
Gene Editing-Based Technologies for Beta-hemoglobinopathies Treatment

 , , ,
, , ,  and
and 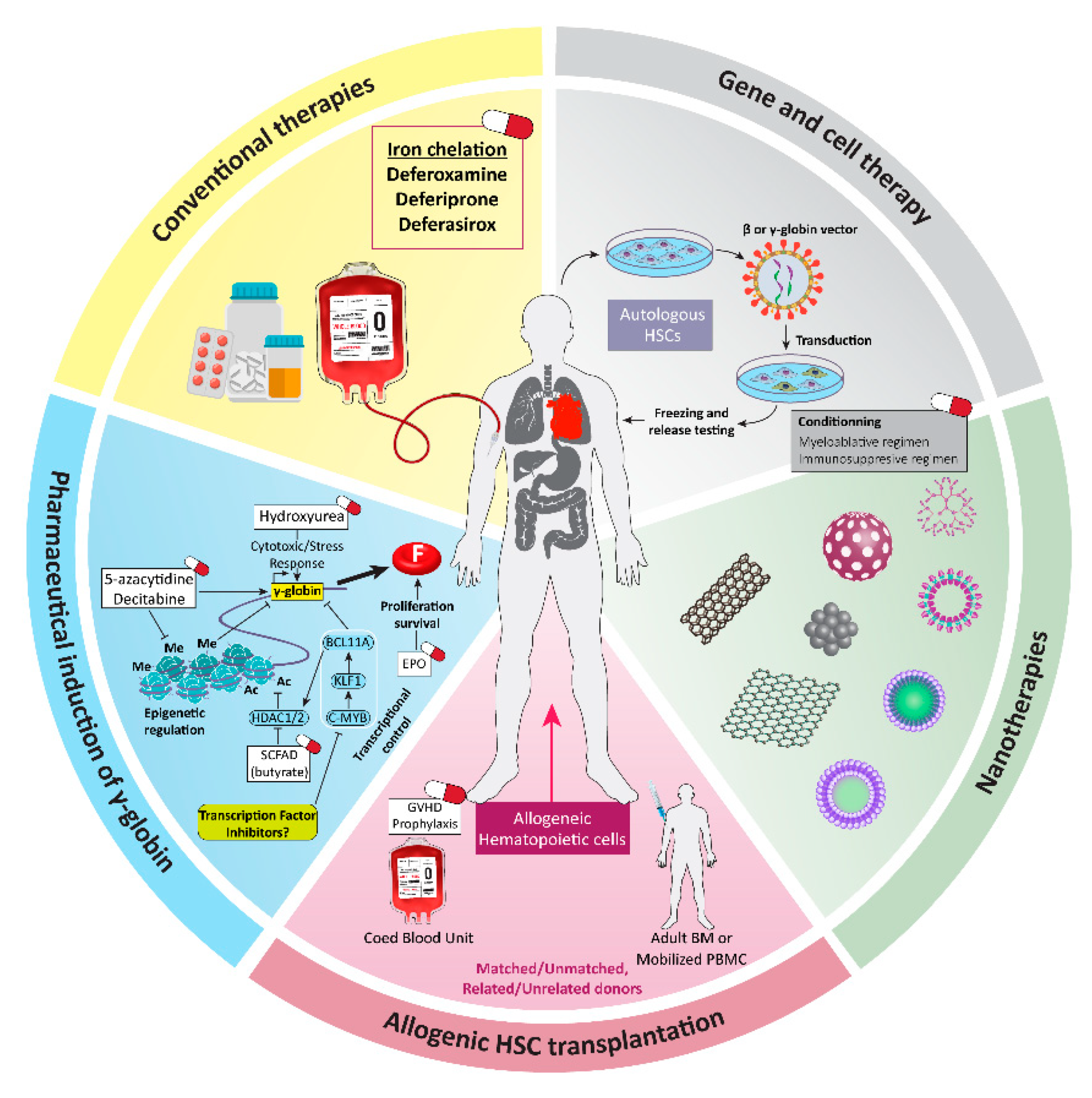{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
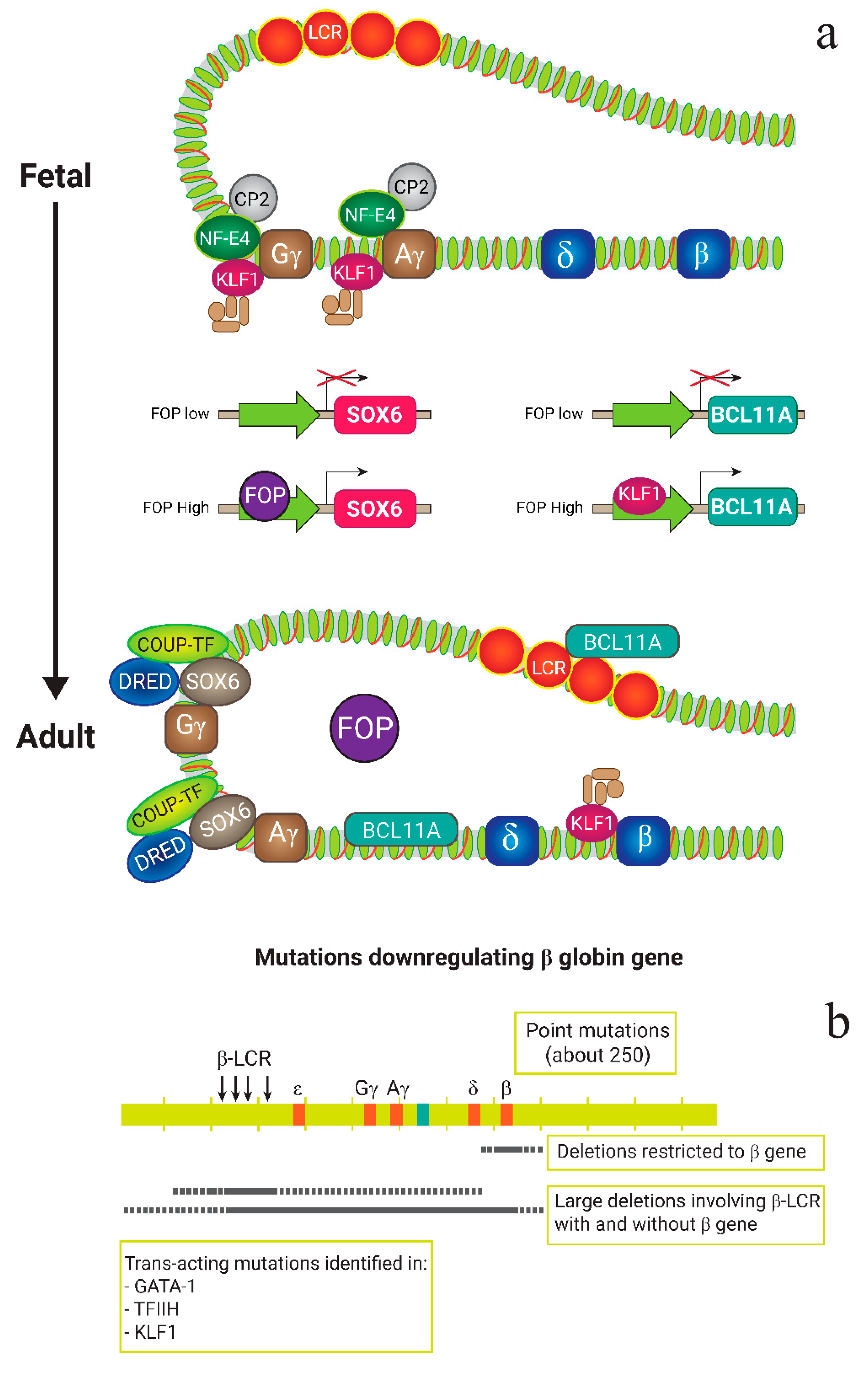
2. β-Thalassemia: Molecular Basics
3. Gene Editing Tools
3.1. Zinc Finger Nucleases
3.2. TALENs
3.3. CRISPR
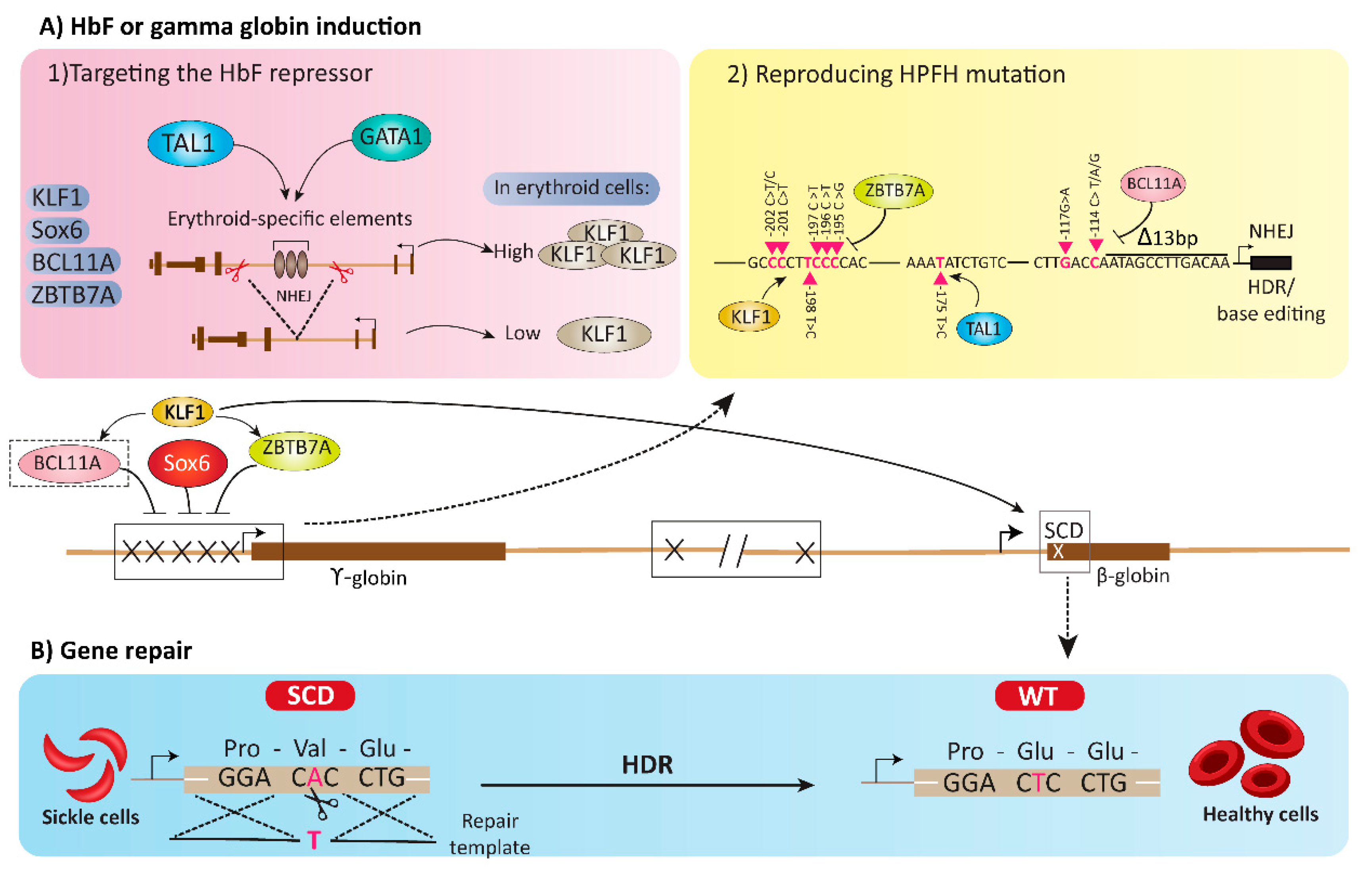
4. HbF or Gamma Globin Induction Using Gene Editing Tools
4.1. Targeting the HbF Repressors
4.1.1. BCL11A
4.1.2. SOX6
4.1.3. LRF/ZBTB7A
4.1.4. KLF1
4.2. Reproducing HPFH Mutations Recapitulates A Mutation Associated with A Benign Genetic Condition
5. Gene Repair Strategies
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BCL11A | BAF Chromatin Remodeling Complex Subunit BCL11A |
| BDDs | blood disease disorders |
| BMT | bone marrow transplantation |
| Cas | CRISPR-associated protein |
| CRISPR | clustered regularly interspaced short palindromic repeat |
| crRNA | sequence-specific CRISPR RNA |
| EPO | erythropoietin |
| FOG | Friend of GATA |
| FOP | Friend of Prmt1 |
| GATA-1 | GATA-binding factor 1 |
| GATA-2 | GATA-binding factor 2 |
| GVHD | graft versus host disease |
| GETs | gene editing tools |
| HbF | hemoglobin F |
| HDR | homology-directed repair |
| HLA | human leukocyte antigen |
| HPFH | hereditary persistence of fetal hemoglobin |
| HSCs | hematopoietic stem cells |
| IDLVs | integrase-defective lentiviruses |
| iPSCs | in pluripotent stem cells |
| KLF1 | Kruppel Like Factor 1 |
| NF-E2 | Nuclear Factor, Erythroid 2 |
| NHEJ | non-homologous end joining |
| rAAV6 | recombinant adeno-associated viral vectors serotype 6 |
| RBCs | red blood cells |
| SCD | sickle cell disease |
| SCL | Stem Cell Leukemia |
| SOX6 | SRY-Box Transcription Factor 6 |
| ssODN | single-stranded oligodeoxynucleotide |
| TALENs | transcription activator-like effector nucleases |
| tracrRNA | trans-activating crRNA |
| ZBTB7A | Zinc Finger and BTB Domain Containing 7A |
| ZFNs | zinc-finger nucleases |
References
- Mansilla-Soto, J.; Riviere, I.; Boulad, F.; Sadelain, M. Cell and Gene Therapy for the Beta-Thalassemias: Advances and Prospects. Hum. Gene Ther. 2016, 27, 295–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Dreuzy, E.; Bhukhai, K.; Leboulch, P.; Payen, E. Current and future alternative therapies for beta-thalassemia major. Biomed. J. 2016, 39, 24–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachmilewitz, E.A.; Giardina, P.J. How I treat thalassemia. Blood 2011, 118, 3479–3488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Origa, R. Beta-Thalassemia. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2022. [Google Scholar] [PubMed]
- Taishikhina, I.; Lokhmatova, M.; Shelikhova, L. Hematopoietic stem cell transplantation in patients with transfu-sion-dependent β-thalassemia. Review article. Pediatric Hematol. Oncol. Immunopathol. 2020, 19, 178–183. [Google Scholar] [CrossRef]
- Khandros, E.; Kwiatkowski, J.L. Beta thalassemia: monitoring and new treatment approaches. Hematol. Oncol. Clin. 2019, 33, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Thein, S.L. Molecular basis of β thalassemia and potential therapeutic targets. Blood Cells Mol. Dis. 2018, 70, 54–65. [Google Scholar] [CrossRef]
- Shah, F.T.; Sayani, F.; Trompeter, S.; Drasar, E.; Piga, A. Challenges of blood transfusions in β-thalassemia. Blood Rev. 2019, 37, 100588. [Google Scholar] [CrossRef]
