
Assessment of Genetic Diversity of the Salangid, Neosalanx taihuensis, Based on the Mitochondrial COI Gene in Different Chinese River Basins

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
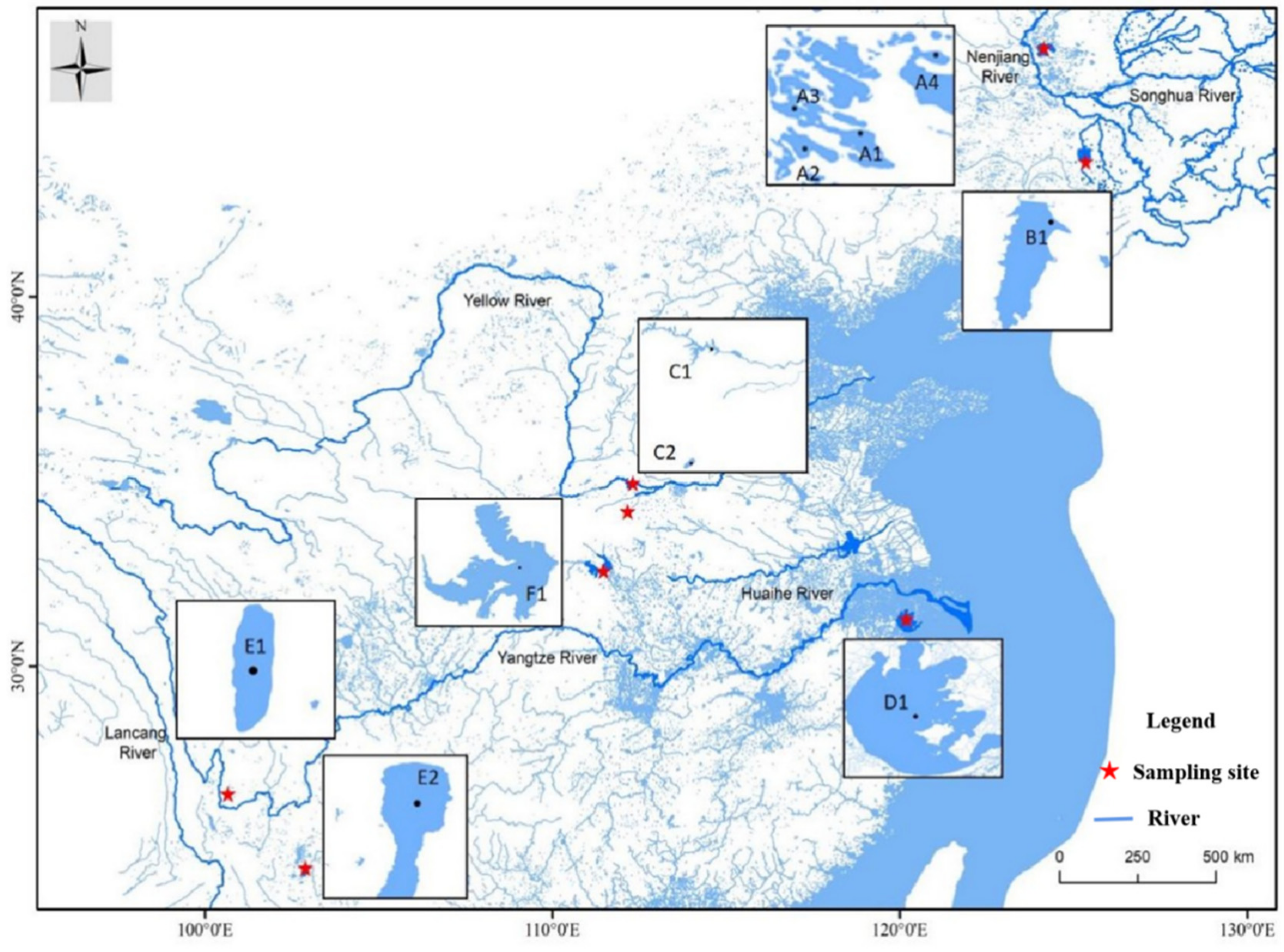
2.1. Sample Collection
2.2. DNA Extraction, PCR Amplification and Sequencing
2.3. Genetic Diversity Analysis
3. Results
3.1. Genetic Diversity
3.2. Population Differentiation
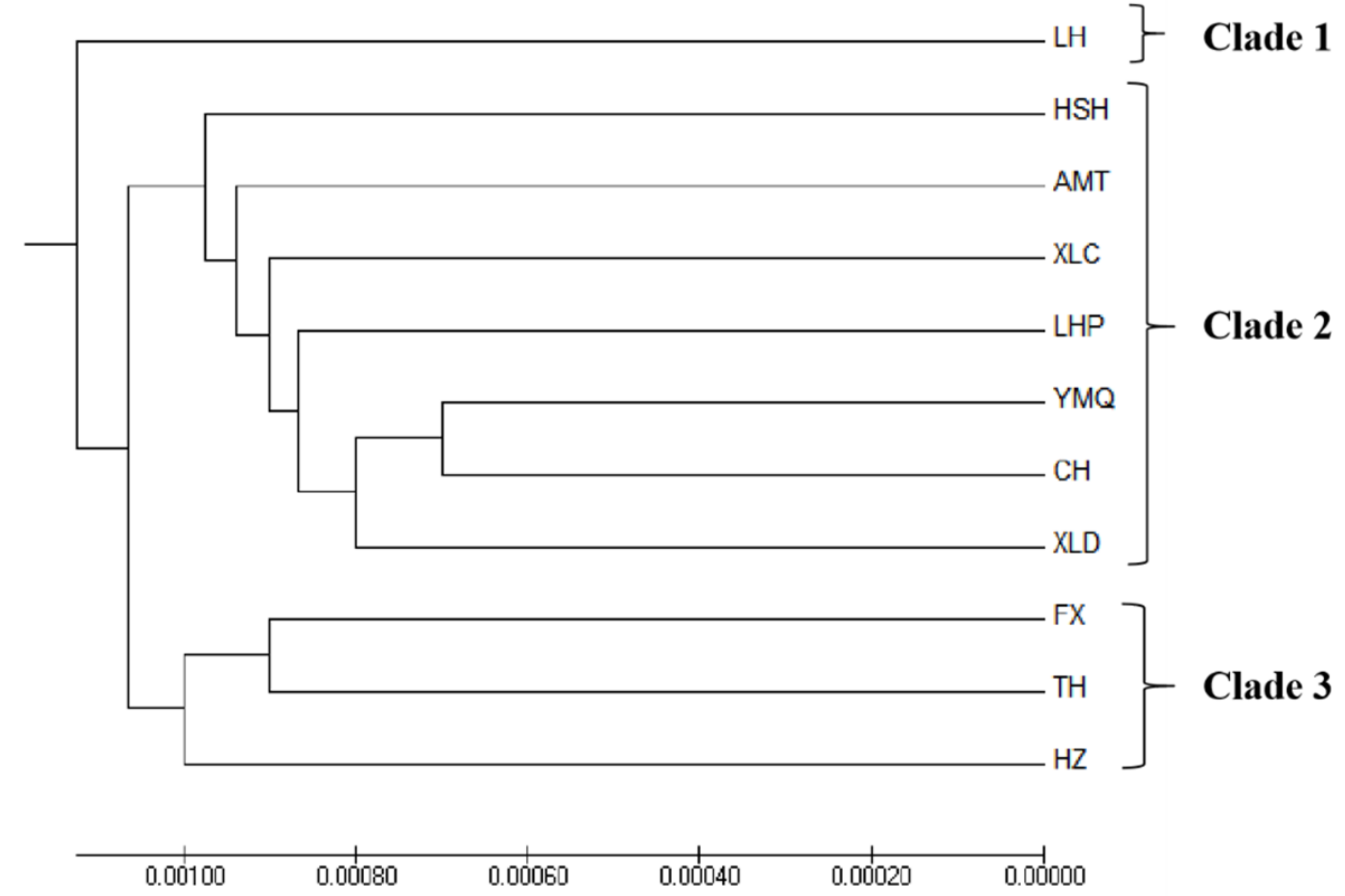
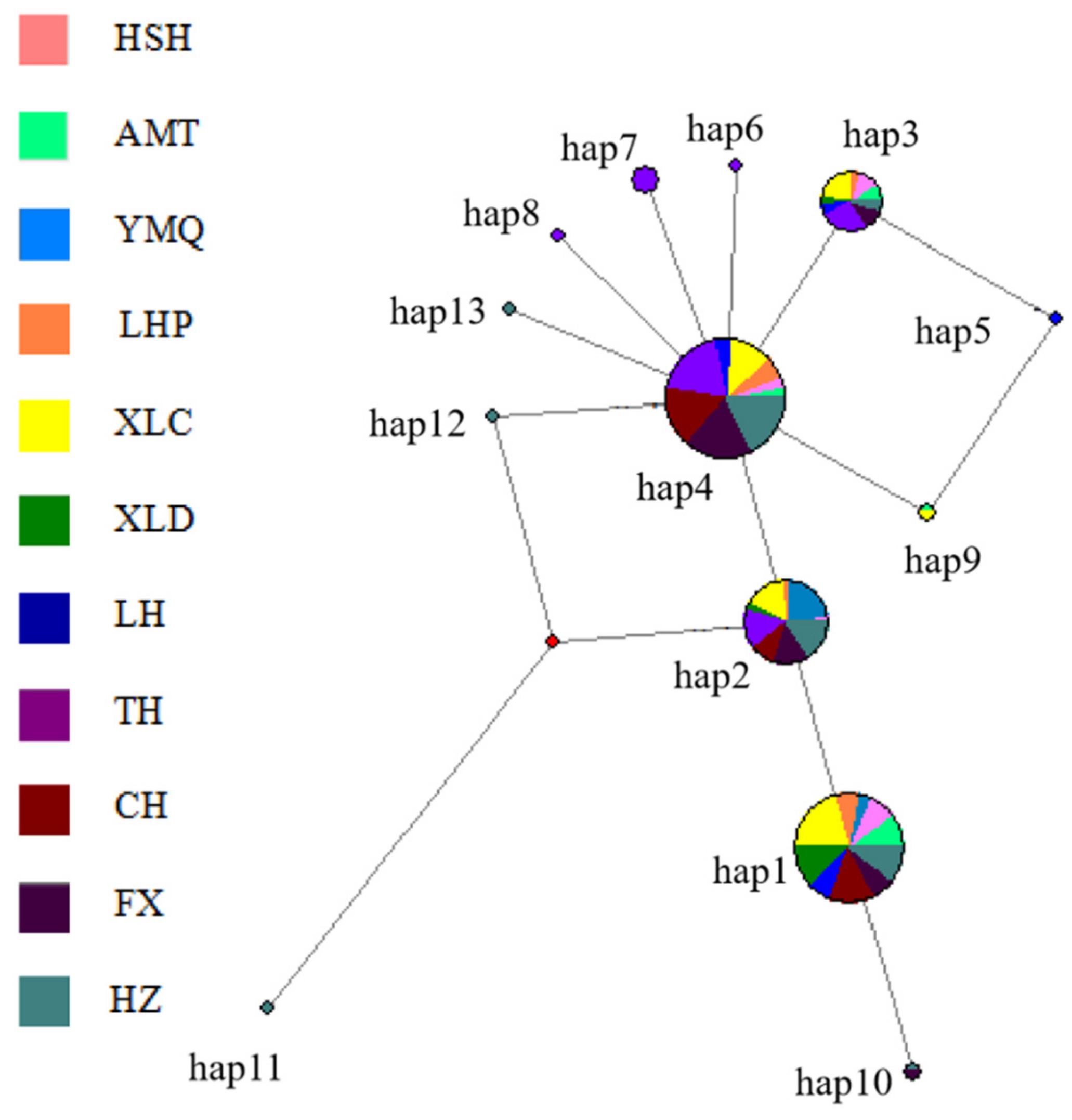
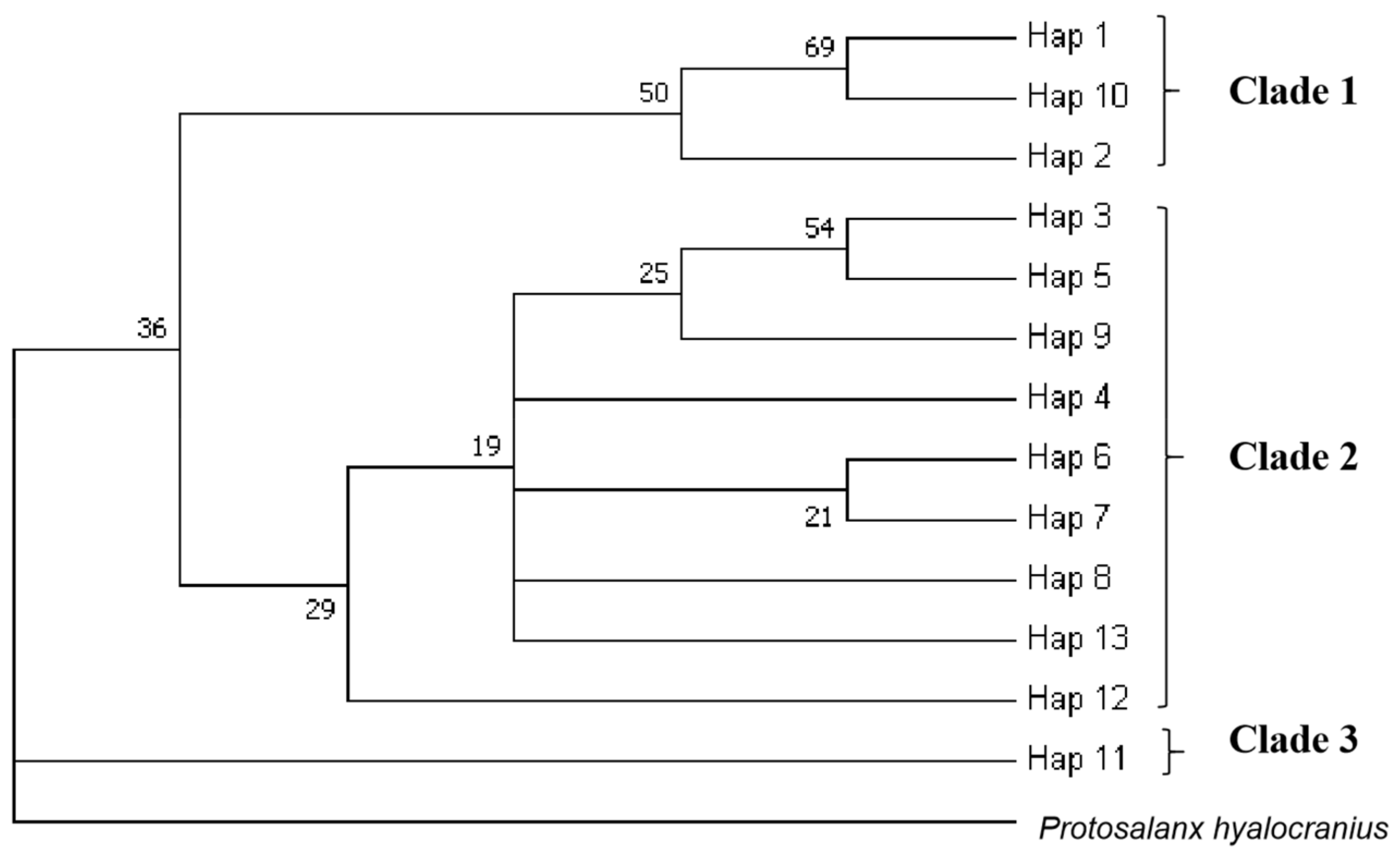
3.3. Population Structure
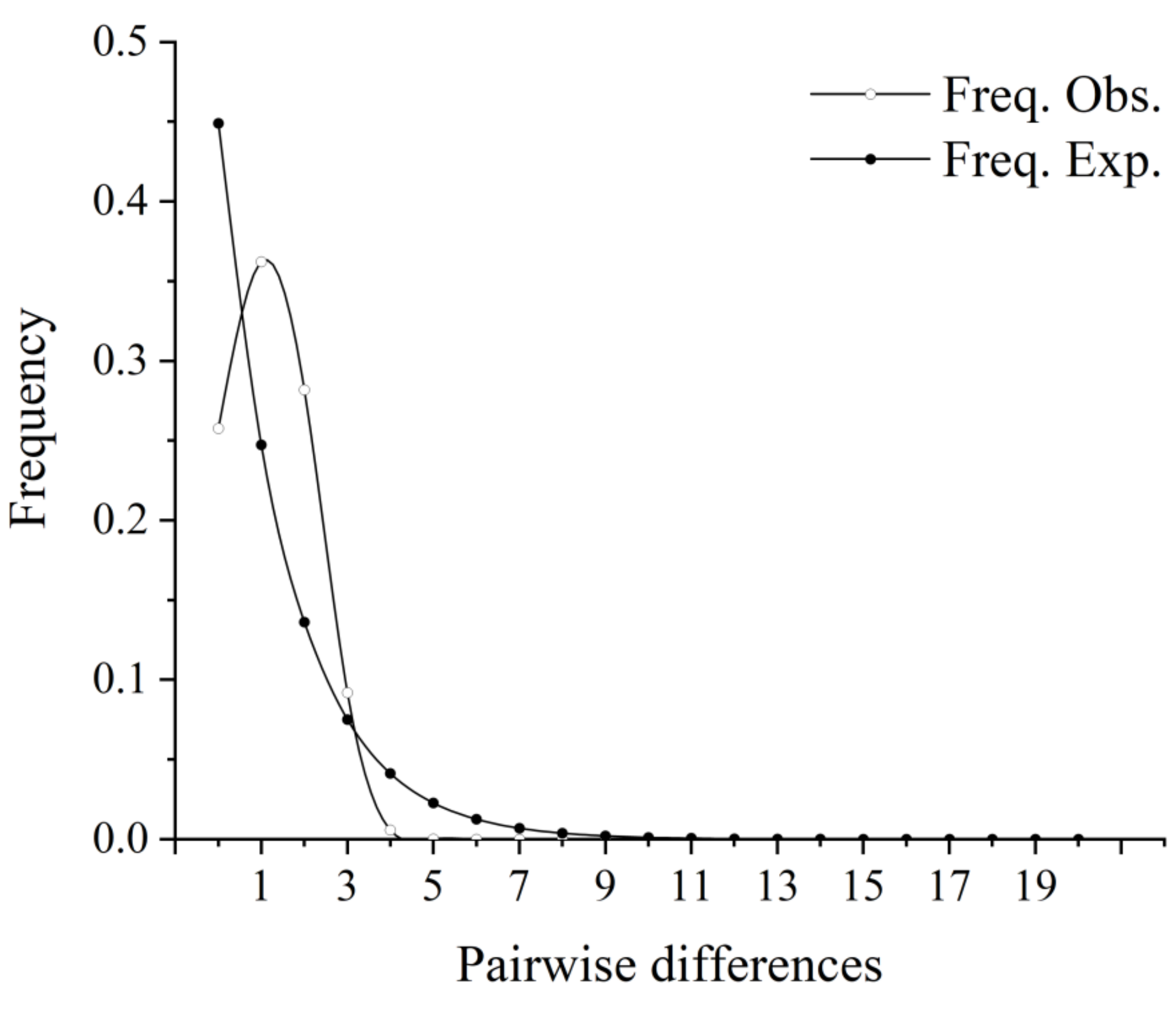
3.4. Historical Demography
4. Discussion
4.1. Genetic Diversity
4.2. Genetic Differentiation
4.3. Population Structure
4.4. Historical Dynamics
4.5. Research Prospective
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Haplotype | Variable Sites | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 35 | 75 | 303 | 315 | 393 | 453 | 483 | 507 | 645 | 646 | 647 | |
| Hap1 | C | T | A | A | A | G | T | T | T | C | C |
| Hap2 | C | T | G | A | A | G | T | T | T | C | C |
| Hap3 | C | T | G | A | A | G | T | C | T | C | C |
| Hap4 | C | T | G | A | A | G | T | T | T | C | C |
| Hap5 | C | T | G | G | A | G | C | C | T | C | C |
| Hap6 | C | T | G | A | G | G | T | T | T | C | C |
| Hap7 | C | C | G | A | A | G | T | T | T | C | C |
| Hap8 | C | T | G | A | A | A | A | T | T | C | C |
| Hap9 | C | T | G | A | A | G | T | T | T | C | C |
| Hap10 | T | T | A | A | A | G | T | T | T | C | C |
| Hap11 | C | T | A | A | A | G | T | T | C | G | A |
| Hap12 | C | T | G | A | A | G | T | T | C | C | C |
| Hap13 | C | T | G | A | A | G | T | T | T | C | T |
| Haplotype | Hap1 | Hap2 | Hap3 | Hap4 | Hap5 | Hap6 | Hap7 | Hap8 | Hap9 | Hap10 | Hap11 | Hap12 | Hap13 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NjRB | 0.2881 | 0.2368 | 0.2105 | 0.1194 | 0.5 | ||||||||
| ShRB | 0.2034 | 0.1316 | 0.2105 | 0.1194 | 0.5 | ||||||||
| YeRB | 0.2034 | 0.0263 | 0.1053 | 0.0448 | 1 | ||||||||
| YzRB | 0.1579 | 0.2632 | 0.1940 | 1 | 1 | 1 | |||||||
| LcRB | 0.2034 | 0.2368 | 0.1053 | 0.3433 | 0.5 | ||||||||
| HhRB | 0.1017 | 0.1579 | 0.1053 | 0.1791 | 0.5 | 1 | 1 | 1 | |||||
| HSH | 0.1017 | 0.0526 | 0.0299 | 0.5 | |||||||||
| AMT | 0.0847 | 0.0263 | 0.1053 | 0.0299 | |||||||||
| YMQ | 0.0339 | 0.2105 | |||||||||||
| LHP | 0.0678 | 0.0263 | 0.0526 | 0.0597 | |||||||||
| XLC | 0.2034 | 0.1579 | 0.2105 | 0.1194 | 0.5 | ||||||||
