
Atrial Fibrillation: Focus on Myocardial Connexins and Gap Junctions


Abstract
:Simple Summary
Abstract
1. Introduction
2. The Genetics of AF
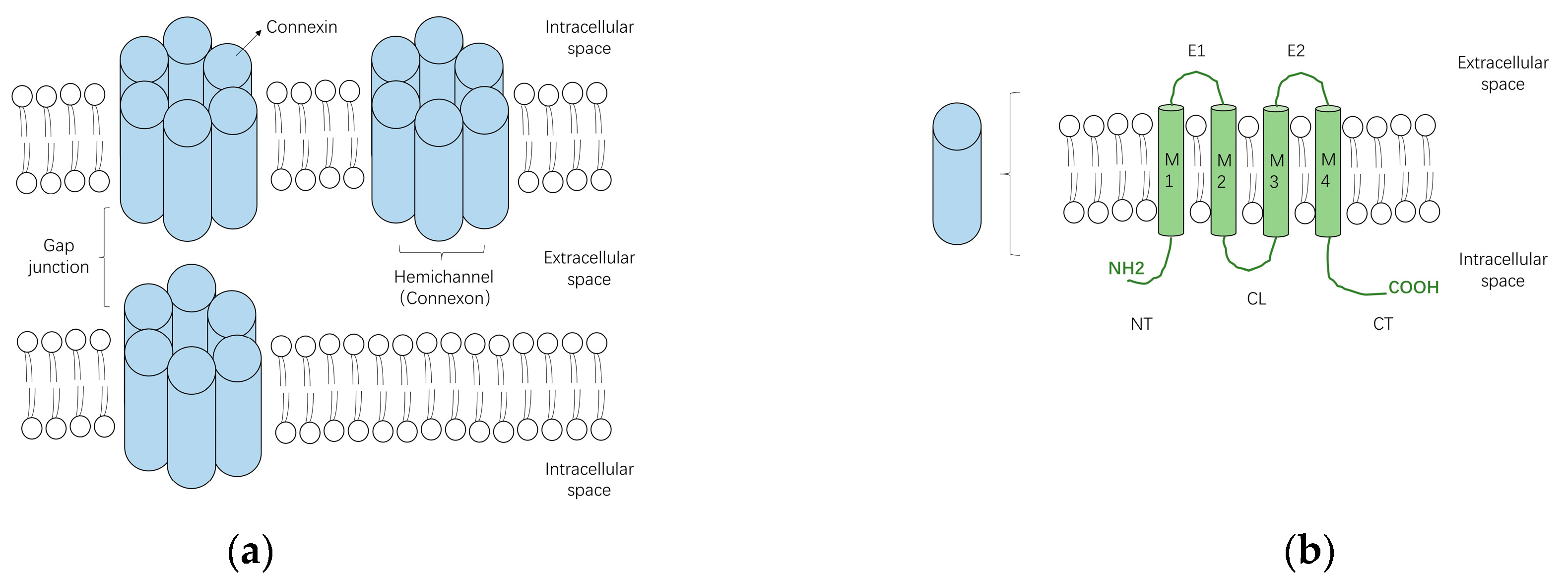
3. Structure and Function of Gap Junctions
4. Subtypes of Cardiac Connexins
5. Changes of Gap Junctions/Connexins in the Pathogenesis of AF
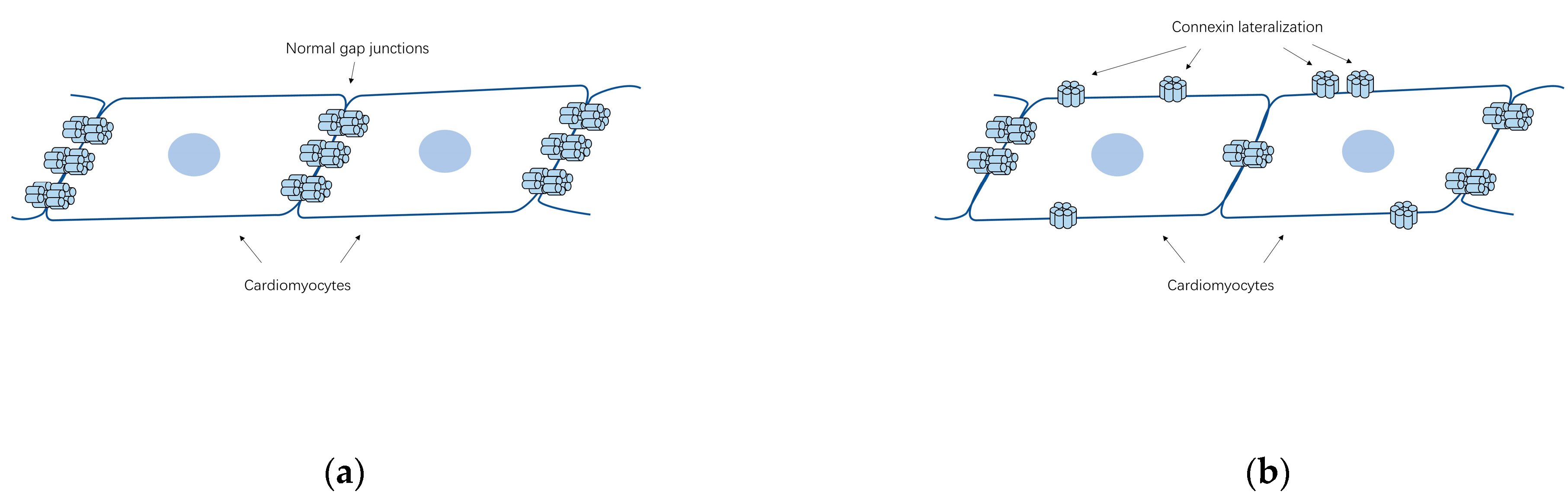
5.1. Gap Junction Remodeling
5.2. Abundance and Distribution of Connexins Associated with AF
5.3. Changes in Atrial Connexin Expression Regulated by Transcription Factors
6. Mutations of Connexins Associated with AF
6.1. Cx40 (GJA5) Mutations and AF
6.2. Cx43 (GJA1) Alterations Implicated in AF
6.3. Cx45 (GJC1) Mutations and Complex Arrhythmias
6.4. A GJA4 (Cx37) Polymorphism and Non-Structural AF
7. Clinical Implications
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef]
- Go, A.S.; Hylek, E.M.; Phillips, K.A.; Chang, Y.; Henault, L.E.; Selby, J.V.; Singer, D.E. Prevalence of diagnosed atrial fibrillation in adults: National implications for rhythm management and stroke prevention: The AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001, 285, 2370–2375. [Google Scholar] [CrossRef] [PubMed]
- Wijesurendra, R.S.; Casadei, B. Mechanisms of atrial fibrillation. Heart 2019, 105, 1860–1867. [Google Scholar] [CrossRef] [PubMed]
- Fuster, V.; Ryden, L.E.; Cannom, D.S.; Crijns, H.J.; Curtis, A.B.; Ellenbogen, K.A.; Halperin, J.L.; Kay, G.N.; Le Huezey, J.Y.; Lowe, J.E.; et al. American College of Cardiology Foundation/American Heart Association Task Force: 2011 ACCF/AHA/HRS focused updates incorporated into the ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrillation: A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2011, 123, e269–e367. [Google Scholar] [PubMed]
- Zimetbaum, P. Atrial Fibrillation. Ann. Intern. Med. 2017, 166, ITC33–ITC48. [Google Scholar] [CrossRef] [PubMed]
- Saffitz, J.E. Connexins, conduction, and atrial fibrillation. N. Engl. J. Med. 2006, 354, 2712–2714. [Google Scholar] [CrossRef]
- January, C.T.; Wann, L.S.; Alpert, J.S.; Calkins, H.; Cigarroa, J.E.; Cleveland, J.C., Jr.; Conti, J.B.; Ellinor, P.T.; Ezekowitz, M.D.; Field, M.E.; et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: A report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation 2014, 130, e199–e267. [Google Scholar] [CrossRef]
