
Soil Microbial Diversity and Community Composition in Rice–Fish Co-Culture and Rice Monoculture Farming System


 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
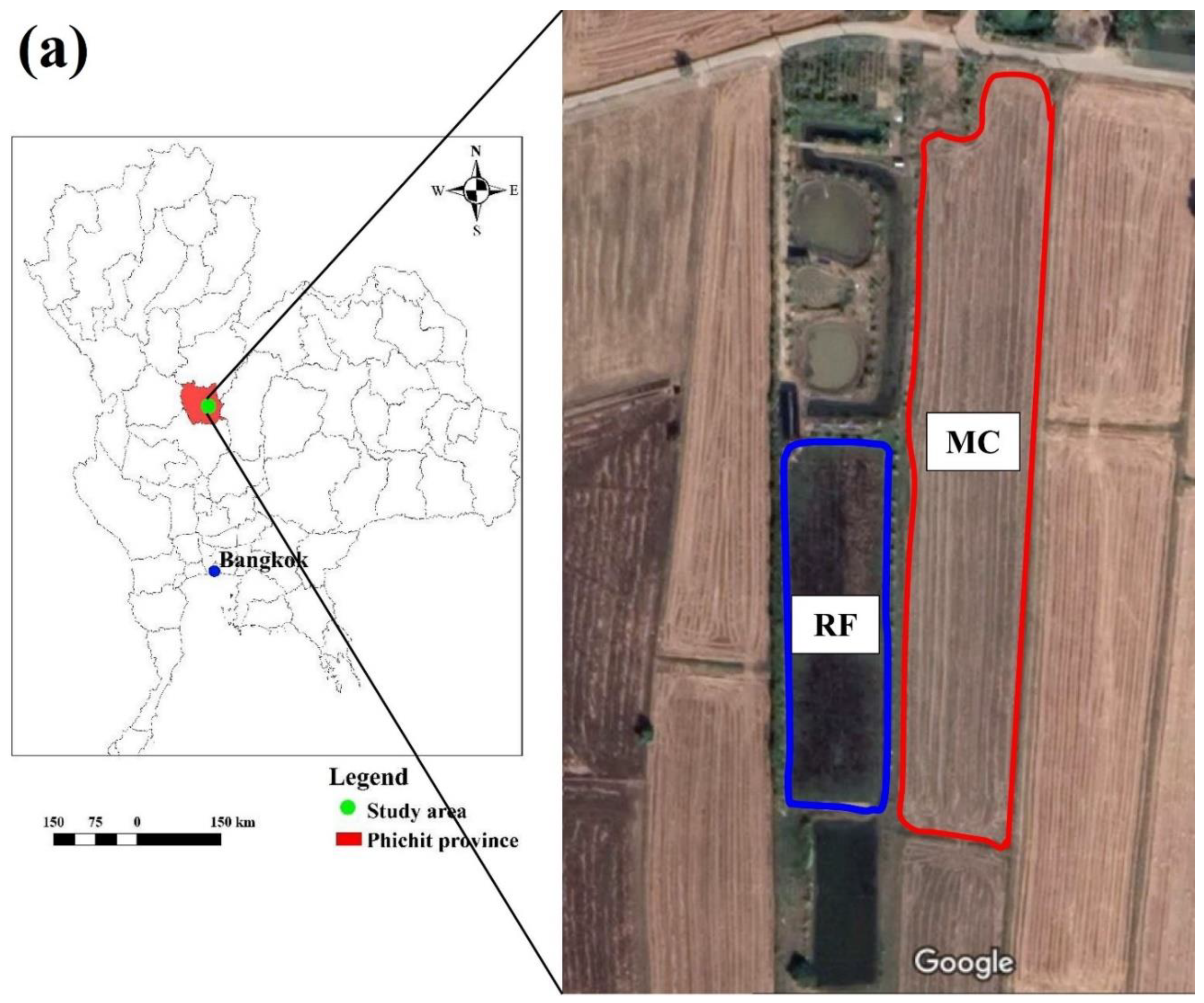
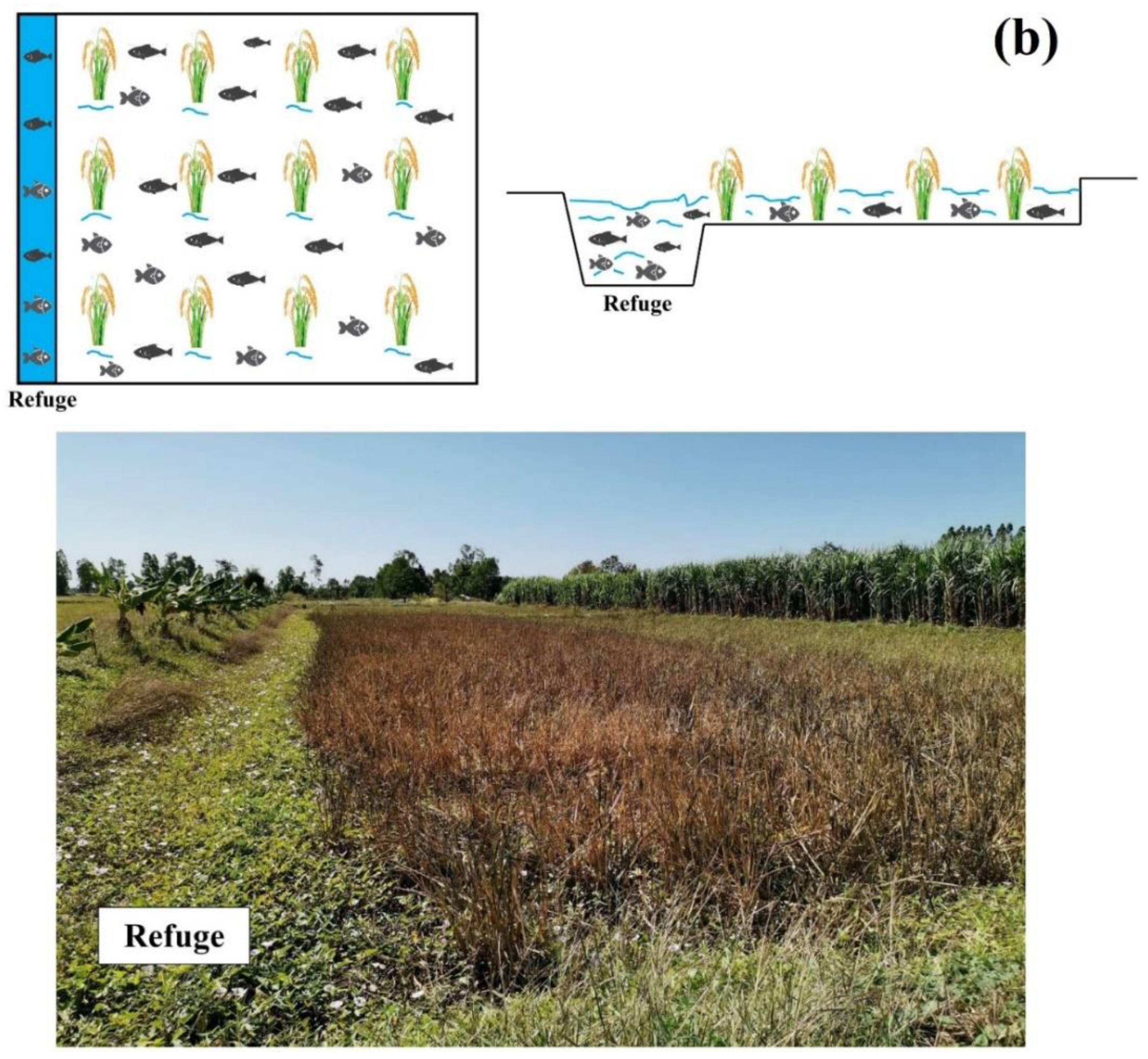
2.1. Description of Study Sites
2.2. Soil Sampling and Measurements
2.3. DNA Extraction and Amplicon Sequencing of 16s rRNA Gene
2.4. Sequencing Analysis and Microbial Taxonomic Identification
2.5. Functional Prediction
2.6. Statistical Analysis
3. Results
3.1. Soil Physicochemical Properties in Rice Monoculture and Rice–Fish Co-Culture Fields
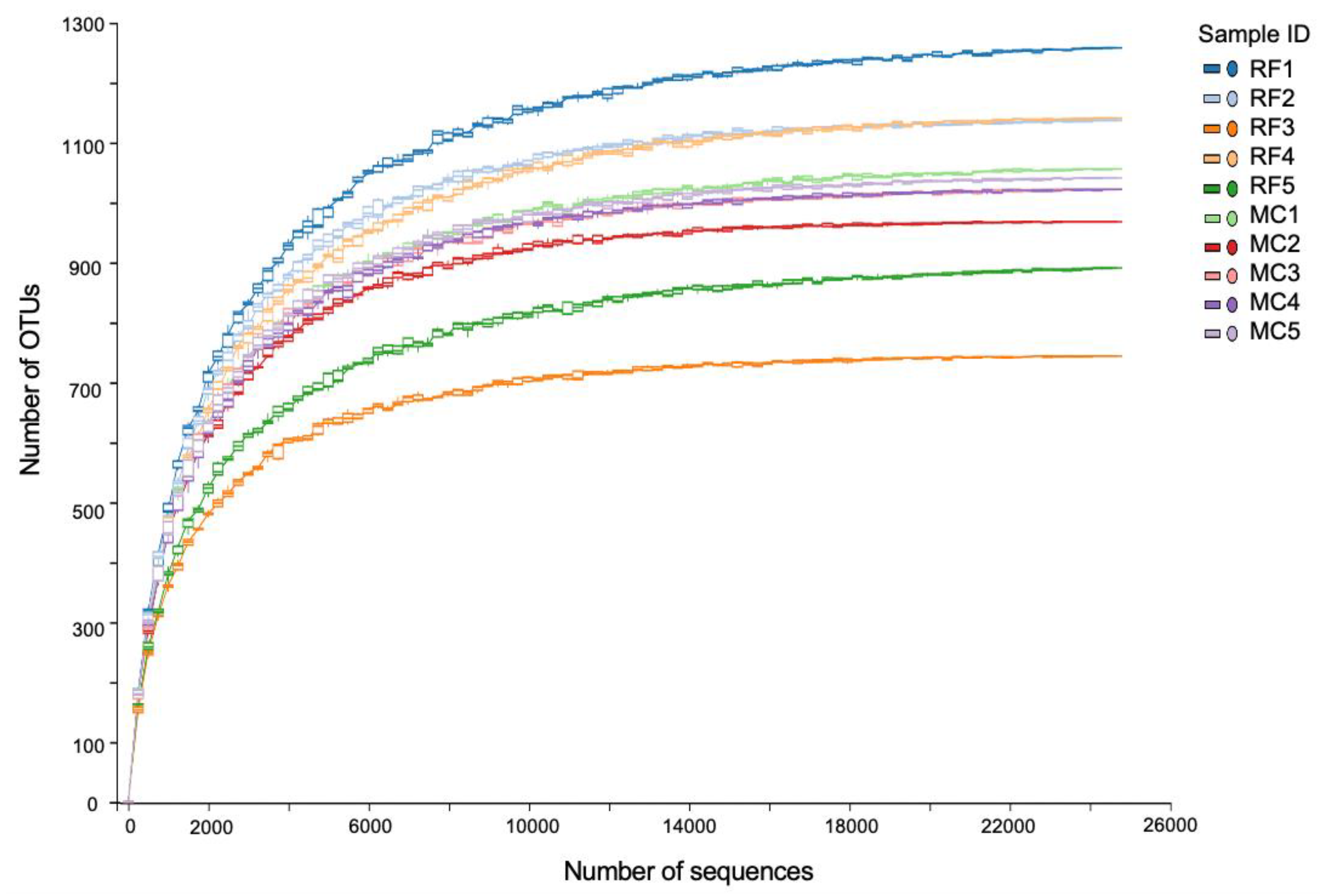
3.2. General Overview of the Sequencing Analysis
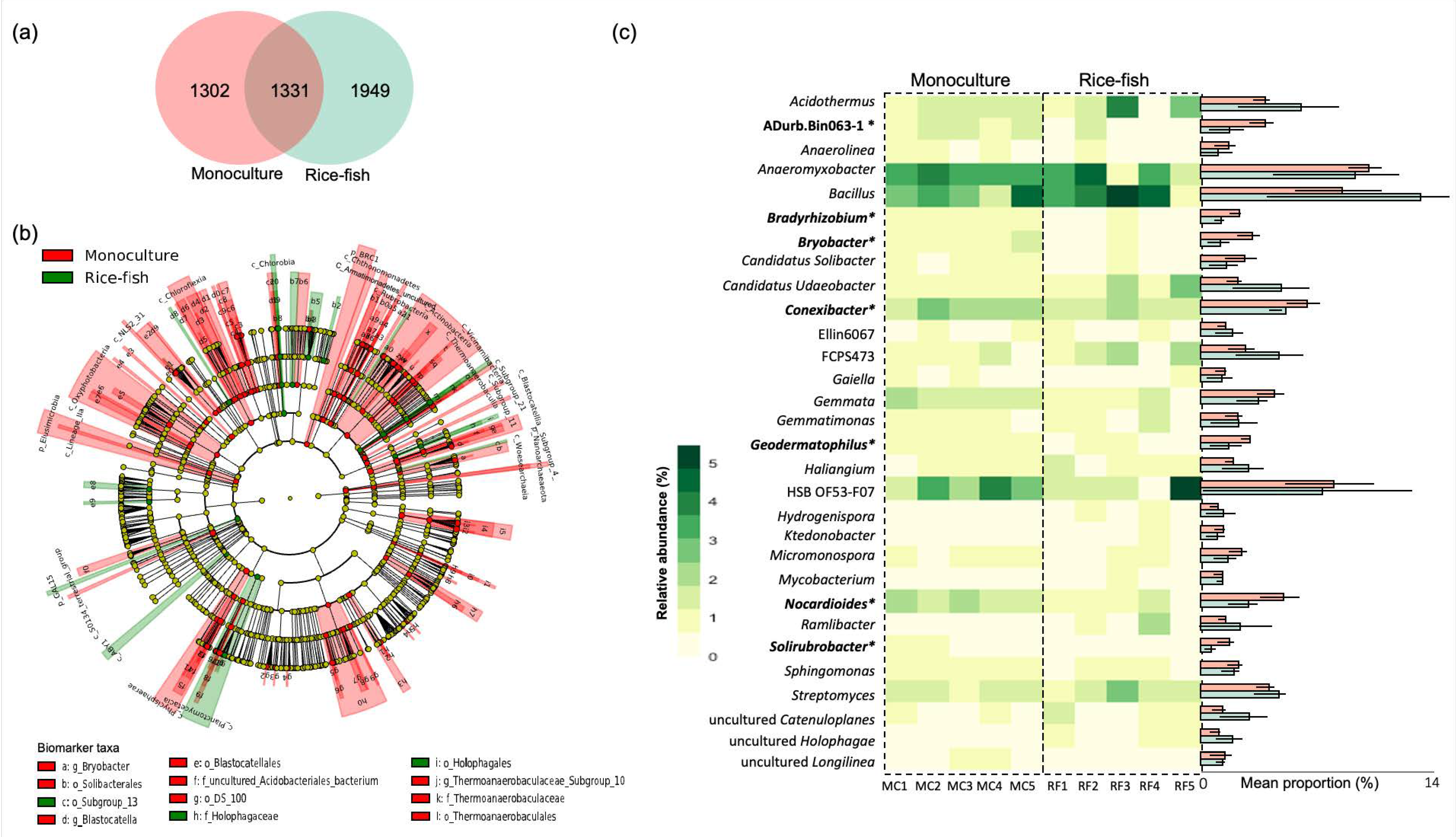
3.3. Taxonomic Distribution and Differential Abundance of Soil Bacteria in Rice Monoculture and Rice–Fish Co-Culture Fields
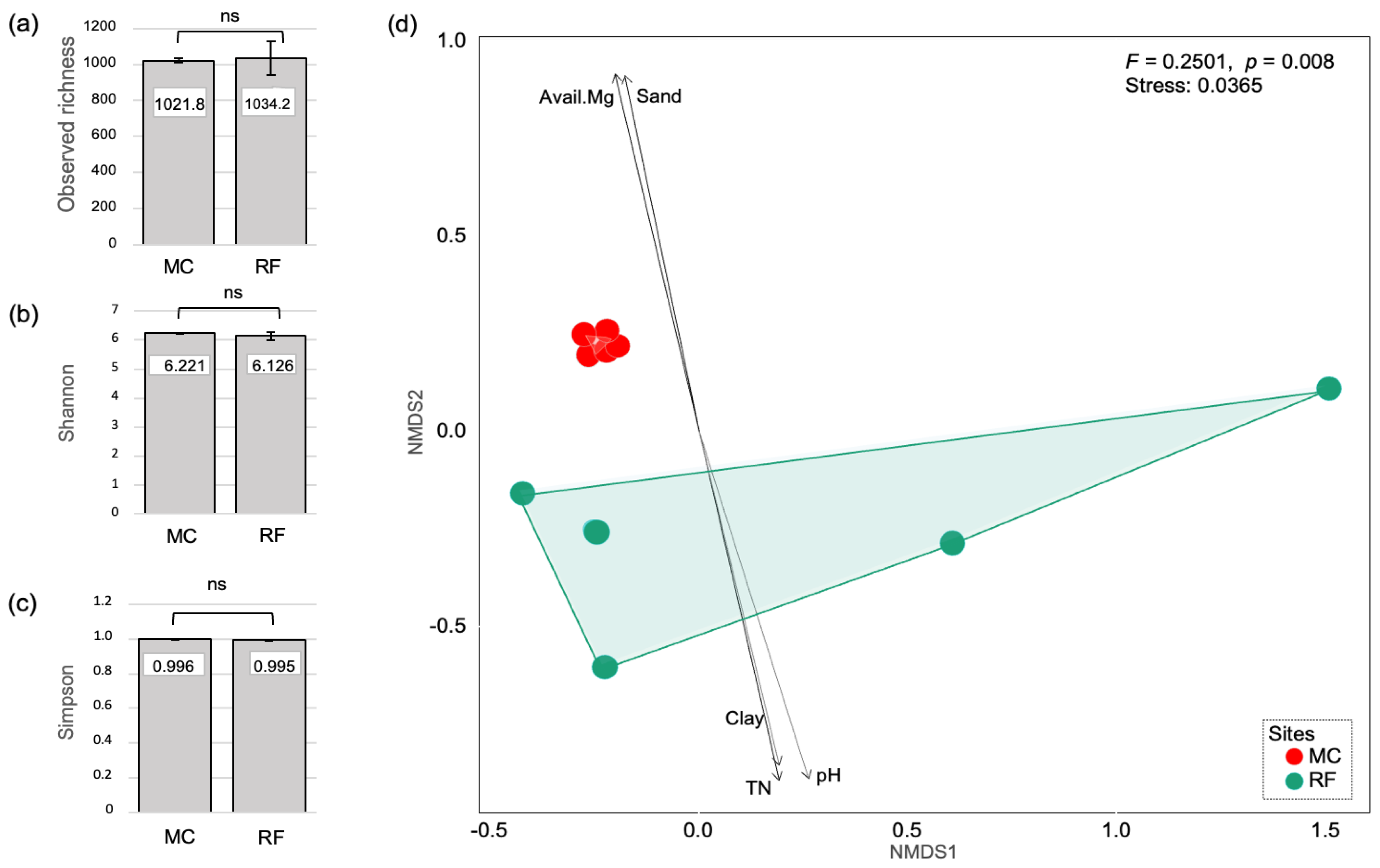
3.4. Richness, Diversity, Community Composition, and Their Correlation to Soil Properties
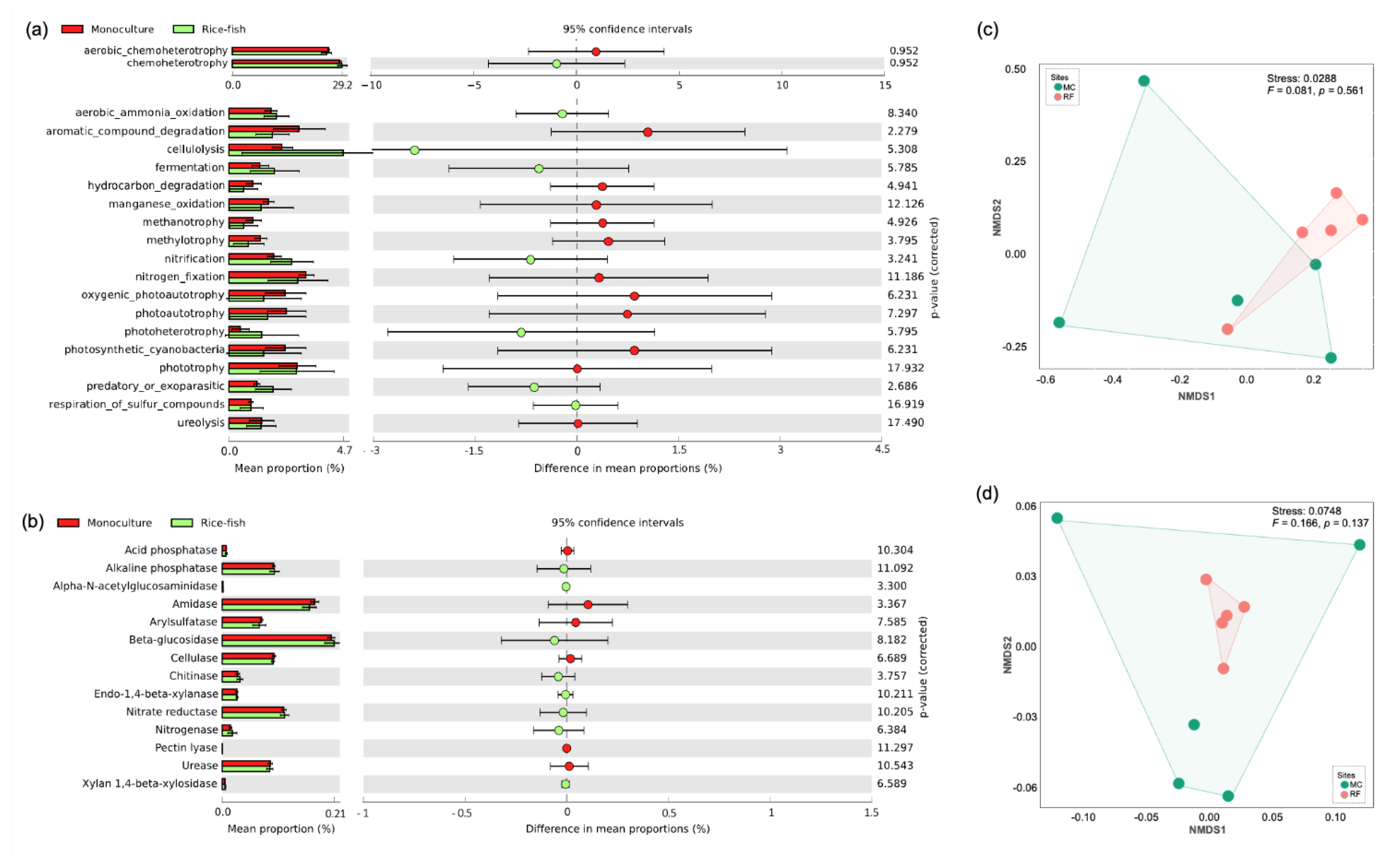
3.5. Predictive Function
4. Discussion
4.1. RF Farming System Can Increase Soil Nutrients
4.2. Microbial Diversity and Community Composition under Rice–Fish Co-Culture and Rice Monoculture
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aislabie, J.; Deslippe, J.; Dymond, J. Soil Microbes and Their Contribution to Soil Services. Ecosystem Services in New Zealand: Conditions and Trends; Manaaki Whenua Press: Lincoln, New Zealand, 2013; pp. 143–161. [Google Scholar]
- Kumari, A.; Kumar, R.; Rani, P.; Beniwal, V.; Kapoor, K.; Sharma, P. Role of Microbes in Sustainable Agriculture. In Microbes in the Service of Mankind; Nagpal, R., Kumar, A., Singh, R., Eds.; JBC Press: Delhi, India, 2014; pp. 178–200. [Google Scholar]
- Jacoby, R.; Peukert, M.; Succurro, A.; Koprivova, A.; Kopriva, S. The Role of Soil Microorganisms in Plant Mineral Nutrition—Current Knowledge and Future Directions. Front. Plant Sci. 2017, 8, 1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falkowski, P.G.; Fenchel, T.; Delong, E.F. The Microbial Engines That Drive Earth’s Biogeochemical Cycles. Science 2008, 320, 1034–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Xiao, Y.; Lv, F.; Hu, L.; Wei, L.; Yuan, Z.; Lin, H. Bacterial Community Structure and Functional Potential of Rhizosphere Soils as Influenced by Nitrogen Addition and Bacterial Wilt Disease under Continuous Sesame Cropping. Appl. Soil Ecol. 2018, 125, 117–127. [Google Scholar] [CrossRef]