| XLD | 0.1356 | 0.0263 | 0.0526 | ||||||||||
| LH | 0.0678 | 0.0526 | 0.0448 | 1 | |||||||||
| TH | 0.1579 | 0.2632 | 0.1940 | 1 | 1 | 1 | |||||||
| CH | 0.1356 | 0.1053 | 0.1642 | ||||||||||
| FX | 0.0678 | 0.1316 | 0.1053 | 0.1791 | 0.5 | ||||||||
| HZ | 0.1017 | 0.1579 | 0.1053 | 0.1791 | 0.5 | 1 | 1 | 1 |
References
- Grummer, J.A.; Beheregaray, L.B.; Bernatchez, L.; Hand, B.K.; Luikart, G.; Narum, S.R.; Taylor, E.B. Aquatic landscape genomics and environmental effects on genetic variation. Trends Ecol. Evol. 2019, 34, 641–654. [Google Scholar] [CrossRef]
- Perdereau, A.C.; Kelleher, C.T.; Douglas, G.C.; Hodkinson, T.R. High levels of gene flow and genetic diversity in irish populations of salix caprea l. Inferred from chloroplast and nuclear ssr markers. BMC Plant Biol. 2014, 14, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujolar, J.M.; Lucarda, A.N.; Simonato, M.; Patarnello, T. Restricted gene flow at the micro- and macro-geographical scale in marble trout based on mtdna and microsatellite polymorphism. Front. Zool. 2011, 8, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, A.R.; Petrosino, G.; Crescenzo, S.; Milana, V.; Talarico, L.; Martinoli, M.; Rakaj, A.; Lorenzoni, M.; Carosi, A.; Ciuffardi, L.; et al. Phylogeography and population structure of squalius lucumonis: A baseline for conservation of an italian endangered freshwater fish. J. Nat. Conserv. 2021, 64, 126085. [Google Scholar] [CrossRef]
- Xie, P.; Chen, Y. Threats to Biodiversity in Chinese Inland Waters. Proc. Jt. Semin. Ecosyst. Res. Manag. China 1999, 28, 674–681. [Google Scholar]
- Liu, Z. The introduction of icefish, neosalanx taihuensis chen, in China with special reference to the subtropical lakes of the yunnan plateau (southwest China). Int. Assoc. Theor. Appl. Limnol. 2002, 27, 3877–3880. [Google Scholar] [CrossRef]
- Fu, C.; Chen, J.; Wu, Q.; Lei, G. Freshwater fish biodiversity in the Yangtze River basin of China: Patterns, threats and conservation. Biodivers. Conserv. 2003, 12, 1649–1685. [Google Scholar] [CrossRef]
- Liu, X.J.; Qin, J.J.; Xu, Y.; Ouyang, S.; Wu, X.P. Biodiversity decline of fish assemblages after the impoundment of the Three Gorges Dam in the Yangtze River Basin, China. Rev. Fish Biol. Fish. 2019, 29, 177–195. [Google Scholar] [CrossRef]
- Chao, Z.; Ren, Z.A.; Xs, A.; Xla, B.; Yc, A. Understanding patterns of taxonomic diversity, functional diversity, and ecological drivers of fish fauna in the Mekong River. Glob. Ecol. Conserv. 2021, 28, 2351. [Google Scholar]
