


Analyzing Predominant Bacterial Species and Potential Short-Chain Fatty Acid-Associated Metabolic Routes in Human Gut Microbiome Using Integrative Metagenomics

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Participants and Fecal Sample Collection
2.2. DNA Extraction and Metagenome Sequencing
2.3. Microbial Taxonomic Analysis and Functional Annotation of 16S rRNA Gene Sequencing Data and WMGS Datasets
2.4. Identification of Predominant Bacterial Species, Potential Metabolic Functions and Associated Routes Involved in Treatment with CMH Using Integrative Analysis
3. Results and Discussion
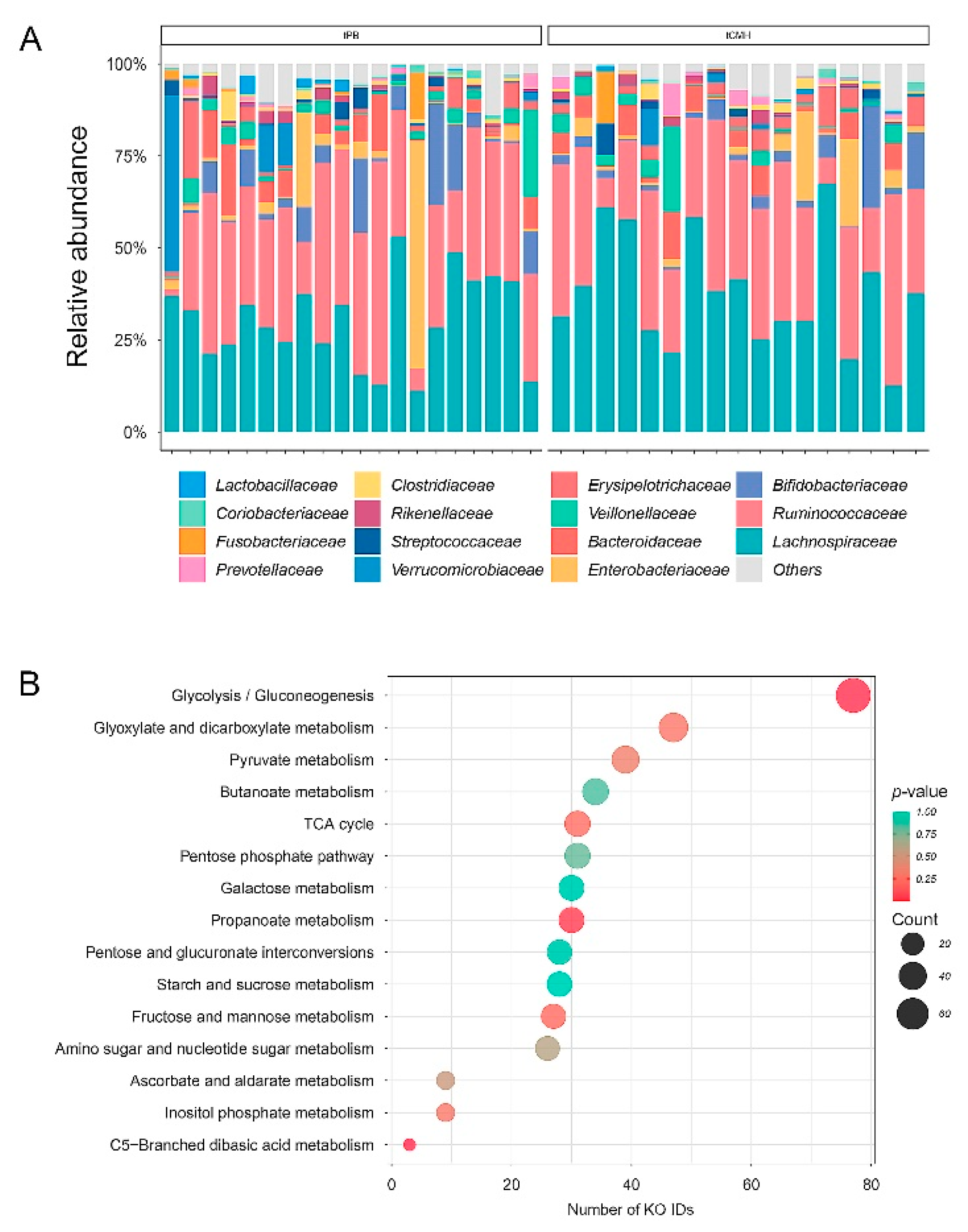
3.1. Assessment of 16S rRNA Gene Datasets on Microbial Composition and Metabolic Function
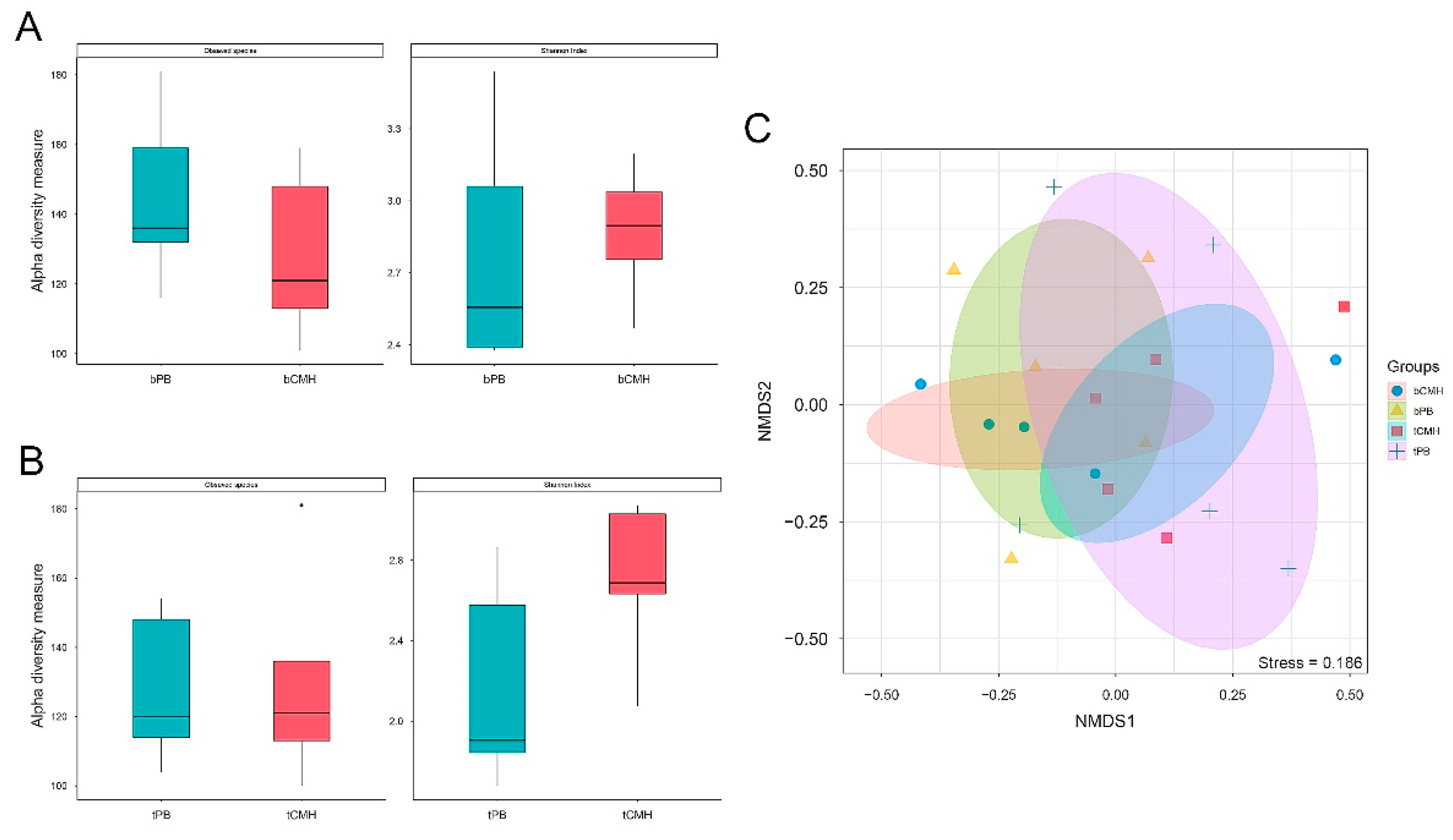
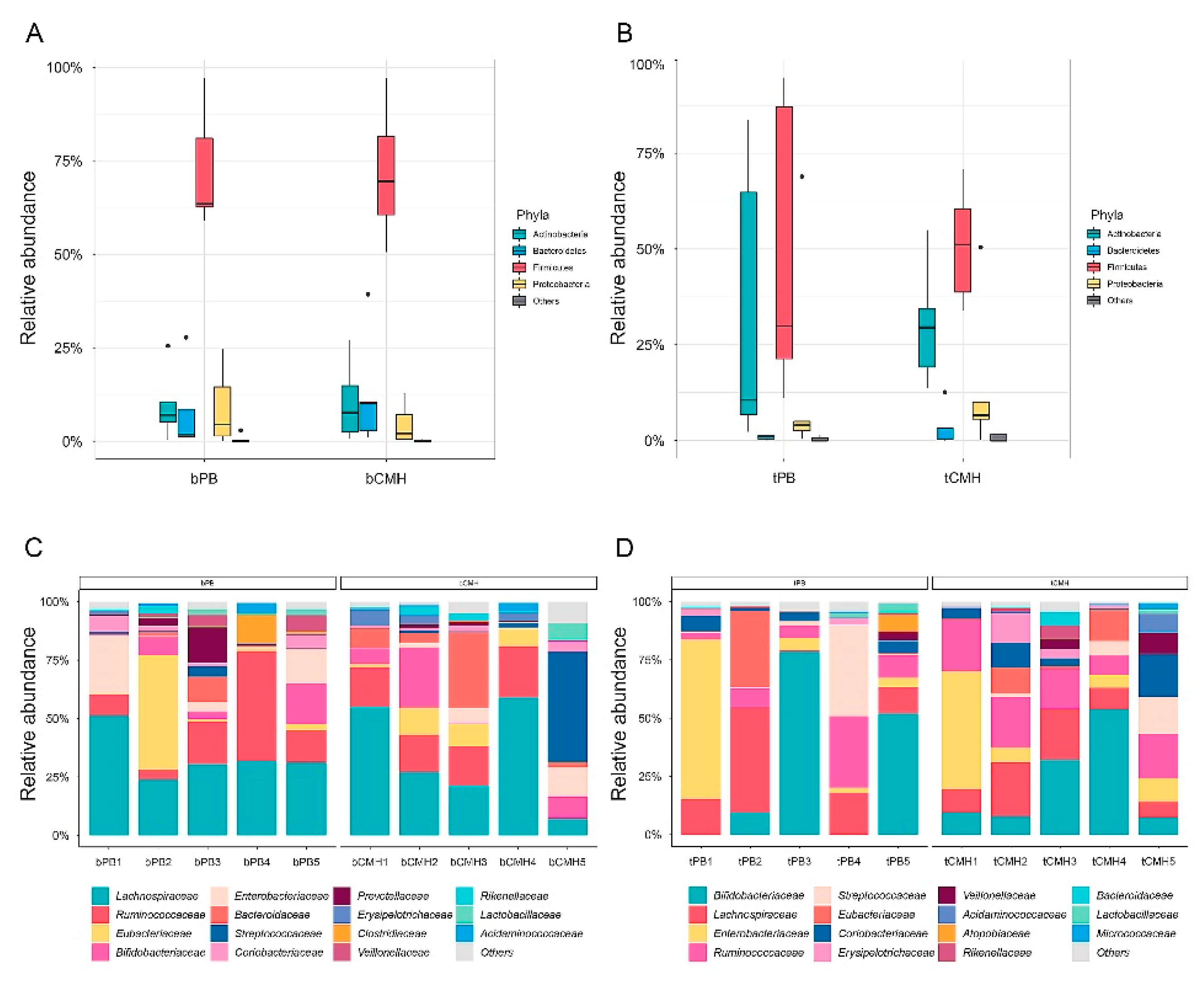
3.2. Assessment of Gemicrobial Diversity and Composition from WMGS Datasets
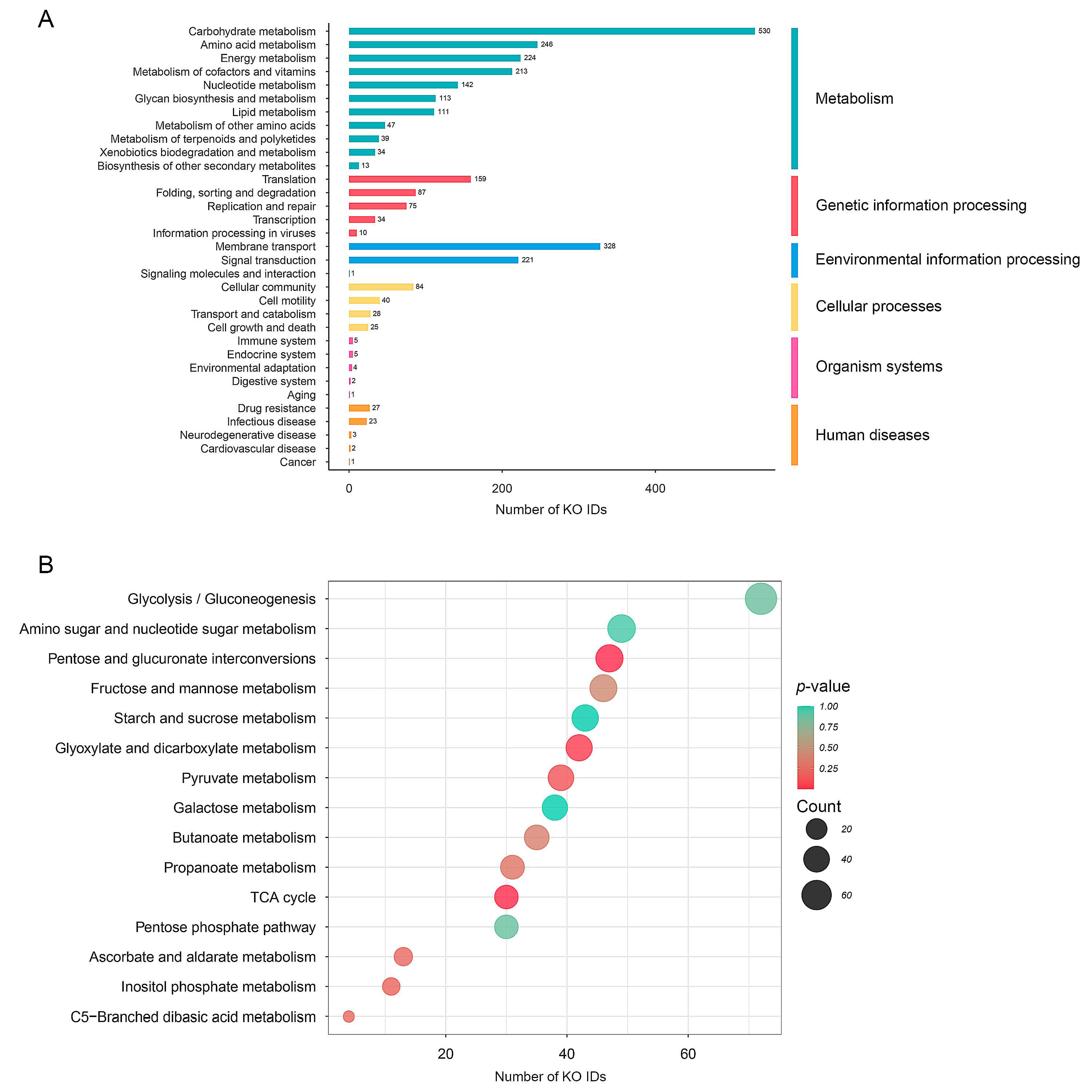
3.3. Assignment of Metabolic Function Underlying KO IDs from WMGS Datasets
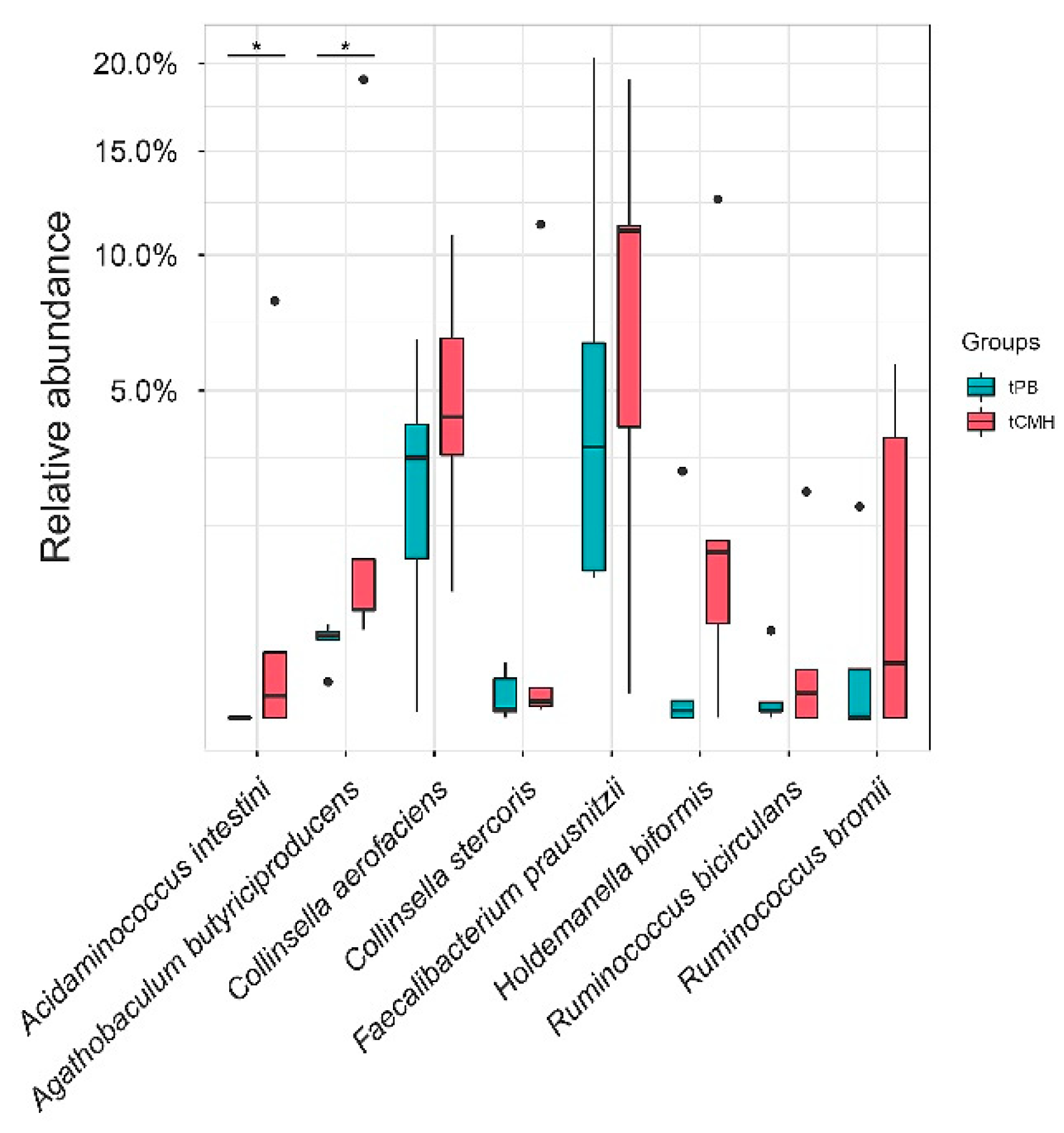
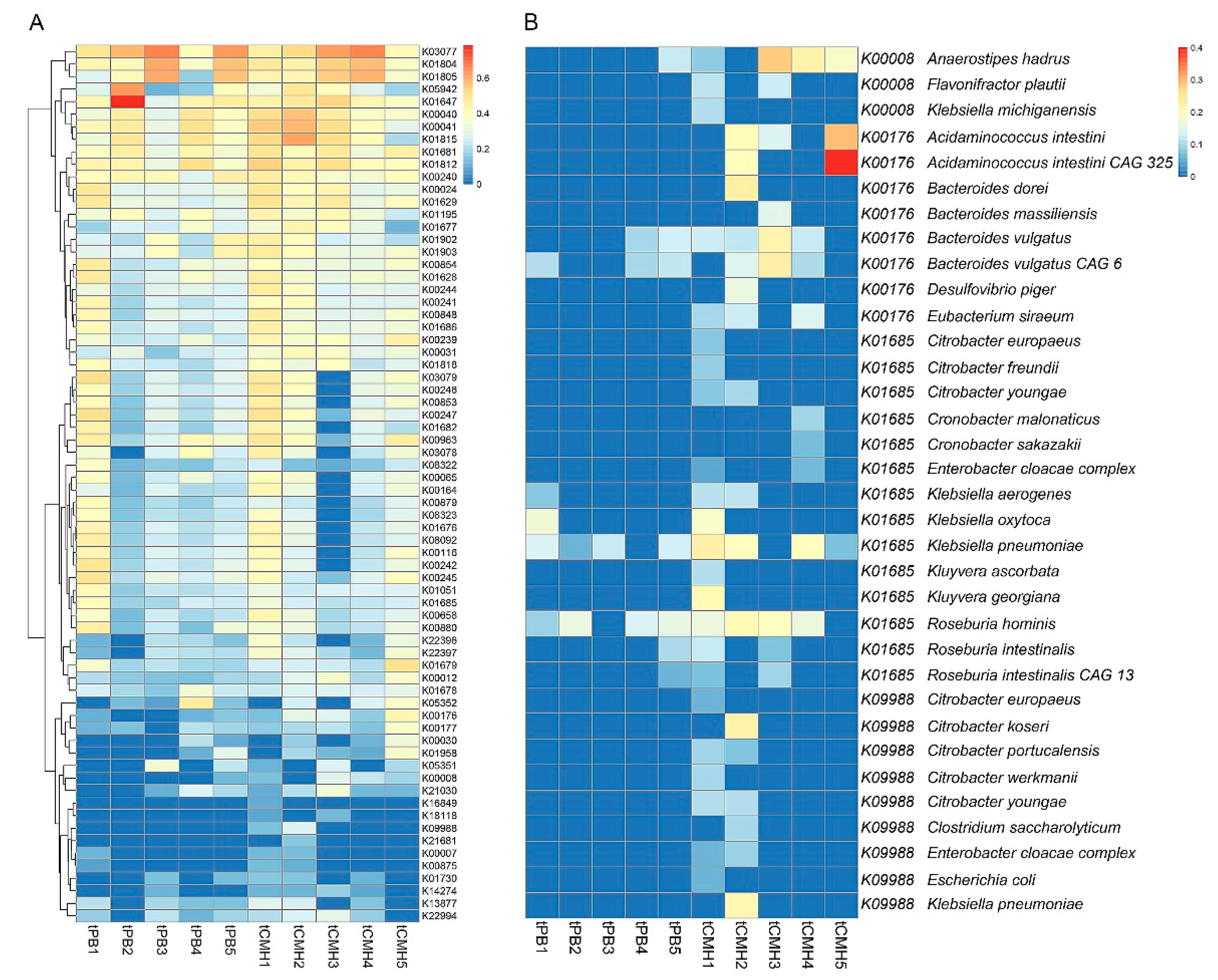
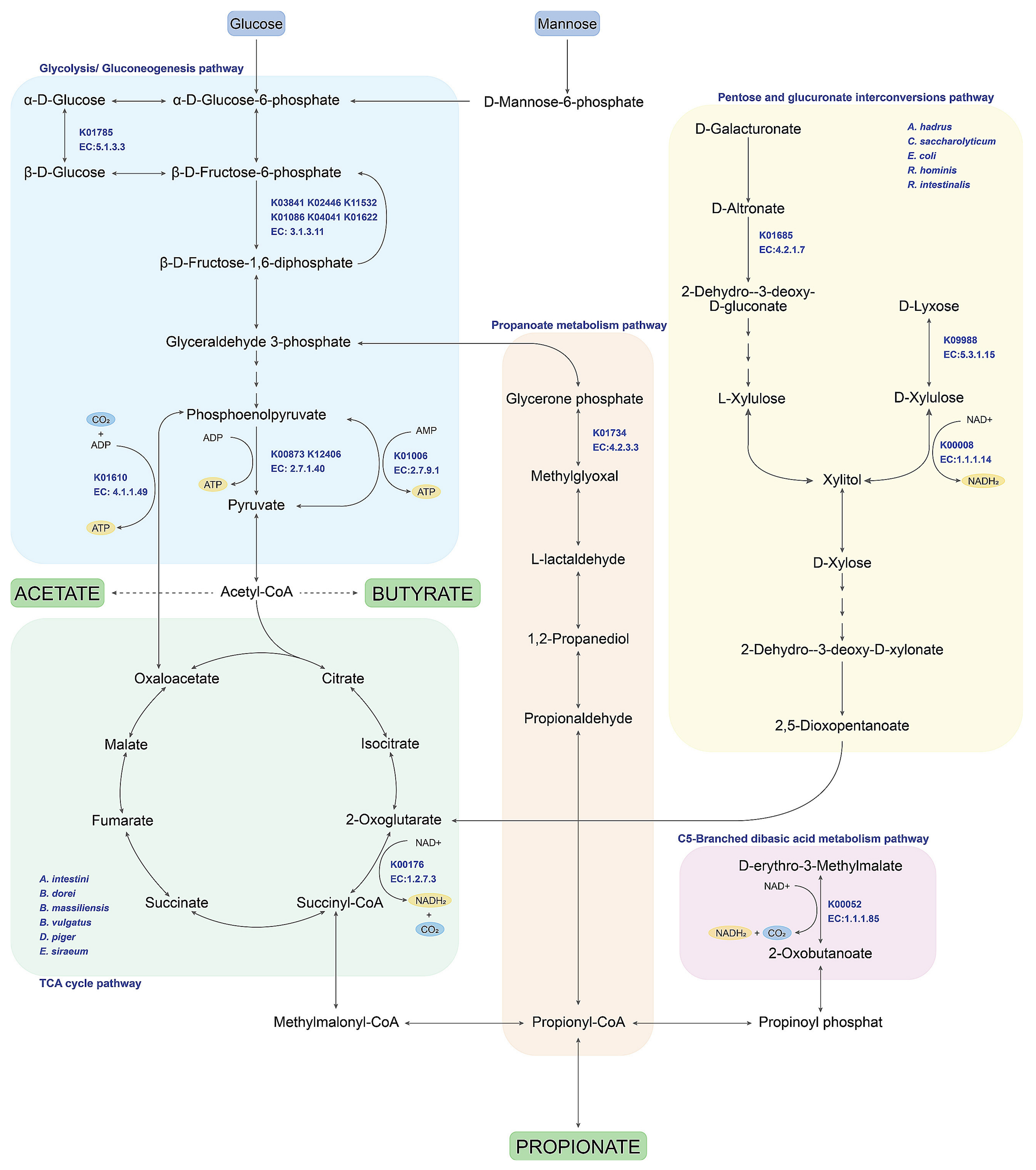