- Gellie, N.J.C.; Mills, J.G.; Breed, M.F.; Lowe, A.J. Revegetation Rewilds the Soil Bacterial Microbiome of an Old Field. Mol. Ecol. 2017, 26, 2895–2904. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Chu, C.B.; Zhou, D.P.; Wang, Q.F.; Wu, S.H.; Zheng, X.Q.; Song, K.; Lv, W.G. Soil bacterial community composition in rice–fish integrated farming systems with different planting years. Sci. Rep. 2021, 11, 10855. [Google Scholar] [CrossRef] [PubMed]
- Gautam, A.; Sekaran, U.; Guzman, J.; Kovács, P.; Hernandez, J.L.G.; Kumar, S. Responses of Soil Microbial Community Structure and Enzymatic Activities to Long-Term Application of Mineral Fertilizer and Beef Manure. Environ. Sustain. Indic. 2020, 8, 100073. [Google Scholar] [CrossRef]
- MacKay, K.T.; Chapman, G.; Sollows, J.; Thongpan, N. Rice-fish culture in Northeast Thailand: Stability and sustainability. In Proceedings of the IFOAM 6th International Scientific Conference; University of California: Santa Cruz, CA, USA, 18–21 August 1986; pp. 355–365. [Google Scholar]
- Halwart, M.; Gupta, M.V. Culture of Fish in Rice Fields; FAO: Rome, Italy; The WorldFish Center: Penang, Malaysia, 2004; pp. 1–83. [Google Scholar]
- Heckman, C.W. Rice Field Ecology in Northeastern Thailand; Springer: Dordrecht, The Netherlands, 1979; Volume 34, pp. 1–228. [Google Scholar]
- Arunrat, N.; Sereenonchai, S.; Chaowiwat, W.; Wang, C.; Hatano, R. Carbon, nitrogen and water footprints of organic rice and conventional rice production over 4 years of cultivation: A case study in the Lower North of Thailand. Agronomy 2022, 12, 380. [Google Scholar] [CrossRef]
- Xie, J.; Hu, L.; Tang, J.; Wu, X.; Li, N.; Yuan, Y.; Yang, H.; Zhang, J.; Luo, S.; Chen, X. Ecological mechanisms underlying the sustainability of the agricultural heritage rice-fish co-culture system. Proc. Natl. Acad. Sci. USA 2011, 8, E1381–E1387. [Google Scholar]
- Zhang, J.; Hu, L.; Ren, W.; Guo, L.; Tang, J.; Shu, M.; Chen, X. Rice-soft shell turtle coculture effects on yield and its environment. Agric. Ecosyst. Environ. 2016, 224, 116–122. [Google Scholar] [CrossRef]
- Arunrat, N.; Sereenonchai, S. Assessing ecosystem services of rice–fish co-culture and rice monoculture in Thailand. Agronomy 2022, 12, 1241. [Google Scholar] [CrossRef]