- Baran, E.; Chum, N.; Fukushima, M.; Hand, T.; Hoetle, K.G.; Jutagate, T.; Kang, B. Fish biodiversity research in the Mekong Basin. Ecol. Res. Monogr, 1st ed.; Springer: Tokyo, Japan, 2012; pp. 149–164. [Google Scholar]
- Chen, W.; Zhao, Y.; Yang, C. Genetic variation and population history of three Carassius auratus populations in Huaihe River, China. Mitochondrial DNA Part A 2016, 27, 4252–4255. [Google Scholar] [CrossRef]
- Wang, Y.C.; Gao, X.W.; Li, T.J.; Yue, Y.; Fang, H.Q.; Ni, J.R. Geocode-based Aquatic Habitats in Hierarchical System of the Yellow River Basin. J. Environ. Inform. 2018, 32, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Zhang, G.; Li, H.; Lu, W. Land use change impacts on hydrology in the Nenjiang River Basin, Northeast China. Forests 2019, 10, 476. [Google Scholar] [CrossRef] [Green Version]
- Zhu, F.Y.; Ye, S.W.; Li, Z.J.; Zhang, T.L.; Yuan, J.; Guo, Z.Q.; Tang, J.F.; Liu, J.S. Growth and reproduction of the non-native icefish neosalanx taihuensis chen, 1956 (salangidae) in a plateau lake, southwestern China. J. Appl. Ichthyol. 2015, 30, 862–869. [Google Scholar] [CrossRef]
- Gong, W.; Wu, L.; Xie, S.; Liu, J.; Murphy, B.R.; Xie, S. Variation in early growth of neosalanx taihuensis between two populations above and below the three gorges dam, China. J. Freshw. Ecol. 2013, 28, 139–146. [Google Scholar] [CrossRef]
- Liu, H.Y.; Li, C.Y.; Xiong, F. Isolation and characterization of 19 polymorphic microsatellite loci from neosalanx taihuensis, a rapidly invasive and adaptative species. Biochem. Syst. Ecol. 2015, 61, 121–123. [Google Scholar] [CrossRef]
- Lai, J.; Zhao, L.; Fan, Y.; Qu, X.; Liu, D.; Guo, Z.; Wang, Y.; Liu, Q.; Chen, Y. Using whole body elemental fingerprint analysis to distinguish different populations of coilia nasus in a large river basin. Biochem. Syst. Ecol. 2015, 60, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Meriam, T.; Wafa, T.; Khawla, T.; Tarek, H.; Abdeljelil, G.; Mhamed, E. Genetic diversity and population structure of Sepia officinalis from the Tunisian cost revealed by mitochondrial COI sequences. Mol. Biol. Rep. 2015, 42, 77–86. [Google Scholar] [CrossRef]
- Sari, A.; Duran, M.; Sen, A.; Bardakci, F. Investigation of Chironomidae (Diptera) relationships using mitochondrial COI gene. Biochem. Syst. Ecol. 2015, 59, 229–238. [Google Scholar] [CrossRef]
