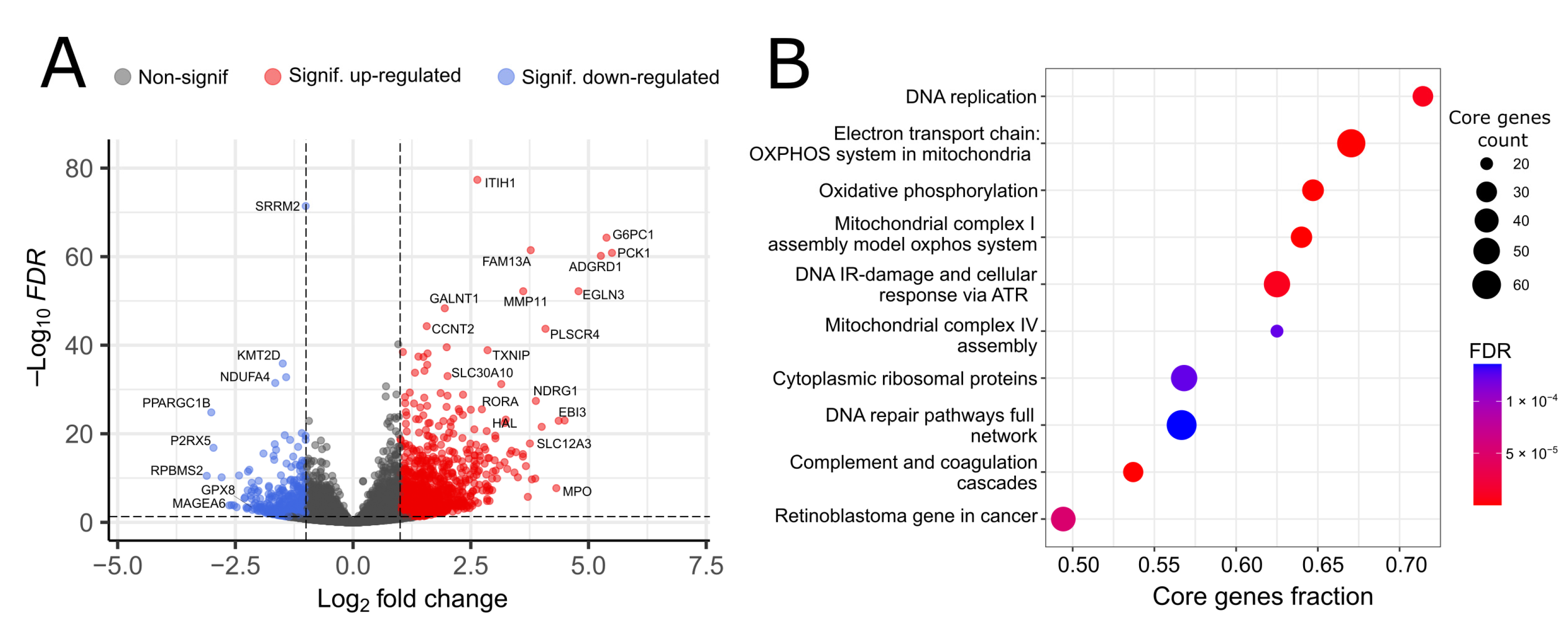
3.1. Transcriptomic Differences
3.2. Proteomic Differences
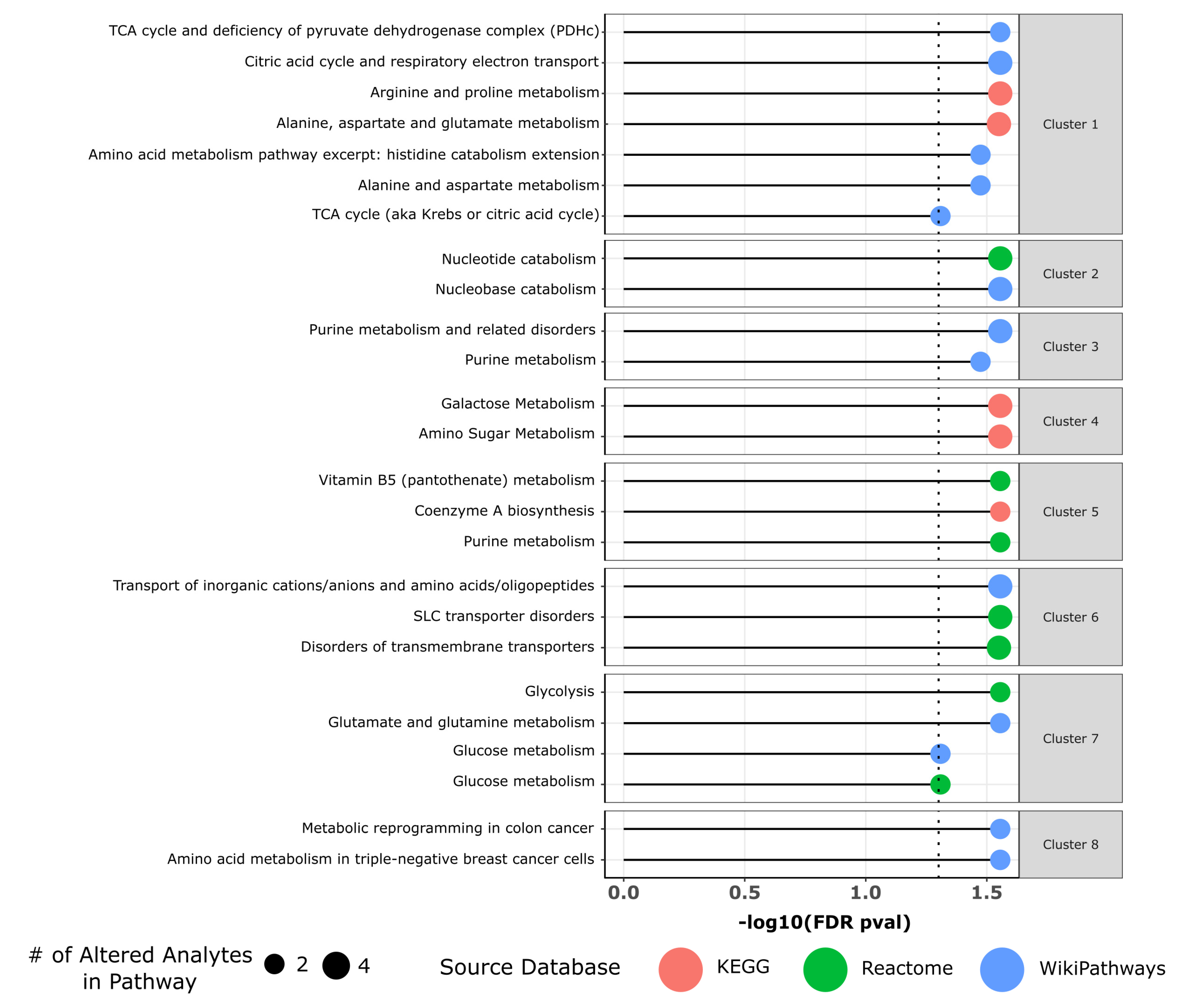
3.3. Metabolic Differences
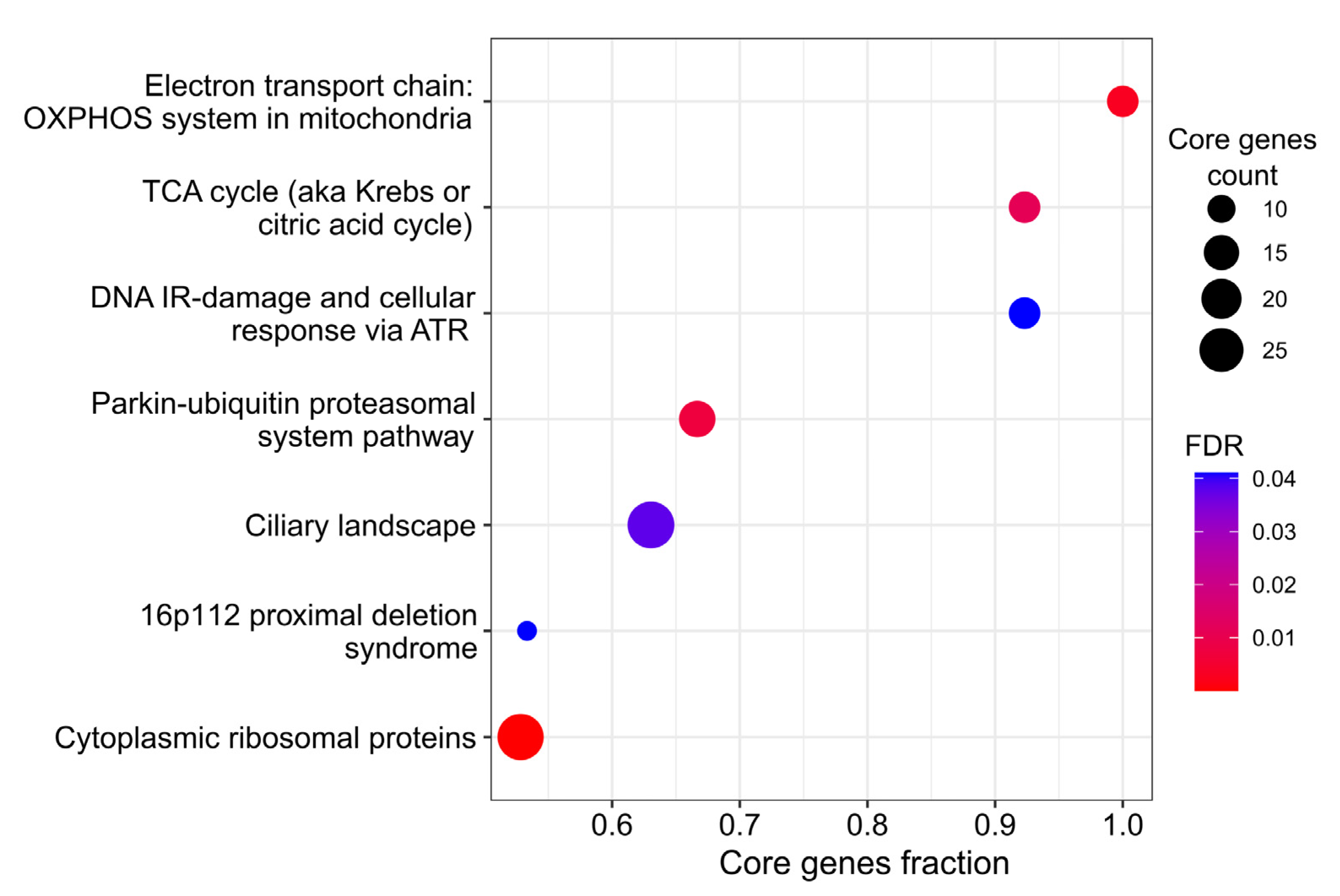
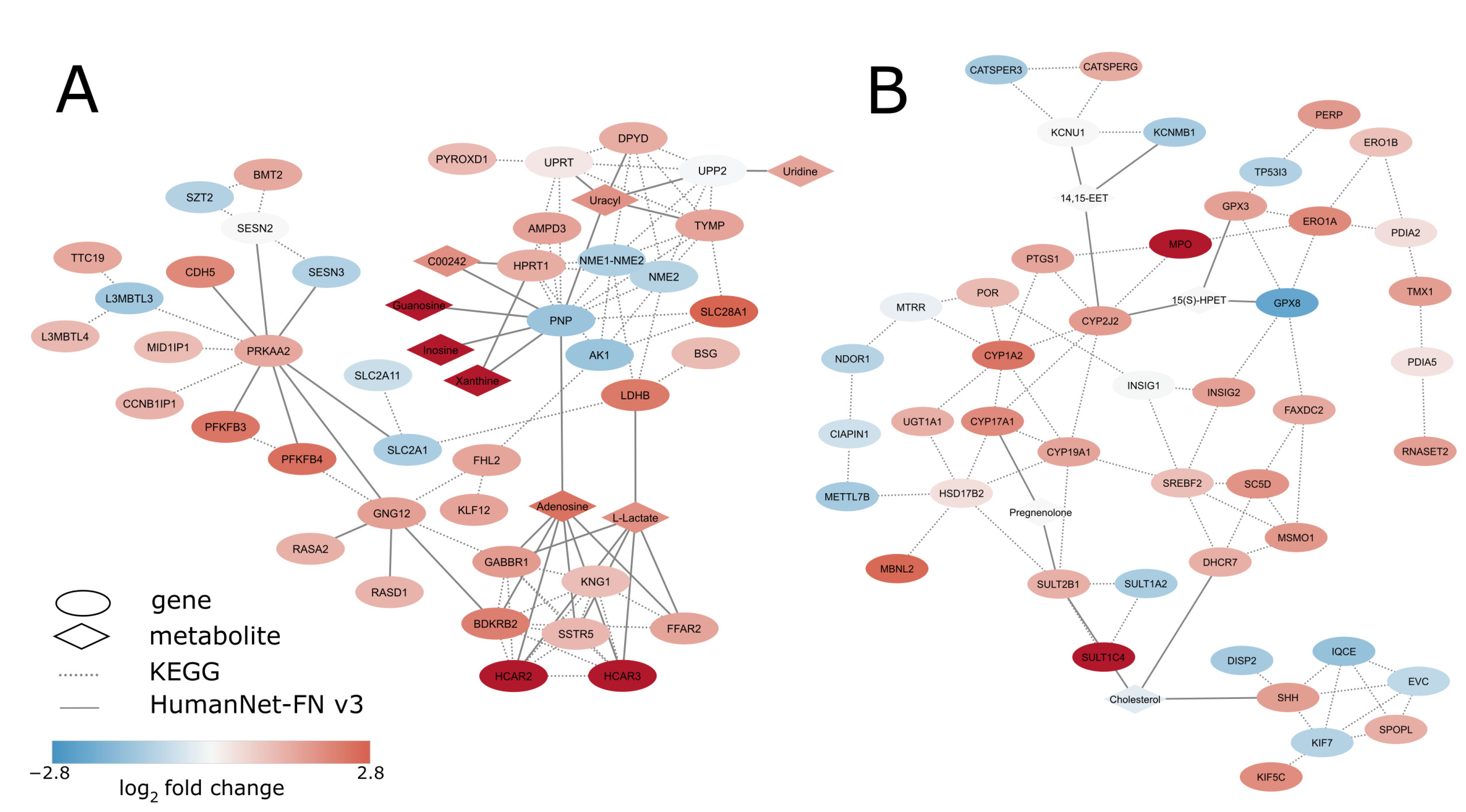
3.4. De Novo Network Enrichment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tomczak, A.; Mortensen, J.M.; Winnenburg, R.; Liu, C.; Alessi, D.T.; Swamy, V.; Vallania, F.; Lofgren, S.; Haynes, W.; Shah, N.H.; et al. Interpretation of Biological Experiments Changes with Evolution of the Gene Ontology and Its Annotations. Sci. Rep. 2018, 8, 5115. [Google Scholar] [CrossRef] [PubMed]
- Rensvold, J.W.; Shishkova, E.; Sverchkov, Y.; Miller, I.J.; Cetinkaya, A.; Pyle, A.; Manicki, M.; Brademan, D.R.; Alanay, Y.; Raiman, J.; et al. Defining Mitochondrial Protein Functions through Deep Multiomic Profiling. Nature 2022, 606, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Nuttall, S.D.; Hanson, B.J.; Mori, M.; Hoogenraad, N.J. hTom34: A Novel Translocase for the Import of Proteins into Human Mitochondria. DNA Cell Biol. 1997, 16, 1067–1074. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial Protein Organization: From Biogenesis to Networks and Function. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Chewawiwat, N.; Yano, M.; Terada, K.; Hoogenraad, N.J.; Mori, M. Characterization of the Novel Mitochondrial Protein Import Component, Tom34, in Mammalian Cells1. J. Biochem. 1999, 125, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, A.; Avramova, L.V.; Weiner, H. Tom34 Unlike Tom20 Does Not Interact with the Leader Sequences of Mitochondrial Precursor Proteins. Arch. Biochem. Biophys. 2002, 400, 97–104. [Google Scholar] [CrossRef]
- Yang, C.-S.; Weiner, H. Yeast Two-Hybrid Screening Identifies Binding Partners of Human Tom34 That Have ATPase Activity and Form a Complex with Tom34 in the Cytosol. Arch. Biochem. Biophys. 2002, 400, 105–110. [Google Scholar] [CrossRef]
- Young, J.C.; Obermann, W.M.; Hartl, F.U. Specific Binding of Tetratricopeptide Repeat Proteins to the C-Terminal 12-KDa Domain of Hsp90. J. Biol. Chem. 1998, 273, 18007–18010. [Google Scholar] [CrossRef] [Green Version]
- Faou, P.; Hoogenraad, N.J. Tom34: A Cytosolic Cochaperone of the Hsp90/Hsp70 Protein Complex Involved in Mitochondrial Protein Import. Biochim. Biophys. Acta 2012, 1823, 348–357. [Google Scholar] [CrossRef]