- Clasen, B.; Loro, V.L.; Murussi, C.R.; Tiecher, T.L.; Moraes, B.; Zanella, R. Bioaccumulation and oxidative stress caused by pesticides in Cyprinus carpio reared in a rice-fish system. Sci. Total Environ. 2018, 626, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Ahring, B.K. Perspectives for anaerobic digestion. Adv. Biochem. Eng. Biotechnol. 2003, 81, 1–30. [Google Scholar] [PubMed]
- Janssen, P.H. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 2006, 72, 1719–2172. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Ma, H.; Zhao, Q.; Yang, J.; Xin, C.; Chen, B. Bacterial Communities in Paddy Soil and Ditch Sediment under Rice-Crab Co-Culture System. AMB Express 2011, 11, 163. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, T.R.; Kong, Y.; Nielsen, P.H. Ecophysiology of abundant denitrifying bacteria in activated sludge. FEMS Microbiol. Ecol. 2007, 60, 370–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Xu, J.; Wan, W.; Xu, Z.; Hu, R.; Zhang, Y.; Zheng, J.; Gu, Z. The Microbiome Structure of a Rice-Cray fish Integrated Breeding Model and Its Association with Cray fish Growth and Water Quality. Microbiol. Spectr. 2022, 10, e02204-21. [Google Scholar] [CrossRef] [PubMed]
- Patricia, B.; Pfisterer, A.B.; Nina, B.; Jing-Shen, H.; Tohru, N.; David, R.; Bernhard, S. Quantifying the evidence for biodiversity effects on ecosystem functioning and services. Ecol. Lett. 2010, 9, 1146–1156. [Google Scholar]
- Richards, L.A. Diagnosis and Improvement of Saline and Alkali Soils. In Agriculture Handbook; United States Department of Agriculture: Washington, DC, USA, 1954; pp. 1–160. [Google Scholar]
- National Soil Survey Center. Soil Survey Laboratory Methods Manual. In Soil Survey Investigations Report No. 42, Version 3.0; Natural Conservation Service: Washington, DC, USA, 1996; pp. 1–716. [Google Scholar]
- Bray, R.A.; Kurtz, L.T. Determination of total organic and available form of phosphorus in soil. Soil Sci. 1945, 59, 39–45. [Google Scholar] [CrossRef]
- Thomas, G.W. Method of Soil Analysis, Part 3: Chemical Methods. In Soil pH and Soil Acidity; Sparks, D.L., Page, A.L., Helmke, P.A., Loeppert, R.H., Soltanpour, P.N., Tabatabai, M.A., Johnston, C.T., Sumner, M.E., Eds.; ASA Inc.: Madison, WI, USA, 1996; pp. 475–490. [Google Scholar]
- Walkley, A.; Black, J.A. An examination of the dichromate method for determining soil organic matter and a proposed modification of the chromic acid titration method. Soil Sci. 1934, 37, 29–38. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods. 