- Zahn, R.J.; Silva, A.J.; Hellberg, R.S. Development of a DNA mini-barcoding protocol targeting COI for the identification of elasmobranch species in shark cartilage pills—ScienceDirect. Food Control. 2020, 109, 106918. [Google Scholar] [CrossRef]
- Satoh, T.P.; Miya, M.; Mabuchi, K.; Nishida, M. Structure and variation of the mitochondrial genome of fishes. BMC Genom. 2016, 17, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Qi, P.Z.; Guo, B.Y.; Xie, C.X.; Wu, C.W.; Lu, S.M.; Duan, Y.J.; Zhou, X.J. Assessing the genetic diversity and population structure of Culter alburnus in China based on mitochondrial 16S rRNA and COI gene sequences. Biochem. Syst. Ecol. 2013, 50, 390–396. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The clustalx windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Librado, P.; Rozas, J. Dnasp v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoyratty, S.; Dupont, J.; Lacoste, S.; Palama, T.L.; Choi, Y.H.; Kim, H.K.; Payet, B.; Grisoni, M.; Fouillaud, M.; Verpoorte, R. Fungal endophytes of Vanilla planifolia across Réunion Island: Isolation, distribution and biotransformation. Phylogenetic Tree Original Data Set (Opens with Mega 5.0 + ms Powerpoint) 2015, 15, 142. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. Online 2004, 1, L418–L421. [Google Scholar] [CrossRef] [Green Version]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Tajima, F.V. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Fu, Y.X.; Li, W.H. Statistical tests of neutrality of mutations. Genetics 1993, 133, 693–709. [Google Scholar] [CrossRef]
- Rogers, A.R.; Harpending, H. Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol. 1992, 9, 552–569. [Google Scholar]
- Zhao, L.S.; Cheng, F.; Zhang, L.; Wang, X.; Zhang, S.Y.; Jiang, G.C.; Xie, S.G. Growth,mortality and evaluation of resource utilization for two salangidae,protosalanx hyalocranius and neosalanx taihuensis in hongze lake. Acta Hydrobiol. Sin. 2018, 42, 240–249. [Google Scholar]
- Smith, G.R. Introgression in fishes: Significance for paleontology, cladistics, and evolutionary rates. Syst. Biol. 1992, 41, 41–57. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under linux and windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Nikolaus, B.; Stafford, D.W. A general method for isolation of high molecular weight DNA from eukaryotes. Nucleic Acids Res. 1976, 3, 2303–2308. [Google Scholar]