- Sadeghi, M.M.; Shariati, L.; Hejazi, Z.; Shahbazi, M.; Tabatabaiefar, M.A.; Khanahmad, H. Inducing indel mutation in the SOX6 gene by zinc finger nuclease for gamma reactivation: An approach towards gene therapy of beta thalassemia. J. Cell. Biochem. 2017, 119, 2512–2519. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Delfi, M.; Hashemi, F.; Zabolian, A.; Saleki, H.; Bagherian, M.; Azami, N.; Farahani, M.V.; Sharifzadeh, S.O.; Hamzehlou, S.; et al. Biomedical application of chi-tosan-based nanoscale delivery systems: Potential usefulness in siRNA delivery for cancer therapy. Carbohydr. Polym. 2021, 260, 117809. [Google Scholar] [CrossRef]
- Kumar, K.S.; Girish, Y.R.; Ashrafizadeh, M.; Mirzaei, S.; Rakesh, K.P.; Gholami, M.H.; Zabolian, A.; Hushmandi, K.; Orive, G.; Kadumudi, F.B.; et al. AIE-featured tetraphenylethylene nanoarchitectures in biomedical application: Bioimaging, drug delivery and disease treatment. Coord. Chem. Rev. 2021, 447, 214135. [Google Scholar] [CrossRef]
- Mirzaei, S.; Gholami, M.H.; Hashemi, F.; Zabolian, A.; Farahani, M.V.; Hushmandi, K.; Zarrabi, A.; Goldman, A.; Ashrafizadeh, M.; Orive, G. Advances in understanding the role of P-gp in doxorubicin resistance: Molecular pathways, therapeutic strategies, and prospects. Drug Discov. Today 2022, 27, 436–455. [Google Scholar] [CrossRef] [PubMed]
- Keservani, R.; Sharma, A.K. Nanoconjugate Nanocarriers for Drug Delivery, 1st ed.; Apple Academic Press: Oakvil, ON, Canada, 2021. [Google Scholar]
- Liu, D.; Zhang, H.; Fontana, F.; Hirvonen, J.T.; Santos, H.A. Current developments and applications of microfluidic technology toward clinical translation of nanomedicines. Adv. Drug Deliv. Rev. 2018, 128, 54–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Wei, M.-Y.; Ma, Q. Nanomedicines: A Potential Treatment for Blood Disorder Diseases. Front. Bioeng. Biotechnol. 2019, 7, 369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, S.; Perche, F.; Pichon, C.; Cabral, H. Nanomedicine-Based Approaches for mRNA Delivery. Mol. Pharm. 2020, 17. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; del Pilar Rodriguez-Torres, M.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [Green Version]
- Kunz, J.B.; Kulozik, A.E. Gene Therapy of the Hemoglobinopathies. HemaSphere 2020, 4, e479. [Google Scholar] [CrossRef]
- Thein, S.L. The molecular basis of β-thalassemia. Cold Spring Harb. Perspect. Med. 2013, 3, a011700. [Google Scholar] [CrossRef] [Green Version]
- Tari, K.; Valizadeh Ardalan, P.; Abbaszadehdibavar, M.; Atashi, A.; Jalili, A.; Gheidishahran, M. Thalassemia an update: molecular basis, clinical features and treatment. Int. J. Biomed. Public Health 2018, 1, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Mettananda, S.; Higgs, D.R. Molecular Basis and Genetic Modifiers of Thalassemia. Hematol. Clin. N. Am. 2018, 32, 177–191. [Google Scholar] [CrossRef]
- McGann, P.T.; Nero, A.C.; Ware, R.E. Clinical features of β-thalassemia and sickle cell disease. In Gene and Cell Therapies for Beta-Globinopathies; Springer: New York, NY, USA, 2017; pp. 1–26. [Google Scholar]
- De Sanctis, V.; Kattamis, C.; Canatan, D.; Soliman, A.T.; Elsedfy, H.; Karimi, M.; Daar, S.; Wali, Y.; Yassin, M.; Soliman, N. β-thalassemia distribution in the old world: an ancient disease seen from a historical standpoint. Mediterr. J. Hematol. Infect. Dis. 2017, 9, e2017018. [Google Scholar] [CrossRef] [Green Version]
- Farashi, S.; Harteveld, C.L. Molecular basis of α-thalassemia. Blood Cells Mol. Dis. 2018, 70, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Shukla, P. Gene editing for cell engineering: trends and applications. Crit. Rev. Biotechnol. 2016, 37, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, B.; Hosseini, N.; Khanahmad, H.; Esfahani, B.N.; Bandehpour, M.; Shariati, L.; Zahedi, N. Targeting of cholera toxin A (ctxA) gene by zinc finger nuclease: pitfalls of using gene editing tools in prokaryotes. Res. Pharm. Sci. 2020, 15, 182–190. [Google Scholar] [CrossRef]