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Ivanova, A.A.; Oshkin, I.Y.; Danilova, O.V.; Philippov, D.A.; Ravin, N.V.; Dedysh, S.N. Rokubacteria in Northern Peatlands: Habitat Preferences and Diversity Patterns. Microorganisms 2021, 10, 11. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Glöckner, F.O.; Yilmaz, P.; Quast, C.; Gerken, J.; Beccati, A.; Ciuprina, A.; Bruns, G.; Yarza, P.; Peplies, J.; Westram, R.; et al. 25 Years of Serving the Community with Ribosomal RNA Gene Reference Databases and Tools. J. Biotechnol. 2017, 261, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Louca, S.; Parfrey, L.W.; Doebeli, M. Decoupling Function and Taxonomy in the Global Ocean Microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef]
- Sansupa, C.; Wahdan, S.F.M.; Hossen, S.; Disayathanoowat, T.; Wubet, T.; Purahong, W. Can We Use Functional Annotation of Prokaryotic Taxa (FAPROTAX) to Assign the Ecological Functions of Soil Bacteria? Appl. Sci. 2021, 11, 688. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for Prediction of Metagenome Functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Hammer, O.; Harper, D.; Ryan, P. PAST: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirhaj, M.; Razzak, M.A.; Wahab, M.A. Comparison of nitrogen balances and efficiencies in rice cum prawn vs. rice cum fish cultures in Mymensingh, North-Eastern Bangladesh. Agric. Syst. 2014, 125, 54–62. [Google Scholar] [CrossRef]
- Bihari, P.; Nayak, A.K.; Gautam, P.; Lal, B.; Shahid, M.; Raja, R.; Tripathi, R.; Bhattacharyya, P.; Panda, B.B.; Mohanty, S.; et al. Long-term effect of rice-based farming systems on soil health. Environ. Monit. Assess. 2015, 187, 296. [Google Scholar] [CrossRef]
- Nayak, P.; Panda, B.; Lal, B.; Gautam, P.; Poonam, A.; Shahid, M.; Tripathi, R.; Kumar, U.; Mohapatra, S.; Jambhulkar, N. Ecological mechanism and diversity in rice based integrated farming system. Ecol. Indic. 2018, 91, 359–375. [Google Scholar] [CrossRef]
- Liang, G.; Liangliang, H.; Lufeng, Z.; Xiaoyu, S.; Zijun, J.; Lilian, D.; Weizheng, R.; Jian, Z.; Jianjun, T.; Xin, C. Coupling Rice with Fish for Sustainable Yields and Soil Fertility in China. Rice Sci. 2020, 27, 175–179. [Google Scholar] [CrossRef]
- Li, P.; Wu, G.; Li, Y.; Hu, C.; Ge, L.; Zheng, X.; Zhang, J.; Chen, J.; Zhang, H.; Bai, N.; et al. Long-term rice-crayfish-turtle co-culture maintains high crop yields by improving soil health and increasing soil microbial community stability. Geoderma 2022, 413, 115745. [Google Scholar] [CrossRef]