- Othman, O. Mitochondrial DNA as a marker for genetic diversity and evolution. Adv. Genet. Eng. Biotechnol. 2012, 1, 1–2. [Google Scholar]
- Byers, D.L. Evolution in heterogeneous environments and the potential of maintenance of genetic variation in traits of adaptive significance. Genetica 2005, 123, 107. [Google Scholar] [CrossRef]
- Grant, W.; Bowen, B. Shallow population histories in deep evolutionary lineages of marine fishes: Insights from sardines and anchovies and lessons for conservation. J. Hered. 1998, 89, 415–426. [Google Scholar] [CrossRef]
- Li, D.M.; Tang, S.K.; Liu, Y.S.; Gu, X.K.; Liu, X.W.; Yin, J.W.; Wang, B.; Ma, H.; Zhang, T.Q.; Pan, J.L. Genetic diversity analysis of four Neosalanx taihuensis populationsin Jiangsu Province based on mitochondrial COI gene sequences. Mar. Fish. 2020, 42, 277–280. [Google Scholar]
- Yang, F. Genetic Diversity Comparison of Coilia Brachygnathus and Neosalanx taihuensis between Populations in the Three GorgesReservoir and Dongting Lake. Master’s Thesis, Southwest University, Chongqing, China, 2019. [Google Scholar]
- Xue, D.X.; Yang, Q.L.; Li, Y.L.; Zong, S.B.; Gao, T.X.; Liu, J.X. Comprehensive assessment of population genetic structure of the overexploited japanese grenadier anchovy (coilia nasus): Implications for fisheries management and conservation. Fish. Res. 2019, 213, 113–120. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, J.; Liu, Z.; Funk, S.M.; Wei, F.; Xu, M.; Li, M. Complex population genetic and demographic history of the salangid, neosalanx taihuensis, based on cytochrome b sequences. BMC Evol. Biol. 2008, 8, 201. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Li, C.; Xiong, F. Population genetic structure of neosalanx taihuensis between invasive and original areas revealed by microsatellite DNA. J. Fish. China 2016, 40, 1521–1530. [Google Scholar]
- Zhang, D.; Lei, G.; Cheng, G.; Wang, Z. Genetic diversity of neosalanx taihuensis based on mitochondrial coi sequences. J. Lake Sci. 2012, 24, 299–306. [Google Scholar]
- Czerniejewski, P. Changes in condition and in carapace length and width of the Chinese mitten crab (Eriocheir sinensis H. Milne Edwards, 1853) harvested in the Odra River estuary in 1999–2007. Oceanol. Hydrobiol. Stud. 2010, 39, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Gong, W.B.; Li, H.T.; Xie, S.Q.; Liu, J.S.; Murphy, B.R.; Xie, S.G. Two spawning stocks of icefish neosalanx taihuensis revealed from annual reproductive cycle analyses. Fish. Sci. 2009, 75, 1157–1165. [Google Scholar] [CrossRef]