- Durech, M.; Trcka, F.; Man, P.; Blackburn, E.A.; Hernychova, L.; Dvorakova, P.; Coufalova, D.; Kavan, D.; Vojtesek, B.; Muller, P. Novel Entropically Driven Conformation-Specific Interactions with Tomm34 Protein Modulate Hsp70 Protein Folding and ATPase Activities. Mol. Cell. Proteom. MCP 2016, 15, 1710–1727. [Google Scholar] [CrossRef] [Green Version]
- Trcka, F.; Durech, M.; Vankova, P.; Vandova, V.; Simoncik, O.; Kavan, D.; Vojtesek, B.; Muller, P.; Man, P. The Interaction of the Mitochondrial Protein Importer TOMM34 with HSP70 Is Regulated by TOMM34 Phosphorylation and Binding to 14-3-3 Adaptors. J. Biol. Chem. 2020, 295, 8928–8944. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Tan, R.; Huang, Y.; Chen, X.; Kong, Q.; Guo, K.; Xu, M. High Expression of Tomm34 and Its Correlations With Clinicopathology in Oral Squamous Cell Carcinoma. Pathol. Oncol. Res. 2021, 27, 641042. [Google Scholar] [CrossRef]
- Zhang, W.; Pan, T. Quantitative Probing of Glycosylated Queuosine Modifications in TRNA. Methods Enzymol. 2021, 658, 73–82. [Google Scholar] [CrossRef]
- Muller, P.; Coates, P.J.; Nenutil, R.; Trcka, F.; Hrstka, R.; Chovanec, J.; Brychtova, V.; Vojtesek, B. Tomm34 Is Commonly Expressed in Epithelial Ovarian Cancer and Associates with Tumour Type and High FIGO Stage. J. Ovarian Res. 2019, 12, 30. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, N.; Yamamoto, S.; Inoue, Y.; Aruga, A.; Yamamoto, M. RT-QPCR Analysis of the Tumor Antigens TOMM34 and RNF43 in Samples Extracted from Paraffin-Embedded Specimens of Colorectal Cancer. Oncol. Lett. 2017, 14, 2281–2287. [Google Scholar] [CrossRef] [Green Version]
- Vasaikar, S.; Huang, C.; Wang, X.; Petyuk, V.A.; Savage, S.R.; Wen, B.; Dou, Y.; Zhang, Y.; Shi, Z.; Arshad, O.A.; et al. Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 2019, 177, 1035–1049. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.A.H.; Ali, M.H.; Abbas, H.H.; Elatrash, G.A.; Foda, A.A.M. Expression of TOMM34 and Its Clinicopathological Correlations in Urothelial Carcinoma of the Bladder. Pathol. Oncol. Res. POR 2020, 26, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, S.; Quader, S.; Ono, R.; Cabral, H.; Aoyagi, K.; Hirose, A.; Yokozaki, H.; Sasaki, H. Molecular Network Profiling in Intestinal- and Diffuse-Type Gastric Cancer. Cancers 2020, 12, 3833. [Google Scholar] [CrossRef]
- Hazama, S.; Nakamura, Y.; Takenouchi, H.; Suzuki, N.; Tsunedomi, R.; Inoue, Y.; Tokuhisa, Y.; Iizuka, N.; Yoshino, S.; Takeda, K.; et al. A Phase I Study of Combination Vaccine Treatment of Five Therapeutic Epitope-Peptides for Metastatic Colorectal Cancer; Safety, Immunological Response, and Clinical Outcome. J. Transl. Med. 2014, 12, 63. [Google Scholar] [CrossRef]