- Campuzano, O.; Perez-Serra, A.; Iglesias, A.; Brugada, R. Genetic basis of atrial fibrillation. Genes Dis. 2016, 3, 257–262. [Google Scholar] [CrossRef] [Green Version]
- Fox, C.S.; Parise, H.; D’Agostino, R.B.; Lloyd-Jones, D.M.; Vasan, R.S.; Wang, T.J.; Levy, D.; Wolf, P.A.; Benjamin, E.J. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. J. Am. Med. Assoc. 2004, 291, 2851–2855. [Google Scholar] [CrossRef]
- Guo, X.J.; Qiu, X.B.; Wang, J.; Guo, Y.H.; Yang, C.X.; Li, L.; Gao, R.F.; Ke, Z.P.; Di, R.M.; Sun, Y.M.; et al. PRRX1 Loss-of-Function Mutations Underlying Familial Atrial Fibrillation. J. Am. Heart Assoc. 2021, 10, e023517. [Google Scholar] [CrossRef]
- Severs, N.J.; Dupont, E.; Thomas, N.; Kaba, R.; Rothery, S.; Jain, R.; Sharpey, K.; Fry, C.H. Alterations in cardiac connexin expression in cardiomyopathies. Adv. Cardiol. 2006, 42, 228–242. [Google Scholar] [PubMed]
- Staerk, L.; Sherer, J.A.; Ko, D.; Benjamin, E.J.; Helm, R.H. Atrial Fibrillation: Epidemiology, Pathophysiology, and Clinical Outcomes. Circ. Res. 2017, 120, 1501–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gollob, M.H. Cardiac connexins as candidate genes for idiopathic atrial fibrillation. Curr. Opin. Cardiol. 2006, 21, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Duffy, H.S.; Wit, A.L. Is there a role for remodeled connexins in AF? No simple answers. J. Mol. Cell. Cardiol. 2008, 44, 4–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozano-Velasco, E.; Franco, D.; Aranega, A.; Daimi, H. Genetics and Epigenetics of Atrial Fibrillation. Int. J. Mol. Sci. 2020, 21, 5717. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, D.F.; Sun, Y.M.; Yang, Y.Q. A novel PITX2c loss-of-function mutation associated with familial atrial fibrillation. Eur. J. Med. Genet. 2014, 57, 25–31. [Google Scholar] [CrossRef]
- Guo, D.F.; Li, R.G.; Yuan, F.; Shi, H.Y.; Hou, X.M.; Qu, X.K.; Xu, Y.J.; Zhang, M.; Liu, X.; Jiang, J.Q.; et al. TBX5 loss-of-function mutation contributes to atrial fibrillation and atypical Holt-Oram syndrome. Mol. Med. Rep. 2016, 13, 4349–4356. [Google Scholar] [CrossRef]