3.4. Identifying Predominant Bacterial Species and Potential Metabolic Routes Using Integrative Metagenomics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goodrich, J.; Waters, J.; Poole, A.; Sutter, J.; Koren, O.; Blekhman, R.; Beaumont, M.; Treuren, W.; Knight, R.; Bell, J.; et al. Human Genetics Shape the Gut Microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [Green Version]
- Ashaolu, T.J.; Ashaolu, J.O.; Adeyeye, S.A.O. Fermentation of prebiotics by human colonic microbiota in vitro and short-chain fatty acids production: A critical review. J. Appl. Microbiol. 2021, 130, 677–687. [Google Scholar] [CrossRef]
- Holscher, H. Dietary Fiber and Prebiotics and the Gastrointestinal Microbiota. Gut Microbes 2017, 8, 172–184. [Google Scholar] [CrossRef]
- Gibson, G.R.; Hutkins, R.; Sanders, M.E.; Prescott, S.L.; Reimer, R.A.; Salminen, S.J.; Scott, K.; Stanton, C.; Swanson, K.S.; Cani, P.D.; et al. Expert consensus document: The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Slavin, J. Fiber and prebiotics: Mechanisms and health benefits. Nutrients 2013, 5, 1417–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Intaratrakul, K.; Nitisinprasert, S.; Nguyen, T.