- Taniguchi, H.; Iwasa, S.; Yamazaki, K.; Yoshino, T.; Kiryu, C.; Naka, Y.; Liew, E.L.; Sakata, Y. Phase 1 Study of OCV-C02, a Peptide Vaccine Consisting of Two Peptide Epitopes for Refractory Metastatic Colorectal Cancer. Cancer Sci. 2017, 108, 1013–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, J.; Kawana, N.; Yamamoto, Y. Pathway Analysis of ChIP-Seq-Based NRF1 Target Genes Suggests a Logical Hypothesis of Their Involvement in the Pathogenesis of Neurodegenerative Diseases. Gene Regul. Syst. Biol. 2013, 7, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Hondius, D.C.; Koopmans, F.; Leistner, C.; Pita-Illobre, D.; Peferoen-Baert, R.M.; Marbus, F.; Paliukhovich, I.; Li, K.W.; Rozemuller, A.J.M.; Hoozemans, J.J.M.; et al. The Proteome of Granulovacuolar Degeneration and Neurofibrillary Tangles in Alzheimer’s Disease. Acta Neuropathol. 2021, 141, 341–358. [Google Scholar] [CrossRef]
- Gerner, M.C.; Niederstaetter, L.; Ziegler, L.; Bileck, A.; Slany, A.; Janker, L.; Schmidt, R.L.J.; Gerner, C.; Del Favero, G.; Schmetterer, K.G. Proteome Analysis Reveals Distinct Mitochondrial Functions Linked to Interferon Response Patterns in Activated CD4+ and CD8+ T Cells. Front. Pharmacol. 2019, 10, 727. [Google Scholar] [CrossRef] [PubMed]
- Poverennaya, E.; Kiseleva, O.; Romanova, A.; Pyatnitskiy, M. Predicting Functions of Uncharacterized Human Proteins: From Canonical to Proteoforms. Genes 2020, 11, 677. [Google Scholar] [CrossRef]
- Terada, K.; Ueno, S.; Yomogida, K.; Imai, T.; Kiyonari, H.; Takeda, N.; Yano, M.; Abe, S.; Aizawa, S.; Mori, M. Expression of Tom34 Splicing Isoforms in Mouse Testis and Knockout of Tom34 in Mice. J. Biochem. 2003, 133, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Chen, Z.; Chen, X.; Chen, X.; Xu, M. TOMM34 Promotes Cell Proliferation, Migration, and Invasion of OSCC and Modulates Mitochondrial Function. J. Oral Pathol. Med. Off. Publ. Int. Assoc. Oral Pathol. Am. Acad. Oral Pathol. 2022, 51, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Pyatnitskiy, M.A.; Arzumanian, V.A.; Radko, S.P.; Ptitsyn, K.G.; Vakhrushev, I.V.; Poverennaya, E.V.; Ponomarenko, E.A. Oxford Nanopore MinION Direct RNA-Seq for Systems Biology. Biology 2021, 10, 1131. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolomics by Gas Chromatography-Mass Spectrometry: Combined Targeted and Untargeted Profiling. Curr. Protoc. Mol. Biol. 2016, 114, 30.4.1–30.4.32. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, U.H.; Cezard, T.; Bridgett, S.; Montazam, A.; Nichols, J.; Blaxter, M.; Gharbi, K. Quality Control of Next-Generation Sequencing Data without a Reference. Front. Genet. 2014, 5, 111. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Kiseleva, O.I.; Kurbatov, I.Y.; Arzumanian, V.A.; Ilgisonis, E.V.; Vakhrushev, I.V.; Lupatov, A.Y.; Ponomarenko, E.A.; Poverennaya, E.V. Exploring Dynamic Metabolome of the HepG2 Cell Line: Rise and Fall. Cells 2022, 11, 3548. [Google Scholar] [CrossRef] [PubMed]
- French, W.R.; Zimmerman, L.J.; Schilling, B.; Gibson, B.W.; Miller, C.A.; Townsend, R.R.; Sherrod, S.D.; Goodwin, C.R.; McLean, J.A.; Tabb, D.L. Wavelet-Based Peak Detection and a New Charge Inference Procedure for MS/MS Implemented in ProteoWizard’s MsConvert. J. Proteome Res. 2015, 14, 1299–1307. [Google Scholar] [CrossRef] [Green Version]
- Barsnes, H.; Vaudel, M. SearchGUI: A Highly Adaptable Common Interface for Proteomics Search and de Novo Engines. J. Proteome Res. 2018, 17, 2552–2555. [Google Scholar] [CrossRef] [PubMed]
- Kiseleva, O.; Poverennaya, E.; Shargunov, A.; Lisitsa, A. Proteomic Cinderella: Customized Analysis of Bulky MS/MS Data in One Night. J. Bioinform. Comput. Biol. 2018, 16, 1740011. [Google Scholar] [CrossRef]
- Bubis, J.A.; Levitsky, L.I.; Ivanov, M.V.; Tarasova, I.A.; Gorshkov, M.V. Comparative Evaluation of Label-Free Quantification Methods for Shotgun Proteomics. Rapid Commun. Mass Spectrom. RCM 2017, 31, 606–612. [Google Scholar] [CrossRef]
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 1 December 2022).
- Ideal: An R/Bioconductor Package for Interactive Differential Expression Analysis|BMC Bioinformatics|Full Text. Available online: https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-03819-5 (accessed on 6 December 2022).
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. ClusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innov. Camb. Mass 2021, 2, 100141. [Google Scholar] [CrossRef]
- Akhmedov, M.; Kedaigle, A.; Chong, R.E.; Montemanni, R.; Bertoni, F.; Fraenkel, E.; Kwee, I. PCSF: An R-Package for Network-Based Interpretation of High-Throughput Data. PLoS Comput. Biol. 2017, 13, e1005694. [Google Scholar] [CrossRef] [Green Version]