- Moore, P.A.; Patrick, W.H. Calcium and Magnesium Availability and Uptake by Rice in Acid Sulfate Soils. Soil Sci. Soc. Am. J. 1989, 53, 816–822. [Google Scholar] [CrossRef]
- Si, G.H.; Peng, C.L.; Yuan, J.F.; Xu, X.Y.; Zhao, S.J.; Xu, D.B.; Wu, J.S. Changes in soil microbial community composition and organic carbon fractions in an integrated rice-crayfish farming system in subtropical China. Sci. Rep. 2017, 7, 28–56. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Wu, J. Effect of introducing frogs and fish on soil phosphorus availability dynamics and their relationship with rice yield in paddy fields. Sci Rep. 2020, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- Hassink, J. Preservation of plant residues in soils differing in unsaturated protectivecapacity. Soil. Sci. Soc. Am. J. 1996, 60, 487–491. [Google Scholar] [CrossRef]
- Yi, X.; Yi, K.; Fang, K.; Gao, H.; Dai, W.; Cao, L. Microbial Community Structures and Important Associations Between Soil Nutrients and the Responses of Specific Taxa to Rice-Frog Cultivation. Front. Microbiol. 2019, 10, 1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Zhang, L.; Guo, X.; Dai, X.; Liu, L.; Xi, L.; Wang, J.; Song, L.; Wang, Y.; Zhu, Y.; et al. Diversity, biogeography, and biodegradation potential of Actinobacteria in the deep-sea sediments along the South-west Indian Ridge. Front. Microbiol. 2016, 7, 1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankratov, T.A.; Dedysh, S.N.; Zavarzin, G.A. The leading role of Actinobacteria in aerobic cellulose degradation in Sphagnum peat bogs. Dokl. Biol. Sci. 2006, 410, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Dolfing, J.; Guo, Z.; Chen, R.; Wu, M.; Li, Z.; Lin, X.; Feng, Y. Important ecophysiological roles of non-dominant actinobacteria in plant residue decomposition, especially in less fertile soils. Microbiome 2021, 9, 84. [Google Scholar] [CrossRef]
- Sorokin, D.Y.; Lücker, S.; Vejmelkova, D.; Kostrikina, N.A.; Kleerebezem, R.; Rijpstra, W.I.C.; Damsté, J.S.S.; Le Paslier, D.; Muyzer, G.; Wagner, M.; et al. Nitrification expanded: Discovery, physiology and genomics of a nitrite-oxidizing bacterium from the phylum Chloroflexi. ISME J. 2012, 6, 2245–2256. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, P.; Anderson, I.C.; Singh, B.K. Microbial modulators of soil carbon storage: Integrating genomic and metabolic knowledge for global prediction. Trends Microbiol. 2013, 21, 641–651. [Google Scholar] [CrossRef]