-H.; Haltrich, D.; Keawsompong, S. Manno-oligosaccharides from copra meal: Optimization of its enzymatic production and evaluation its potential as prebiotic. Bioact. Carbohydr. Diet. Fibre 2022, 27, 100292. [Google Scholar] [CrossRef]
- Prayoonthien, P.; Rastall, R.A.; Kolida, S.; Nitisinprasert, S.; Keawsompong, S. In vitro fermentation of copra meal hydrolysate by human fecal microbiota. 3 Biotech 2019, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Sathitkowitchai, W.; Suratannon, N.; Keawsompong, S.; Weerapakorn, W.; Patumcharoenpol, P.; Nitisinprasert, S.; Nakphaichit, M. A randomized trial to evaluate the impact of copra meal hydrolysate on gastrointestinal symptoms and gut microbiome. PeerJ 2021, 9, e12158. [Google Scholar] [CrossRef] [PubMed]
- Nakphaichit, M. Effect of Increasing Dietary Protein from Soybean Meal on Intestinal Microbiota and Their Fatty Acids Production in Broiler Chicken. Adv. Anim. Vet. Sci. 2014, 2, 337–343. [Google Scholar] [CrossRef]
- Raethong, N.; Nakphaichit, M.; Suratannon, N.; Sathitkowitchai, W.; Weerapakorn, W.; Keawsompong, S.; Vongsangnak, W. Analysis of human gut microbiome: Taxonomy and metabolic functions in Thai adults. Genes 2021, 12, 331. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Patumcharoenpol, P.; Nakphaichit, M.; Panagiotou, G.; Senavonge, A.; Suratannon, N.; Vongsangnak, W. MetGEMs Toolbox: Metagenome-scale models as integrative toolbox for uncovering metabolic functions and routes of human gut microbiome. PLoS Comput. Biol. 2021, 17, e1008487. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’hara, R.R.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.5-6. 2019. Available online: https://CRAN.R-project.org/package=vegan (accessed on 25 June 2021).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Truong, D.T.; Franzosa, E.A.; Tickle, T.L.; Scholz, M.; Weingart, G.; Pasolli, E.; Tett, A.; Huttenhower, C.; Segata, N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef] [PubMed]
- Beghini, F.; McIver, L.J.; Blanco-Míguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Manghi, P.; Scholz, M.; Thomas, A.M.; et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 2021, 10, e65088. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 2020, 29, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M. KEGG mapping tools for uncovering hidden features in biological data. Protein Sci. 2022, 31, 47–53. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- La-Ongkham, O.; Nakphaichit, M.; Nakayama, J.; Keawsompong, S.; Nitisinprasert, S. Age-related changes in the gut microbiota and the core gut microbiome of healthy Thai humans. 3 Biotech 2020, 10, 276. [Google Scholar] [CrossRef]
- Nogal, A.; Valdes, A.M.; Menni, C. The role of short-chain fatty acids in the interplay between gut microbiota and diet in cardio-metabolic health. Gut Microbes 2021, 13, 1897212. [Google Scholar] [CrossRef]
- Portincasa, P.; Bonfrate, L.; Vacca, M.; De Angelis, M.; Farella, I.; Lanza, E.; Khalil, M.; Wang, D.Q.-H.; Sperandio, M.; Di Ciaula, A. Gut Microbiota and Short Chain Fatty Acids: Implications in Glucose Homeostasis. Int. J. Mol. Sci. 2022, 23, 1105. [Google Scholar] [CrossRef]
- Tsukuda, N.; Yahagi, K.; Hara, T.; Watanabe, Y.; Matsumoto, H.; Mori, H.; Higashi, K.; Tsuji, H.; Matsumoto, S.; Kurokawa, K.; et al. Key bacterial taxa and metabolic pathways affecting gut short-chain fatty acid profiles in early life. ISME J. 2021, 15, 2574–2590. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Palmas, V.; Pisanu, S.; Madau, V.; Casula, E.; Deledda, A.; Cusano, R.; Uva, P.; Vascellari, S.; Loviselli, A.; Manzin, A.; et al. Gut microbiota markers associated with obesity and overweight in Italian adults. Sci. Rep. 2021, 11, 5532. [Google Scholar] [CrossRef]
- Mirzaei, R.; Bouzari, B.; Hosseini-Fard, S.R.; Mazaheri, M.; Ahmadyousefi, Y.; Abdi, M.; Jalalifar, S.; Karimitabar, Z.; Teimoori, A.; Keyvani, H.; et al. Role of microbiota-derived short-chain fatty acids in nervous system disorders. Biomed. Pharmacother. 2021, 139, 111661. [Google Scholar] [CrossRef]
- Markowiak-Kopeć, P.; Śliżewska, K. The Effect of Probiotics on the Production of Short-Chain Fatty Acids by Human Intestinal Microbiome. Nutrients 2020, 12, 1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macy, J.M.; Ljungdahl, L.G.; Gottschalk, G. Pathway of succinate and propionate formation in Bacteroides fragilis. J. Bacteriol. 1978, 134, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Prayoonthien, P.; Nitisinprasert, S.; Keawsompong, S. In vitro fermentation of copra meal hydrolysate by chicken microbiota. 3 Biotech 2018, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Ye, K.; Li, M.; Ying, J.; Wang, H.; Han, J.; Shi, L.; Xiao, J.; Shen, Y.; Feng, X.; et al. Xylitol enhances synthesis of propionate in the colon via cross-feeding of gut microbiota. Microbiome 2021, 9, 62. [Google Scholar] [CrossRef]
- Kircher, B.; Woltemate, S.; Gutzki, F.; Schlüter, D.; Geffers, R.; Bähre, H.; Vital, M. Predicting butyrate- and propionate-forming bacteria of gut microbiota from sequencing data. bioRxiv 2022. [Google Scholar] [CrossRef] [PubMed]
- Van der Hee, B.; Wells, J.M. Microbial Regulation of Host Physiology by Short-chain Fatty Acids. Trends Microbiol. 2021, 29, 700–712. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Raw Reads (Mb) | Clean Reads (Mb) | Effective Rates * (%) |
|---|---|---|---|
| Baseline | |||
| bPB | 273.40 # | 271.29 | 99.23 |
| bCMH | 276.56 # | 273.71 | 98.97 |
| Treatment | |||
| tPB | 301.76 | 299.58 | 99.28 |
| tCMH | 290.02 | 287.83 | 99.24 |