- Martens, M.; Ammar, A.; Riutta, A.; Waagmeester, A.; Slenter, D.N.; Hanspers, K.; Miller, R.A.; Digles, D.; Lopes, E.N.; Ehrhart, F.; et al. WikiPathways: Connecting Communities. Nucleic Acids Res. 2021, 49, D613–D621. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Huo, C.; Zhou, T.; Xie, S. M1A RNA Modification in Gene Expression Regulation. Genes 2022, 13, 910. [Google Scholar] [CrossRef] [PubMed]
- Balsa, E.; Marco, R.; Perales-Clemente, E.; Szklarczyk, R.; Calvo, E.; Landázuri, M.O.; Enríquez, J.A. NDUFA4 Is a Subunit of Complex IV of the Mammalian Electron Transport Chain. Cell Metab. 2012, 16, 378–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Moody, J.P.; Edgerly, C.K.; Bordiuk, O.L.; Cormier, K.; Smith, K.; Beal, M.F.; Ferrante, R.J. Mitochondrial Loss, Dysfunction and Altered Dynamics in Huntington’s Disease. Hum. Mol. Genet. 2010, 19, 3919–3935. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Jiang, J.; Zhou, L.; Huang, Z.; Qin, S.; Chen, H.-N.; Peng, L.; Zhang, Z.; Li, B.; Luo, M.; et al. Disrupting Metformin Adaptation of Liver Cancer Cells by Targeting the TOMM34/ATP5B Axis. EMBO Mol. Med. 2022, 14, e16082. [Google Scholar] [CrossRef] [PubMed]
- Sagnol, S.; Yang, Y.; Bessin, Y.; Allemand, F.; Hapkova, I.; Notarnicola, C.; Guichou, J.-F.; Faure, S.; Labesse, G.; de Santa Barbara, P. Homodimerization of RBPMS2 through a New RRM-Interaction Motif Is Necessary to Control Smooth Muscle Plasticity. Nucleic Acids Res. 2014, 42, 10173–10184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hapkova, I.; Skarda, J.; Rouleau, C.; Thys, A.; Notarnicola, C.; Janikova, M.; Bernex, F.; Rypka, M.; Vanderwinden, J.-M.; Faure, S.; et al. High Expression of the RNA-Binding Protein RBPMS2 in Gastrointestinal Stromal Tumors. Exp. Mol. Pathol. 2013, 94, 314–321. [Google Scholar] [CrossRef]
- Finck, B.N.; Kelly, D.P. PGC-1 Coactivators: Inducible Regulators of Energy Metabolism in Health and Disease. J. Clin. Investig. 2006, 116, 615–622. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Liu, Y.; Liu, X.; Zhu, L.; Cui, Y.; Cui, A.; Qiao, A.; Kong, X.; Liu, Y.; Chen, Q.; et al. PGC-1 Beta-Regulated Mitochondrial Biogenesis and Function in Myotubes Is Mediated by NRF-1 and ERR Alpha. Mitochondrion 2010, 10, 516–527. [Google Scholar] [CrossRef]
- Blesa, J.R.; Prieto-Ruiz, J.A.; Abraham, B.A.; Harrison, B.L.; Hegde, A.A.; Hernández-Yago, J. NRF-1 Is the Major Transcription Factor Regulating the Expression of the Human TOMM34 Gene. Biochem. Cell Biol. Biochim. Biol. Cell. 2008, 86, 46–56. [Google Scholar] [CrossRef]