- Carroll, D. Genome engineering with zinc-finger nucleases. Genetics 2011, 188, 773–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, M.; Yang, Y. Targeted genome editing tools for disease modeling and gene therapy. Curr. Gene Ther. 2014, 14, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Ousterout, D.G.; Gersbach, C.A. The Development of TALE Nucleases for Biotechnology. TALENs 2016, 1338, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Zhao, H. Transcription activator-like effector nucleases (TALENs): A highly efficient and versatile tool for genome editing. Biotechnol. Bioeng. 2013, 110, 1811–1821. [Google Scholar] [CrossRef]
- Mohammadinejad, R.; Sassan, H.; Pardakhty, A.; Hashemabadi, M.; Ashrafizadeh, M.; Dehshahri, A.; Mandegary, A. ZEB1 and ZEB2 gene editing mediated by CRISPR/Cas9 in A549 cell line. Bratisl. Med. J. 2020, 121, 31–36. [Google Scholar] [CrossRef]
- Mohammadinejad, R.; Dehshahri, A.; Sassan, H.; Behnam, B.; Ashrafizadeh, M.; Gholami, A.S.; Pardakhty, A.; Mandegary, A. Preparation of carbon dot as a potential CRISPR/Cas9 plasmid delivery system for lung cancer cells. Minerva Biotecnol. 2020, 32, 106–113. [Google Scholar] [CrossRef]
- Barrangou, R.; Horvath, P. A decade of discovery: CRISPR functions and applications. Nat. Microbiol. 2017, 2, 17092. [Google Scholar] [CrossRef]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.M.; Musunuru, K. Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J. Clin. Investig. 2014, 124, 4154–4161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, G.; Tariq, M.A.; Shahid, K.; Ahmad, F.; Akram, J. Advances in genome editing: the technology of choice for precise and efficient β-thalassemia treatment. Gene Ther. 2020, 28, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Sripichai, O.; Fucharoen, S. Fetal hemoglobin regulation in β-thalassemia: heterogeneity, modifiers and therapeutic ap-proaches. Expert Rev. Hematol. 2016, 9, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Sankaran, V.G.; Cappellini, M.D.; Duca, L.; Nathan, D.G.; Taher, A.T. Fetal hemoglobin levels and morbidity in untransfused patients with β-thalassemia intermedia. Blood 2012, 119, 364–367. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, M.H. Targeting fetal hemoglobin expression to treat β hemoglobinopathies. Expert Opin. Ther. Targets 2022, 26, 347–359. [Google Scholar] [CrossRef]
- Demirci, S.; Leonard, A.; Tisdale, J.F. Genome editing strategies for fetal hemoglobin induction in beta-hemoglobinopathies. Hum. Mol. Genet. 2020, 29, R100–R106. [Google Scholar] [CrossRef]
- Cui, S.; Engle, J.D. Reactivation of fetal hemoglobin for treating β-thalassemia and sickle cell disease. In Gene and Cell Therapies for Beta-Globinopathies; Malik, P.T.J., Ed.; Springer: New York, NY, USA, 2017; Volume 1013, pp. 177–202. [Google Scholar]
- Rivers, A.; Molokie, R.; Lavelle, D. A new target for fetal hemoglobin reactivation. Haematologica 2019, 104, 2325–2327. [Google Scholar] [CrossRef] [Green Version]
- Topfer, S.K.; Feng, R.; Huang, P.; Ly, L.C.; Martyn, G.E.; Blobel, G.A.; Weiss, M.J.; Quinlan, K.G.R.; Crossley, M. Dis-rupting the adult globin promoter alleviates promoter competition and reactivates fetal globin gene expression. Blood J. Am. Soc. Hematol. 2022, 139, 2107–2118. [Google Scholar]
- Ravi, N.S.; Wienert, B.; Wyman, S.K.; Bell, H.W.; George, A.; Mahalingam, G.; Vu, J.T.; Prasad, K.; Bandlamudi, B.P.; Devaraju, N. Identification of novel HPFH-like mutations by CRISPR base editing that elevate the expression of fetal he-moglobin. Elife 2022, 11, e65421. [Google Scholar] [CrossRef]