- Liu, J.; Sui, Y.; Yu, Z.; Shi, Y.; Chu, H.; Jin, J.; Liu, X.; Wang, G. High throughput sequencing analysis of biogeographical distribution of bacterial communities in the black soils of northeast China. Soil Biol. Biochem. 2014, 70, 113–122. [Google Scholar] [CrossRef]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microb. 2009, 75, 5111–5120. [Google Scholar] [CrossRef] [Green Version]
- Dedysh, S.N.; Ivanova, A.A. Planctomycetes in boreal and subarctic wetlands: Diversity patterns and potential ecological functions. FEMS Microbiol. Ecol. 2019, 95, fiy227. [Google Scholar] [CrossRef] [Green Version]
- García-Orenes, F.; Morugán-Coronado, A.; Zornoza, R.; Scow, K. Changes in soil microbial community structure influenced by agricultural management practices in a Mediterranean agro-ecosystem. PLoS ONE 2013, 8, e80522. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, C.; Li, X.L.; Yang, X.S.; Zhao, L.B.; Liu, L.; Zhu, C.; Li, R.H. Linking plant ecological stoichiometry with soil nutrient and bacterial communities in apple orchards. Appl. Soil Ecol. 2018, 126, 1–10. [Google Scholar] [CrossRef]
- Fu, X.; Wang, J.; Sainju, U.M.; Zhao, F.; Liu, W. Soil microbial community and carbon and nitrogen fractions responses to mulching under winter wheat. Appl. Soil Ecol. 2019, 139, 64–68. [Google Scholar] [CrossRef]
- Saxena, A.K.; Kumar, M.; Chakdar, H.; Anuroopa, N.; Bagyaraj, D.J. Bacillus species in soil as a natural resource for plant health and nutrition. J. Appl. Microbiol. 2019, 128, 1583–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, Y.; Yamanaka, H.; Xu, Z.-X.; Shiratori, Y.; Aono, T.; Amachi, S.; Senoo, K.; Itoh, H. Diazotrophic Anaeromyxobacter isolates from soils. Appl. Environ. Microbiol. 2020, 86, e00956-20. [Google Scholar] [CrossRef] [PubMed]
- Xue, P.-P.; Carrillo, Y.; Pino, V.; Minasny, B.; McBratney, A.B. Soil Properties Drive Microbial Community Structure in a Large Scale Transect in South Eastern Australia. Sci. Rep. 2018, 8, 11725. [Google Scholar] [CrossRef] [Green Version]
- Sui, X.; Zhang, R.; Frey, B.; Yang, L.; Liu, Y.; Ni, H.; Li, M.-H. Soil Physicochemical Properties Drive the Variation in Soil Microbial Communities along a Forest Successional Series in a Degraded Wetland in Northeastern China. Ecol. Evol. 2021, 11, 2194–2208. [Google Scholar] [CrossRef]
- Viruel, E.; Fontana, C.A.; Puglisi, E.; Nasca, J.A.; Banegas, N.R.; Pier, S.; Cocconcelli, P.S. Land-use change affects the diversity and functionality of soil bacterial communities in semi-arid Chaco region, Argentina. Appl. Soil Ecol. 2022, 172, 104362. [Google Scholar] [CrossRef]