- Mohsenzadegan, M.; Razmi, M.; Vafaei, S.; Abolhasani, M.; Madjd, Z.; Saeednejad Zanjani, L.; Sharifi, L. Co-Expression of Cancer-Testis Antigens of MAGE-A6 and MAGE-A11 Is Associated with Tumor Aggressiveness in Patients with Bladder Cancer. Sci. Rep. 2022, 12, 599. [Google Scholar] [CrossRef] [PubMed]
- Ramming, T.; Hansen, H.G.; Nagata, K.; Ellgaard, L.; Appenzeller-Herzog, C. GPx8 Peroxidase Prevents Leakage of H2O2 from the Endoplasmic Reticulum. Free Radic. Biol. Med. 2014, 70, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Chauvet, C.; Vanhoutteghem, A.; Duhem, C.; Saint-Auret, G.; Bois-Joyeux, B.; Djian, P.; Staels, B.; Danan, J.-L. Control of Gene Expression by the Retinoic Acid-Related Orphan Receptor Alpha in HepG2 Human Hepatoma Cells. PLoS ONE 2011, 6, e22545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuoka, H.; Shima, A.; Kuramoto, D.; Kikumoto, D.; Matsui, T.; Michihara, A. Phosphoenolpyruvate Carboxykinase, a Key Enzyme That Controls Blood Glucose, Is a Target of Retinoic Acid Receptor-Related Orphan Receptor α. PLoS ONE 2015, 10, e0137955. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Ning, J.; Cao, W.; Wang, S.; Du, T.; Jiang, J.; Feng, X.; Zhang, B. Research Progress of TXNIP as a Tumor Suppressor Gene Participating in the Metabolic Reprogramming and Oxidative Stress of Cancer Cells in Various Cancers. Front. Oncol. 2020, 10, 568574. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial Electron Transport Chain: Oxidative Phosphorylation, Oxidant Production, and Methods of Measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Hilton, B.A.; Li, Z.; Musich, P.R.; Wang, H.; Cartwright, B.M.; Serrano, M.; Zhou, X.Z.; Lu, K.P.; Zou, Y. ATR Plays a Direct Antiapoptotic Role at Mitochondria, Which Is Regulated by Prolyl Isomerase Pin1. Mol. Cell 2015, 60, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Seirafi, M.; Kozlov, G.; Gehring, K. Parkin Structure and Function. Febs J. 2015, 282, 2076–2088. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 Binding to the TOM Complex and Alternate Intracellular Membranes in Recruitment and Activation of the E3 Ligase Parkin. Dev. Cell 2012, 22, 320–333. [Google Scholar] [CrossRef]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.A.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-Dependent Recruitment of Parkin to Mitochondria in Mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braisted, J.; Patt, A.; Tindall, C.; Sheils, T.; Neyra, J.; Spencer, K.; Eicher, T.; Mathé, E.A. RaMP-DB 2.0: A Renovated Knowledgebase for Deriving Biological and Chemical Insight from Genes, Proteins, and Metabolites. Bioinformatics 2023, 39, btac726. [Google Scholar] [CrossRef] [PubMed]