- Stopar, K.; Ramšak, A.; Trontelj, P.; Malej, A. Lack of genetic structure in the jellyfish pelagia noctiluca (cnidaria: Scyphozoa: Semaeostomeae) across european seas. Mol. Phylogenetics Evol. 2010, 57, 417–428. [Google Scholar] [CrossRef]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef]
- Schneider, S.; Excoffier, L. Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: Application to human mitochondrial DNA. Genetics 1999, 152, 1079–1089. [Google Scholar] [CrossRef]
- Lambeck, K.; Esat, T.M.; Potter, E.-K. Links between climate and sea levels for the past three million years. Nature 2002, 419, 199–206. [Google Scholar] [CrossRef]
- Yang, Y.; Qi, Y. A study on bioecological & economic effect of neosalanx taihuensis chen in taihu. J. Southwest Natl. Coll. 1999, 3, 269–273. [Google Scholar]
- Bai, R.; Liu, X.; Liu, X.; Liu, L.; Wang, J.; Liao, S.; Zhu, A.; Li, Z. The development of biodiversity conservation measures in China’s hydro projects: A review. Environ. Int. 2017, 108, 285–298. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, Y.; Yu, R.; Xu, X.; Xu, M.; Li, G.; Wang, W.; Yang, Y. China’s biodiversity conservation in the process of implementing the sustainable development goals (sdgs). J. Clean. Prod. 2022, 338, 130595. [Google Scholar] [CrossRef]
- Chen, T.; Wang, Y.; Gardner, C.; Wu, F. Threats and protection policies of the aquatic biodiversity in the Yangtze river. J. Nat. Conserv. 2020, 58, 125931. [Google Scholar] [CrossRef]





| Populations Codes | T (%) | C (%) | A (%) | G (%) | T + A (%) | C+G (%) |
|---|---|---|---|---|---|---|
| Nenjiang River Basin | ||||||
| A1 (Huoshaohei lake, HSH) | 21.45 | 19.20 | 26.15 | 33.20 | 47.60 | 52.40 |
| A2 (Amuta lake, AMT) | 21.45 | 19.20 | 26.13 | 33.21 | 47.58 | 52.41 |
| A3 (Yamenqi lake, YMQ) | 21.48 | 19.17 | 26.15 | 33.19 | 47.63 | 52.36 |
| A4 (Longhupao lake, LHP) | 21.47 | 19.19 | 26.11 | 33.25 | 47.58 | 52.44 |
| Songhua River Basin | ||||||
| B1 (Xinlicheng reservoir, XLC) | 21.46 | 19.20 | 26.12 | 33.24 | 47.58 | 52.44 |
| Yellow River Basin | ||||||
| C1 (Xiaolangdi reservoir, XLD) | 21.47 | 19.18 | 26.21 | 33.14 | 47.68 | 52.32 |
| C2 (Luhun reservoir, LH) | 21.43 | 19.22 | 26.05 | 33.30 | 47.48 | 52.52 |
| Yangtze River Basin | ||||||
| D1 (Taihu lake, TH) | 21.44 | 19.21 | 25.99 | 33.37 | 47.43 | 52.58 |