- Hartmann, M.; Frey, B.; Mayer, J.; Mäder, P.; Widmer, F. Distinct soil microbial diversity under long-term organic and conventional farming. ISME J. 2015, 9, 1177–1194. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Cheng, X.; Li, J.; Dong, Y. Magnesium and nitrogen drive soil bacterial community structure under long-term apple orchard cultivation systems. Appl. Soil Ecol. 2021, 167, 104103. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, Q.; Zhang, M.; Guo, J.; Zheng, J.; Chen, Z.; Jia, Y.; Zhang, J.; Li, Z.; Zhang, H. Mg2+ distribution in activated sludge and its effects on the nitrifying activity and the characteristics of extracellular polymeric substances and sludge flocs. Process Biochem. 2020, 88, 120–128. [Google Scholar] [CrossRef]
- Chen, H.; Ma, K.; Huang, Y.; Yao, Z.; Chu, C. Stable Soil Microbial Functional Structure Responding to Biodiversity Loss Based on Metagenomic Evidences. Front. Microbiol. 2021, 12, 716764. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive Functional Profiling of Microbial Communities Using 16S RRNA Marker Gene Sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Soil Properties | Rice Monoculture | Rice–fish Co-Culture | T | Sig. |
|---|---|---|---|---|
| pH (1:2.5) | 4.7 ± 0.3 | 6.0 ± 0.2 | 8.031 | ** |
| BD (Mg m−3) | 1.4 ± 0.02 | 1.4 ± 0.02 | −2.414 | * |
| OM (%) | 2.1 ± 0.4 | 3.4 ± 0.2 | 6.997 | ** |
| SOC (Mg C ha−1) | 51.0 ± 9.2 | 80.9 ± 3.5 | 13.878 | ** |
| TN (%) | 0.3 ± 0.01 | 0.5 ± 0.2 | 15.637 | ** |
| ECe (dS m−1) | 1.0 ± 0.4 | 0.4 ± 0.01 | −3.636 | ** |
| Avail. P (mg kg−1) | 13.6 ± 1.9 | 20.0 ± 0.9 | 6.928 | ** |
| Avail. K (mg kg−1) | 162.8 ± 6.2 | 170.0 ± 4.1 | 2.182 | NS |
| Avail. Ca (mg kg−1) | 2554.4 ± 85.2 | 2279.0 ± 90.0 | −4.967 | ** |
| Avail. Mg (mg kg−1) | 225.0 ± 5.6 | 175.1 ± 3.6 | −16.709 | ** |
| Sand (%) | 17.4 ± 0.9 | 10.1 ± 0.8 | −14.141 | ** |
| Silt (%) | 42.0 ± 1.3 | 43.6 ± 1.0 | 2.123 | NS |
| Clay (%) | 40.6 ± 0.9 | 46.3 ± 0.9 | 10.486 | ** |
| Soil texture | Silty Clay | Silty Clay | - | - |
| Soil Properties | pH | BD | SOC | TN | ECe | P | K | Ca | Mg | %Sand | %Silt | %Clay |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pH | −0.718 | 0.391 | −0.387 | −0.086 | −0.302 | −0.613 | 0.275 | −0.401 | 0.061 | −0.056 | 0.010 | |
| BD | −0.562 | 0.106 | −0.015 | −0.160 | 0.558 | 0.216 | −0.662 | 0.204 | −0.197 | −0.313 | 0.537 | |