- Weber, L.; Frati, G.; Felix, T.; Hardouin, G.; Casini, A.; Wollenschlaeger, C.; Meneghini, V.; Masson, C.; De Cian, A.; Chalumeau, A.; et al. Editing a γ-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci. Adv. 2020, 6, eaay9392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavazzana, M.; Mavilio, F. Gene Therapy for Hemoglobinopathies. Hum. Gene Ther. 2018, 29, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Uda, M.; Galanello, R.; Sanna, S.; Lettre, G.; Sankaran, V.G.; Chen, W.; Usala, G.; Busonero, F.; Maschio, A.; Albai, G. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phe-notype of β-thalassemia. Proc. Natl. Acad. Sci. USA 2008, 105, 1620–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Peng, C.; Sankaran, V.G.; Shao, Z.; Esrick, E.B.; Chong, B.G.; Ippolito, G.C.; Fujiwara, Y.; Ebert, B.L.; Tucker, P.W.; et al. Correction of Sickle Cell Disease in Adult Mice by Interference with Fetal Hemoglobin Silencing. Science 2011, 334, 993–996. [Google Scholar] [CrossRef] [Green Version]
- Basak, A.; Hancarova, M.; Ulirsch, J.C.; Balci, T.B.; Trkova, M.; Pelisek, M.; Vlckova, M.; Muzikova, K.; Cermak, J.; Trka, J.; et al. BCL11A deletions result in fetal hemoglobin persis-tence and neurodevelopmental alterations. J. Clin. Investig. 2015, 125, 2363–2368. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Keller, J.R.; Ortiz, M.; Tessarollo, L.; Rachel, R.A.; Nakamura, T.; Jenkins, N.A.; Copeland, N.G. Bcl11a is essential for normal lymphoid development. Nat. Immunol. 2003, 4, 525–532. [Google Scholar] [CrossRef]
- Sankaran, V.G.; Xu, J.; Ragoczy, T.; Ippolito, G.C.; Walkley, C.; Maika, S.D.; Fujiwara, Y.; Ito, M.; Groudine, M.; Bender, M.A.; et al. Developmental and species-divergent globin switching are driven by BCL11A. Nature 2009, 460, 1093–1097. [Google Scholar] [CrossRef] [Green Version]
- Tsang, J.C.H.; Yu, Y.; Burke, S.; Buettner, F.; Wang, C.; Kolodziejczyk, A.A.; Teichmann, S.A.; Lu, L.; Liu, P. Single-cell transcriptomic reconstruction reveals cell cycle and multi-lineage differentiation defects in Bcl11a-deficient hematopoietic stem cells. Genome Biol. 2015, 16, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Luc, S.; Huang, J.; McEldoon, J.L.; Somuncular, E.; Li, D.; Rhodes, C.; Mamoor, S.; Hou, S.; Xu, J.; Orkin, S.H. Bcl11a De-ficiency Leads to Hematopoietic Stem Cell Defects with an Aging-like Phenotype. Cell Rep. 2016, 16, 3181–3194. [Google Scholar] [CrossRef] [Green Version]
- Canver, M.C.; Smith, E.C.; Sher, F.; Pinello, L.; Sanjana, N.E.; Shalem, O.; Chen, D.D.; Schupp, P.G.; Vinjamur, D.S.; Garcia, S.P.; et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 2015, 527, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Vierstra, J.; Reik, A.; Chang, K.-H.; Stehling-Sun, S.; Zhou, Y.-Y.; Hinkley, S.J.; Paschon, D.E.; Zhang, L.; Psatha, N.; Bendana, Y.R.; et al. Functional footprinting of regulatory DNA. Nat. Methods 2015, 12, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Bauer, D.E.; Kamran, S.C.; Lessard, S.; Xu, J.; Fujiwara, Y.; Lin, C.; Shao, Z.; Canver, M.C.; Smith, E.C.; Pinello, L.; et al. An Erythroid Enhancer of BCL11A Subject to Genetic Variation Determines Fetal Hemoglobin Level. Science 2013, 342, 253–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.-H.; Smith, S.E.; Sullivan, T.; Chen, K.; Zhou, Q.; West, J.A.; Liu, M.; Liu, Y.; Vieira, B.F.; Sun, C.; et al. Long-Term Engraftment and Fetal Globin Induction upon BCL11A Gene Editing in Bone-Marrow-Derived CD34 + Hematopoietic Stem and Progenitor Cells. Mol. Ther. Methods Clin. Dev. 2017, 4, 137–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khosravi, M.A.; Abbasalipour, M.; Concordet, J.-P.; Berg, J.V.; Zeinali, S.; Arashkia, A.; Azadmanesh, K.; Buch, T.; Karimipoor, M. Targeted deletion of BCL11A gene by CRISPR-Cas9 system for fetal hemoglobin reactivation: A promising approach for gene therapy of beta thalassemia disease. Eur. J. Pharmacol. 2019, 854, 398–405. [Google Scholar] [CrossRef]