| Lanchang River Basin | ||||||
| E1 (Chenghai, CH) | 21.36 | 19.30 | 25.95 | 33.41 | 47.31 | 52.71 |
| E2 (Fuxian lake, FX) | 21.39 | 19.27 | 25.92 | 33.42 | 47.31 | 52.69 |
| Huaihe River Basin | ||||||
| F1 (Hongze lake, HZ) | 21.42 | 19.20 | 25.99 | 33.40 | 47.41 | 52.60 |
| Total average | 21.43 | 19.22 | 26.04 | 33.32 | 47.47 | 52.54 |
| Population | H1 | H2 | H3 | H4 | H5 | H6 | H7 | H8 | H9 | H10 | H11 | H12 | H13 | N | Hd | s | k | π |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nenjiang River Basin | 40 | 0.724 | 4 | 1.186 | 0.0018 | |||||||||||||
| A1 (Huoshaohei lake) | 6 | 1 | 2 | 1 | 10 | 0.644 | 4 | 1.467 | 0.0022 | |||||||||
| A2 (Amuta lake) | 5 | 1 | 2 | 2 | 10 | 0.733 | 3 | 1.444 | 0.0022 | |||||||||
| A3 (Yamenqi lake) | 2 | 8 | 10 | 0.356 | 1 | 0.356 | 0.0006 | |||||||||||
| A4 (Longhupao lake) | 4 | 1 | 1 | 4 | 10 | 0.733 | 3 | 1.289 | 0.0020 | |||||||||
| Songhua River Basin | 31 | 0.753 | 4 | 1.290 | 0.0020 | |||||||||||||
| B1 (Xinlicheng reservoir) | 12 | 6 | 4 | 8 | 1 | 31 | 0.753 | 4 | 1.290 | 0.0020 | ||||||||
| Yellow River Basin | 19 | 0.754 | 5 | 1.684 | 0.0026 | |||||||||||||
| C1 (Xiaolangdi reservoir) | 8 | 1 | 1 | 10 | 0.533 | 4 | 0.956 | 0.0015 | ||||||||||
| C2 (Luhun reservoir) | 4 | 1 | 3 | 1 | 9 | 0.861 | 5 | 2.222 | 0.0034 | |||||||||
| Yangtze River Basin | 30 | 0.789 | 6 | 1.120 | 0.0017 | |||||||||||||
| D1 (Taihu lake) | 6 | 5 | 13 | 1 | 4 | 1 | 30 | 0.789 | 6 | 1.120 | 0.0017 | |||||||
| Lanchang River Basin | 47 | 0.826 | 6 | 1.576 | 0.0024 | |||||||||||||
| E1 (Chenghai) | 8 | 4 | 11 | 23 | 0.771 | 3 | 1.296 | 0.0020 | ||||||||||
| E2 (Fuxian lake) | 4 | 5 | 2 | 12 | 1 | 24 | 0.880 | 6 | 1.819 | 0.0028 | ||||||||
| Huaihe River Basin | 30 | 0.777 | 7 | 1.407 | 0.0022 | |||||||||||||
| F1 (Hongze lake) | 6 | 6 | 2 | 12 | 1 | 1 | 1 | 1 | 30 | 0.777 | 7 | 1.407 | 0.0022 | |||||
| Entire region (all samples) | 59 | 38 | 19 | 67 | 1 | 1 | 4 | 1 | 2 | 2 | 1 | 1 | 1 | 197 | 0.724 | 9 | 1.237 | 0.0022 |
| Populations | NjRB | ShRB | YeRB | YzRB | LcRB | HhRB | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| A1 (HSH) | A2 (AMT) | A3 (YMQ) | A4 (LHP) | B1 (XLC) | C1 (XLD) | C2 (LH) | D1 (TH) | E1 (CH) | E2 (FX) | F1 (HZ) | |
| Nenjiang River Basin | |||||||||||
| A1 (HSH) | 0.0021 | 0.0018 | 0.0020 | 0.0021 | 0.0018 | 0.0023 | 0.0026 | 0.0019 | 0.0022 | 0.0024 | |
| A2 (AMT) | −0.0862 | 0.0017 | 0.0020 | 0.0020 | 0.0018 | 0.0023 | 0.0024 | 0.0020 | 0.0020 | 0.0023 | |