- Hewton, K.G.; Johal, A.S.; Parker, S.J. Transporters at the Interface between Cytosolic and Mitochondrial Amino Acid Metabolism. Metabolites 2021, 11, 112. [Google Scholar] [CrossRef]
- De Vitto, H.; Arachchige, D.B.; Richardson, B.C.; French, J.B. The Intersection of Purine and Mitochondrial Metabolism in Cancer. Cells 2021, 10, 2603. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.Y.; Baek, S.; Cha, J.; Yang, S.; Kim, E.; Marcotte, E.M.; Hart, T.; Lee, I. HumanNet v3: An Improved Database of Human Gene Networks for Disease Research. Nucleic Acids Res. 2022, 50, D632–D639. [Google Scholar] [CrossRef]
- Gómez-Romero, L.; López-Reyes, K.; Hernández-Lemus, E. The Large Scale Structure of Human Metabolism Reveals Resilience via Extensive Signaling Crosstalk. Front. Physiol. 2020, 11, 588012. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New Perspectives on Genomes, Pathways, Diseases and Drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Soltis, A.R.; Kennedy, N.J.; Xin, X.; Zhou, F.; Ficarro, S.B.; Yap, Y.S.; Matthews, B.J.; Lauffenburger, D.A.; White, F.M.; Marto, J.A.; et al. Hepatic Dysfunction Caused by Consumption of a High-Fat Diet. Cell Rep. 2017, 21, 3317–3328. [Google Scholar] [CrossRef] [Green Version]
- Blondel, V.D.; Guillaume, J.-L.; Lambiotte, R.; Lefebvre, E. Fast Unfolding of Communities in Large Networks. J. Stat. Mech. Theory Exp. 2008, 2008, P10008. [Google Scholar] [CrossRef] [Green Version]
- Stoeckler, J.D.; Poirot, A.F.; Smith, R.M.; Parks, R.E.; Ealick, S.E.; Takabayashi, K.; Erion, M.D. Purine Nucleoside Phosphorylase. 3. Reversal of Purine Base Specificity by Site-Directed Mutagenesis. Biochemistry 1997, 36, 11749–11756. [Google Scholar] [CrossRef]
- Arpaia, E.; Benveniste, P.; Di Cristofano, A.; Gu, Y.; Dalal, I.; Kelly, S.; Hershfield, M.; Pandolfi, P.P.; Roifman, C.M.; Cohen, A. Mitochondrial Basis for Immune Deficiency. J. Exp. Med. 2000, 191, 2197–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, S.; Chiyomaru, T.; Kawakami, K.; Yoshino, H.; Enokida, H.; Nohata, N.; Fuse, M.; Ichikawa, T.; Naya, Y.; Nakagawa, M.; et al. Tumour Suppressors MiR-1 and MiR-133a Target the Oncogenic Function of Purine Nucleoside Phosphorylase (PNP) in Prostate Cancer. Br. J. Cancer 2012, 106, 405–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buj, R.; Chen, C.