- Psatha, N.; Reik, A.; Phelps, S.; Zhou, Y.; Dalas, D.; Yannaki, E.; Levasseur, D.N.; Urnov, F.D.; Holmes, M.C.; Papayannopoulou, T. Disruption of the BCL11A Erythroid Enhancer Reactivates Fetal Hemoglobin in Erythroid Cells of Patients with β-Thalassemia Major. Mol. Ther. Methods Clin. Dev. 2018, 10, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and beta-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef]
- Ma, S.-P.; Gao, X.-X.; Zhou, G.-Q.; Zhang, H.-K.; Yang, J.-M.; Wang, W.-J.; Song, X.-M.; Chen, H.-Y.; Lu, D.-R. Reactivation of γ-globin expression using a minicircle DNA system to treat β-thalassemia. Gene 2022, 820, 146289. [Google Scholar] [CrossRef]
- Xu, J.; Sankaran, V.G.; Ni, M.; Menne, T.F.; Puram, R.V.; Kim, W.; Orkin, S.H. Transcriptional silencing of γ-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes Dev. 2010, 24, 783–798. [Google Scholar] [CrossRef] [Green Version]
- Yi, Z.; Cohen-Barak, O.; Hagiwara, N.; Kingsley, P.D.; Fuchs, D.A.; Erickson, D.T.; Epner, E.M.; Palis, J.; Brilliant, M.H. Sox6 directly silences epsilon globin expression in definitive erythropoiesis. PLoS Genet 2006, 2, e14. [Google Scholar] [CrossRef]
- Shariati, L.; Rohani, F.; Heidari Hafshejani, N.; Kouhpayeh, S.; Boshtam, M.; Mirian, M.; Rahimmanesh, I.; Hejazi, Z.; Mo-darres, M.; Pieper, I.L. Disruption of SOX6 gene using CRISPR/Cas9 technology for gamma-globin reactivation: An ap-proach towards gene therapy of β-thalassemia. J. Cell. Biochem. 2018, 119, 9357–9363. [Google Scholar] [CrossRef]
- Maeda, T.; Ito, K.; Merghoub, T.; Poliseno, L.; Hobbs, R.M.; Wang, G.; Dong, L.; Maeda, M.; Dore, L.C.; Zelent, A.; et al. LRF Is an Essential Downstream Target of GATA1 in Erythroid Development and Regulates BIM-Dependent Apoptosis. Dev. Cell 2009, 17, 527–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunardi, A.; Guarnerio, J.; Wang, G.; Maeda, T.; Pandolfi, P.P. Role of LRF/Pokemon in lineage fate decisions. Blood 2013, 121, 2845–2853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, T.; Wang, X.; Maeda, M.; Canver, M.C.; Sher, F.; Funnell, A.P.W.; Fisher, C.; Suciu, M.; Martyn, G.E.; Norton, L.J.; et al. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science 2016, 351, 285–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Liu, K.; Sun, C.-W.; Pawlik, K.M.; Townes, T.M. KLF1 regulates BCL11A expression and γ-to β-globin gene switching. Nat. Genet. 2010, 42, 742–744. [Google Scholar] [CrossRef]
- Shariati, L.; Khanahmad, H.; Salehi, M.; Hejazi, Z.; Rahimmanesh, I.; Tabatabaiefar, M.A.; Modarressi, M.H. Genetic dis-ruption of the KLF1 gene to overexpress the γ-globin gene using the CRISPR/Cas9 system. J. Gene Med. 2016, 18, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Lamsfus-Calle, A.; Daniel-Moreno, A.; Antony, J.S.; Epting, T.; Heumos, L.; Baskaran, P.; Admard, J.; Casadei, N.; Latifi, N.; Siegmund, D.M. Comparative targeting analysis of KLF1, BCL11A, and HBG1/2 in CD34+ HSPCs by CRISPR/Cas9 for the induction of fetal hemoglobin. Sci. Rep. 2020, 10, 10133. [Google Scholar] [CrossRef] [PubMed]