| Total | 1141.74 | 1132.41 | 99.18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kingkaw, A.; Raethong, N.; Patumcharoenpol, P.; Suratannon, N.; Nakphaichit, M.; Keawsompong, S.; Roytrakul, S.; Vongsangnak, W. Analyzing Predominant Bacterial Species and Potential Short-Chain Fatty Acid-Associated Metabolic Routes in Human Gut Microbiome Using Integrative Metagenomics. Biology 2023, 12, 21. https://doi.org/10.3390/biology12010021
Kingkaw A, Raethong N, Patumcharoenpol P, Suratannon N, Nakphaichit M, Keawsompong S, Roytrakul S, Vongsangnak W. Analyzing Predominant Bacterial Species and Potential Short-Chain Fatty Acid-Associated Metabolic Routes in Human Gut Microbiome Using Integrative Metagenomics. Biology. 2023; 12(1):21. https://doi.org/10.3390/biology12010021
Chicago/Turabian StyleKingkaw, Amornthep, Nachon Raethong, Preecha Patumcharoenpol, Narissara Suratannon, Massalin Nakphaichit, Suttipun Keawsompong, Sittiruk Roytrakul, and Wanwipa Vongsangnak. 2023. "Analyzing Predominant Bacterial Species and Potential Short-Chain Fatty Acid-Associated Metabolic Routes in Human Gut Microbiome Using Integrative Metagenomics" Biology 12, no. 1: 21. https://doi.org/10.3390/biology12010021