-W.; Dahl, E.S.; Leon, K.E.; Kuskovsky, R.; Maglakelidze, N.; Navaratnarajah, M.; Zhang, G.; Doan, M.T.; Jiang, H.; et al. Suppression of P16 Induces MTORC1-Mediated Nucleotide Metabolic Reprogramming. Cell Rep. 2019, 28, 1971–1980.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinelli, J.B.; Haigis, M.C. The Multifaceted Contributions of Mitochondria to Cellular Metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Vert, G.; Chory, J. Crosstalk in Cellular Signaling: Background Noise or the Real Thing? Dev. Cell 2011, 21, 985–991. [Google Scholar] [CrossRef] [Green Version]
- Sever, R.; Brugge, J.S. Signal Transduction in Cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [Green Version]
- Mechteridis, K.; Lauber, M.; Baumbach, J.; List, M. KeyPathwayMineR: De Novo Pathway Enrichment in the R Ecosystem. Front. Genet. 2021, 12, 812853. [Google Scholar] [CrossRef]
- Shi, W.J.; Zhuang, Y.; Russell, P.H.; Hobbs, B.D.; Parker, M.M.; Castaldi, P.J.; Rudra, P.; Vestal, B.; Hersh, C.P.; Saba, L.M.; et al. Unsupervised Discovery of Phenotype-Specific Multi-Omics Networks. Bioinforma. Oxf. Engl. 2019, 35, 4336–4343. [Google Scholar] [CrossRef]
- Nguyen, T.-M.; Shafi, A.; Nguyen, T.; Draghici, S. Identifying Significantly Impacted Pathways: A Comprehensive Review and Assessment. Genome Biol. 2019, 20, 203. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poverennaya, E.V.; Pyatnitskiy, M.A.; Dolgalev, G.V.; Arzumanian, V.A.; Kiseleva, O.I.; Kurbatov, I.Y.; Kurbatov, L.K.; Vakhrushev, I.V.; Romashin, D.D.; Kim, Y.S.; et al. Exploiting Multi-Omics Profiling and Systems Biology to Investigate Functions of TOMM34. Biology 2023, 12, 198. https://doi.org/10.3390/biology12020198
Poverennaya EV, Pyatnitskiy MA, Dolgalev GV, Arzumanian VA, Kiseleva OI, Kurbatov IY, Kurbatov LK, Vakhrushev IV, Romashin DD, Kim YS, et al. Exploiting Multi-Omics Profiling and Systems Biology to Investigate Functions of TOMM34. Biology. 2023; 12(2):198. https://doi.org/10.3390/biology12020198
Chicago/Turabian StylePoverennaya, Ekaterina V., Mikhail A. Pyatnitskiy, Georgii V. Dolgalev, Viktoria A. Arzumanian, Olga I. Kiseleva, Ilya Yu. Kurbatov, Leonid K. Kurbatov, Igor V. Vakhrushev, Daniil D. Romashin, Yan S. Kim, and et al. 2023. "Exploiting Multi-Omics Profiling and Systems Biology to Investigate Functions of TOMM34" Biology 12, no. 2: 198. https://doi.org/10.3390/biology12020198