- Siatecka, M.; Bieker, J.J. The multifunctional role of EKLF/KLF1 during erythropoiesis. Blood 2011, 118, 2044–2054. [Google Scholar] [CrossRef] [Green Version]
- Arnaud, L.; Saison, C.; Helias, V.; Lucien, N.; Steschenko, D.; Giarratana, M.-C.; Prehu, C.; Foliguet, B.; Montout, L.; de Brevern, A.G.; et al. A Dominant Mutation in the Gene Encoding the Erythroid Transcription Factor KLF1 Causes a Congenital Dyserythropoietic Anemia. Am. J. Hum. Genet. 2010, 87, 721–727. [Google Scholar] [CrossRef] [Green Version]
- Wienert, B.; Martyn, G.E.; Funnell, A.P.W.; Quinlan, K.G.R.; Crossley, M. Wake-up Sleepy Gene: Reactivating Fetal Globin for beta-Hemoglobinopathies. Trends Genet. TIG 2018, 34, 927–940. [Google Scholar] [CrossRef]
- Forget, B.G. Molecular Basis of Hereditary Persistence of Fetal Hemoglobin. Ann. N. Y. Acad. Sci. 1998, 850, 38–44. [Google Scholar] [CrossRef]
- Wienert, B.; Martyn, G.; Kurita, R.; Nakamura, Y.; Quinlan, K.G.R.; Crossley, M. KLF1 drives the expression of fetal hemoglobin in British HPFH. Blood 2017, 130, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Wienert, B.; Funnell, A.P.W.; Norton, L.; Pearson, R.C.M.; Wilkinson-White, L.E.; Lester, K.; Vadolas, J.; Porteus, M.H.; Matthews, J.; Quinlan, K.; et al. Editing the genome to introduce a beneficial naturally occurring mutation associated with increased fetal globin. Nat. Commun. 2015, 6, 7085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Beshlawy, A.; Mostafa, A.; Youssry, I.; Gabr, H.; Mansour, I.M.; El-Tablawy, M.; Aziz, M.; Hussein, I.R. Correction of aberrant pre-mRNA splicing by antisense oligonucleotides in beta-thalassemia Egyptian patients with IVSI-110 mutation. J. Pediatr. Hematol. Oncol. 2008, 30, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Gabr, H.; El Ghamrawy, M.K.; Almaeen, A.H.; Abdelhafiz, A.S.; Hassan, A.O.S.; El Sissy, M.H. CRISPR-mediated gene modification of hematopoietic stem cells with beta-thalassemia IVS-1-110 mutation. Stem Cell Res. Ther. 2020, 11, 390. [Google Scholar] [CrossRef] [PubMed]
- Martyn, G.; Wienert, B.; Yang, L.; Shah, M.; Norton, L.J.; Burdach, J.; Kurita, R.; Nakamura, Y.; Pearson, R.C.M.; Funnell, A.P.W.; et al. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat. Genet. 2018, 50, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Giardine, B.; van Baal, S.; Kaimakis, P.; Riemer, C.; Miller, W.; Samara, M.; Kollia, P.; Anagnou, N.P.; Chui, D.H.; Wajcman, H.; et al. HbVar database of human hemoglobin variants and thalassemia mutations: 2007 update. Hum. Mutat. 2007, 28, 206. [Google Scholar] [CrossRef] [PubMed]
- Voit, R.A.; Hendel, A.; Pruett-Miller, S.M.; Porteus, M.H. Nuclease-mediated gene editing by homologous recombination of the human globin locus. Nucleic Acids Res. 2013, 42, 1365–1378. [Google Scholar] [CrossRef] [Green Version]
- Broeders, M.; Herrero-Hernandez, P.; Ernst, M.P.; van der Ploeg, A.T.; Pijnappel, W.P. Sharpening the Molecular Scissors: Advances in Gene-Editing Technology. iScience 2019, 23, 100789. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.-D.; Gao, F.; Liu, M.-J.; Fan, Q.-L.; Chen, D.-K.; Ma, W.-T. Methods for Enhancing Clustered Regularly Interspaced Short Palindromic Repeats/Cas9-Mediated Homology-Directed Repair Efficiency. Front. Genet. 2019, 10, 551. [Google Scholar] [CrossRef] [Green Version]