| A3 (YMQ) | 0.2146 ** | 0.1818 ** | 0.0016 | 0.0016 | 0.0014 | 0.0021 | 0.0021 | 0.0014 | 0.0016 | 0.0018 | |
| A4 (LHP) | −0.0598 | −0.0847 | 0.2094 ** | 0.0019 | 0.0019 | 0.0022 | 0.0021 | 0.0017 | 0.0018 | 0.0020 | |
| Songhua River Basin | |||||||||||
| B1 (XLC) | −0.0285 | −0.0545 | 0.1278 * | −0.0633 | 0.0019 | 0.0022 | 0.0022 | 0.0018 | 0.0019 | 0.0021 | |
| C1 (XLD) | 0.0253 | 0.0678 * | 0.3687 ** | 0.1756 ** | 0.1446 * | 0.0023 | 0.0031 | 0.0018 | 0.0023 | 0.0024 | |
| C2 (LH) | −0.0702 | −0.0871 | 0.2454 ** | −0.0916 | −0.0472 | 0.1617 ** | 0.0024 | 0.0021 | 0.0022 | 0.0024 | |
| Yangtze River Basin | |||||||||||
| D1 (TH) | 0.3035 ** | 0.2458 ** | 0.4443 ** | 0.1610 ** | 0.1966 ** | 0.5387 ** | 0.1544 ** | 0.0019 | 0.0018 | 0.0021 | |
| Lanchang River Basin | |||||||||||
| E1 (CH) | 0.0074 | −0.0103 | 0.1920 ** | −0.0633 | −0.0187 | 0.2391 * | −0.0127 | 0.1908 ** | 0.0016 | 0.0019 | |
| E2 (FX) | 0.0869 * | 0.0399 | 0.2441 ** | −0.0321 | 0.0157 | 0.3401 ** | −0.0049 | 0.0718 * | −0.0052 | 0.0019 | |
| Huaihe River Basin | |||||||||||
| F1 (HZ) | 0.0524 * | 0.0155 | 0.1614 ** | −0.0391 | 0.0054 | 0.2584 ** | −0.0102 | 0.0948 * | −0.0137 | −0.0286 | |
| Source of Variation | d. f. | Sum of Squares | Variance Components | Percentage of Variation |
|---|---|---|---|---|
| Among population | 10 | 16.673 | 0.06215Va | 9.63 |
| Within population | 186 | 108.450 | 0.58307Vb | 90.37 |
| Total | 196 | 125.122 | 0.6452 | |
| Fixation | Fst | 0.6452 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, D.-A.; He, M.; Ren, Y.-F.; Luo, H.; Zhou, Y.-F.; Jiang, S.-L.; You, Y. Assessment of Genetic Diversity of the Salangid, Neosalanx taihuensis, Based on the Mitochondrial COI Gene in Different Chinese River Basins. Biology 2022, 11, 968. https://doi.org/10.3390/biology11070968
Fang D-A, He M, Ren Y-F, Luo H, Zhou Y-F, Jiang S-L, You Y. Assessment of Genetic Diversity of the Salangid, Neosalanx taihuensis, Based on the Mitochondrial COI Gene in Different Chinese River Basins. Biology. 2022; 11(7):968. https://doi.org/10.3390/biology11070968
Chicago/Turabian StyleFang, Di-An, Miao He, Ya-Fei Ren, Hui Luo, Yan-Feng Zhou, Shu-Lun Jiang, and Yang You. 2022. "Assessment of Genetic Diversity of the Salangid, Neosalanx taihuensis, Based on the Mitochondrial COI Gene in Different Chinese River Basins" Biology 11, no. 7: 968. https://doi.org/10.3390/biology11070968