| SOC | −0.316 | −0.187 | −0.787 | −0.485 | 0.744 | −0.367 | −0.293 | 0.328 | 0.247 | −0.887 | 0.796 | |
| TN | −0.373 | 0.775 | −0.454 | −0.157 | −0.434 | 0.773 | −0.226 | −0.516 | −0.709 | 0.871 | −0.361 | |
| ECe | −0.087 | 0.524 | −0.904 | 0.741 | −0.565 | −0.493 | 0.803 | 0.231 | 0.626 | 0.180 | −0.770 | |
| P | −0.101 | −0.515 | −0.243 | −0.316 | 0.149 | 0.194 | −0.549 | 0.587 | 0.121 | −0.819 | 0.831 | |
| K | 0.258 | 0.618 | −0.266 | 0.512 | 0.307 | −0.868 | −0.452 | −0.026 | −0.543 | 0.377 | 0.056 | |
| Ca | 0.700 | −0.745 | 0.046 | −0.277 | −0.323 | 0.049 | −0.120 | 0.292 | 0.733 | 0.128 | −0.808 | |
| Mg | 0.474 | 0.198 | −0.551 | 0.618 | 0.498 | −0.435 | 0.668 | 0.445 | 0.791 | −0.681 | 0.069 | |
| %Sand | −0.126 | 0.811 | −0.618 | 0.917 | 0.808 | −0.446 | 0.744 | −0.333 | 0.691 | −0.546 | −0.274 | |
| %Silt | −0.333 | −0.559 | 0.341 | −0.535 | −0.355 | 0.827 | −0.992 | 0.022 | −0.756 | −0.762 | −0.656 | |
| %Clay | 0.631 | 0.051 | 0.090 | −0.090 | −0.258 | −0.818 | 0.774 | 0.295 | 0.468 | 0.173 | −0.770 |
| Parameter | NMDS1 | NMDS2 | r | p-Value |
|---|---|---|---|---|
| pH | 0.284 | −0.959 | 0.857 | 0.024 * |
| BD | −0.523 | 0.853 | 0.697 | 0.108 |
| OM | 0.286 | −0.958 | 0.762 | 0.084 |
| TN | 0.211 | −0.978 | 0.834 | 0.012 * |
| ECe | −0.238 | 0.971 | 0.563 | 0.516 |
| Avail. P | 0.266 | −0.964 | 0.662 | 0.240 |
| Avail. K | 0.176 | −0.984 | 0.261 | 1.000 |
| Avail. Ca | −0.088 | 0.996 | 0.649 | 0.240 |
| Avail. Mg | −0.214 | 0.977 | 0.880 | 0.024 * |
| Sand | −0.190 | 0.982 | 0.866 | 0.036 * |
| Silt | 0.104 | −0.995 | 0.460 | 1.000 |
| Clay | 0.220 | −0.976 | 0.764 | 0.048 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arunrat, N.; Sansupa, C.; Kongsurakan, P.; Sereenonchai, S.; Hatano, R. Soil Microbial Diversity and Community Composition in Rice–Fish Co-Culture and Rice Monoculture Farming System. Biology 2022, 11, 1242. https://doi.org/10.3390/biology11081242
Arunrat N, Sansupa C, Kongsurakan P, Sereenonchai S, Hatano R. Soil Microbial Diversity and Community Composition in Rice–Fish Co-Culture and Rice Monoculture Farming System. Biology. 2022; 11(8):1242. https://doi.org/10.3390/biology11081242
Chicago/Turabian StyleArunrat, Noppol, Chakriya Sansupa, Praeploy Kongsurakan, Sukanya Sereenonchai, and Ryusuke Hatano. 2022. "Soil Microbial Diversity and Community Composition in Rice–Fish Co-Culture and Rice Monoculture Farming System" Biology 11, no. 8: 1242. https://doi.org/10.3390/biology11081242