- Schiroli, G.; Conti, A.; Ferrari, S.; DELLA Volpe, L.; Jacob, A.; Albano, L.; Beretta, S.; Calabria, A.; Vavassori, V.; Gasparini, P.; et al. Precise Gene Editing Preserves Hematopoietic Stem Cell Function following Transient p53-Mediated DNA Damage Response. Cell Stem Cell 2019, 24, 551–565.e8. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Exline, C.M.; Declercq, J.J.; Llewellyn, G.N.; Hayward, S.B.; Li, P.W.-L.; Shivak, D.A.; Surosky, R.T.; Gregory, P.; Holmes, M.C.; et al. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat. Biotechnol. 2015, 33, 1256–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattabhi, S.; Lotti, S.N.; Berger, M.P.; Singh, S.; Lux, C.; Jacoby, K.; Lee, C.; Negre, O.; Scharenberg, A.M.; Rawlings, D.J. In Vivo Outcome of Homology-Directed Repair at the HBB Gene in HSC Using Alternative Donor Template Delivery Methods. Mol. Ther. Nucleic Acids 2019, 17, 277–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, Z.; Lomova, A.; Said, S.; Miggelbrink, A.; Kuo, C.Y.; Campo-Fernandez, B.; Hoban, M.D.; Masiuk, K.E.; Clark, D.N.; Long, J.; et al. Editing the Sickle Cell Disease Mutation in Human Hematopoietic Stem Cells: Comparison of Endonucleases and Homologous Donor Templates. Mol. Ther. 2019, 27, 1389–1406. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.M.; Ikeda, K.; Cromer, M.K.; Uchida, N.; Nishimura, T.; Romano, R.; Tong, A.J.; Lemgart, V.T.; Camarena, J.; Pavel-Dinu, M.; et al. Highly Efficient and Marker-free Genome Editing of Human Pluripotent Stem Cells by CRISPR-Cas9 RNP and AAV6 Donor-Mediated Homologous Recombination. Cell Stem Cell 2019, 24, 821–828.e5. [Google Scholar] [CrossRef]
- Park, S.; Gianotti-Sommer, A.; Molina-Estevez, F.J.; Vanuytsel, K.; Skvir, N.; Leung, A.; Rozelle, S.S.; Shaikho, E.; Weir, I.; Jiang, Z.; et al. A Comprehensive, Ethnically Diverse Library of Sickle Cell Disease-Specific Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 8, 1076–1085. [Google Scholar] [CrossRef] [Green Version]
- Hoban, M.D.; Cost, G.J.; Mendel, M.C.; Romero, Z.; Kaufman, M.L.; Joglekar, A.V.; Ho, M.; Lumaquin, D.; Gray, D.; Lill, G.R.; et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood 2015, 125, 2597–2604. [Google Scholar] [CrossRef]
- DeWitt, M.A.; Magis, W.; Bray, N.L.; Wang, T.; Berman, J.R.; Urbinati, F.; Heo, S.-J.; Mitros, T.; Muñoz, D.P.; Boffelli, D.; et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci. Transl. Med. 2016, 8, 360ra134. [Google Scholar] [CrossRef] [Green Version]
- Vakulskas, C.A.; Dever, D.P.; Rettig, G.R.; Turk, R.; Jacobi, A.M.; Collingwood, M.A.; Bode, N.M.; McNeill, M.S.; Yan, S.; Camarena, J.; et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018, 24, 1216–1224. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Lee, C.; Dever, D.P.; Davis, T.H.; Camarena, J.; Srifa, W.; Zhang, Y.; Paikari, A.; Chang, A.K.; Porteus, M.H.; et al. Highly efficient editing of the β-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res. 2019, 47, 7955–7972. [Google Scholar] [CrossRef]
- Magis, W.; DeWitt, M.A.; Wyman, S.K.; Vu, J.T.; Heo, S.-J.; Shao, S.J.; Hennig, F.; Romero, Z.G.; Campo-Fernandez, B.; Said, S.; et al. High-level correction of the sickle mutation is amplified in vivo during erythroid differentiation. iScience 2022, 25. [Google Scholar] [CrossRef]
- Xie, F.; Ye, L.; Chang, J.C.; Beyer, A.I.; Wang, J.; Muench, M.O.; Kan, Y.W. Seamless gene correction of β-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res. 2014, 24, 1526–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, B.; Fan, Y.; He, W.; Zhu, D.; Niu, X.; Wang, D.; Ou, Z.; Luo, M.; Sun, X. Improved hematopoietic differentiation effi-ciency of gene-corrected beta-thalassemia induced pluripotent stem cells by CRISPR/Cas9 system. Stem Cells Dev. 2015, 24, 1053–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Tong, Y.; Liu, X.-Z.; Wang, T.-T.; Cheng, L.; Wang, B.-Y.; Lv, X.; Huang, Y.; Liu, D.-P. Both TALENs and CRISPR/Cas9 directly target the HBB IVS2–654 (C > T) mutation in β-thalassemia-derived iPSCs. Sci. Rep. 2015, 5, srep12065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, X.; He, W.; Song, B.; Ou, Z.; Fan, D.; Chen, Y.; Fan, Y.; Sun, X. Combining Single Strand Oligodeoxynucleotides and CRISPR/Cas9 to Correct Gene Mutations in β-Thalassemia-induced Pluripotent Stem Cells. J. Biol. Chem. 2016, 291, 16576–16585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yang, Y.; Kang, X.; Lin, B.; Yu, Q.; Song, B.; Gao, G.; Chen, Y.; Sun, X.; Li, X.; et al. One-Step Biallelic and Scarless Correction of a beta-Thalassemia Mutation in Patient-Specific iPSCs without Drug Selection. Mol. Ther. Nucleic Acids. 2017, 6, 57–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wattanapanitch, M.; Damkham, N.; Potirat, P.; Trakarnsanga, K.; Janan, M.; Kheolamai, P.; Klincumhom, N.; Is-saragrisil, S. One-step genetic correction of hemoglobin E/beta-thalassemia patient-derived iPSCs by the CRISPR/Cas9 sys-tem. Stem Cell Res. Ther. 2018, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Bai, H.; Mahairaki, V.; Gao, Y.; He, C.; Wen, Y.; Jin, Y.-C.; Wang, Y.; Pan, R.L.; Qasba, A.; et al. A Universal Approach to Correct Various HBB Gene Mutations in Human Stem Cells for Gene Therapy of Beta-Thalassemia and Sickle Cell Disease. Stem Cells Transl. Med. 2017, 7, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Luk, K.; Yao, Q.; Shen, A.H.; Zeng, J.; Wu, Y.; Luo, H.-Y.; Brendel, C.; Pinello, L.; Chui, D.H.K.; et al. Editing aberrant splice sites efficiently restores β-globin expression in β-thalassemia. Blood 2019, 133, 2255–2262. [Google Scholar] [CrossRef]
- Patsali, P.; Turchiano, G.; Papasavva, P.; Romito, M.; Loucari, C.C.; Stephanou, C.; Christou, S.; Sitarou, M.; Mussolino, C.; Cornu, T.I.; et al. Correction of IVS I-110(G>A) β-thalassemia by CRISPR/Cas-and TALEN-mediated disruption of aberrant regulatory elements in human hematopoietic stem and progenitor cells. Haematologica 2019, 104, e497–e501. [Google Scholar] [CrossRef] [Green Version]
- Ma, N.; Liao, B.; Zhang, H.; Wang, L.; Shan, Y.; Xue, Y.; Huang, K.; Chen, S.; Zhou, X.; Chen, Y.; et al. Tran-scription activator-like effector nuclease (TALEN)-mediated gene correction in integration-free β-thalassemia induced plu-ripotent stem cells. J. Biol. Chem. 2013, 288, 34671–34679. [Google Scholar] [CrossRef] [Green Version]
- Cosenza, L.C.; Gasparello, J.; Romanini, N.; Zurlo, M.; Zuccato, C.; Gambari, R.; Finotti, A. Efficient CRISPR-Cas9-based genome editing of β-globin gene on erythroid cells from homozygous β039-thalassemia patients. Mol. Ther. Methods Clin. Dev. 2021, 21, 507–523. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahimmanesh, I.; Boshtam, M.; Kouhpayeh, S.; Khanahmad, H.; Dabiri, A.; Ahangarzadeh, S.; Esmaeili, Y.; Bidram, E.; Vaseghi, G.; Haghjooy Javanmard, S.; et al. Gene Editing-Based Technologies for Beta-hemoglobinopathies Treatment. Biology 2022, 11, 862. https://doi.org/10.3390/biology11060862
Rahimmanesh I, Boshtam M, Kouhpayeh S, Khanahmad H, Dabiri A, Ahangarzadeh S, Esmaeili Y, Bidram E, Vaseghi G, Haghjooy Javanmard S, et al. Gene Editing-Based Technologies for Beta-hemoglobinopathies Treatment. Biology. 2022; 11(6):862. https://doi.org/10.3390/biology11060862
Chicago/Turabian StyleRahimmanesh, Ilnaz, Maryam Boshtam, Shirin Kouhpayeh, Hossein Khanahmad, Arezou Dabiri, Shahrzad Ahangarzadeh, Yasaman Esmaeili, Elham Bidram, Golnaz Vaseghi, Shaghayegh Haghjooy Javanmard, and et al. 2022. "Gene Editing-Based Technologies for Beta-hemoglobinopathies Treatment" Biology 11, no. 6: 862. https://doi.org/10.3390/biology11060862