- Li, N.; Wang, Z.S.; Wang, X.H.; Xu, Y.J.; Qiao, Q.; Li, X.M.; Di, R.M.; Guo, X.J.; Li, R.G.; Zhang, M.; et al. A SHOX2 loss-of-function mutation underlying familial atrial fibrillation. Int. J. Med. Sci. 2018, 15, 1564–1572. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.H.; Wang, X.H.; Xu, Y.J.; Gu, J.N.; Yang, C.X.; Qiao, Q.; Guo, X.J.; Guo, Y.H.; Qiu, X.B.; Jiang, W.F.; et al. ISL1 loss-of-function variation causes familial atrial fibrillation. Eur. J. Med. Genet. 2020, 63, 104029. [Google Scholar] [CrossRef]
- Li, N.; Xu, Y.J.; Shi, H.Y.; Yang, C.X.; Guo, Y.H.; Li, R.G.; Qiu, X.B.; Yang, Y.Q.; Zhang, M. KLF15 Loss-of-Function Mutation Underlying Atrial Fibrillation as well as Ventricular Arrhythmias and Cardiomyopathy. Genes 2021, 12, 408. [Google Scholar] [CrossRef]
- Cheng, C.; Liu, H.; Tan, C.; Tong, D.; Zhao, Y.; Liu, X.; Si, W.; Wang, L.; Liang, L.; Li, J.; et al. Mutation in NPPA causes atrial fibrillation by activating inflammation and cardiac fibrosis in a knock-in rat model. FASEB J. 2019, 33, 8878–8891. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Zhao, M.; Cheng, C.; Huang, Y.; Han, S.; Li, W.; Tu, X.; Luo, X.; Yu, X.; Liu, Y.; et al. Lamin A mutation impairs interaction with nucleoporin NUP155 and disrupts nucleocytoplasmic transport in atrial fibrillation. Hum. Mutat. 2019, 40, 310–325. [Google Scholar] [CrossRef] [PubMed]
- Goodenough, D.A.; Paul, D.L. Gap junctions. Cold Spring Harb. Perspect. Biol. 2009, 1, a002576. [Google Scholar] [CrossRef] [PubMed]
- Saez, J.C.; Leybaert, L. Hunting for connexin hemichannels. FEBS Lett. 2014, 588, 1205–1211. [Google Scholar] [CrossRef]
- Chaldoupi, S.M.; Loh, P.; Hauer, R.N.; de Bakker, J.M.; van Rijen, H.V. The role of connexin40 in atrial fibrillation. Cardiovasc. Res. 2009, 84, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.S.; Axelsen, L.N.; Sorgen, P.L.; Verma, V.; Delmar, M.; Holstein-Rathlou, N.H. Gap junctions. Compr. Physiol. 2012, 2, 1981–2035. [Google Scholar]
- Beyer, E.C.; Berthoud, V.M. Gap junction gene and protein families: Connexins, innexins, and pannexins. Biochim. Biophys. Acta Biomembr. 2018, 1860, 5–8. [Google Scholar] [CrossRef]
- Severs, N.J.; Bruce, A.F.; Dupont, E.; Rothery, S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc. Res. 2008, 80, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Ortega, A.; Tarazon, E.; Gil-Cayuela, C.; Garcia-Manzanares, M.; Martinez-Dolz, L.; Lago, F.; Gonzalez-Juanatey, J.R.; Cinca, J.; Jorge, E.; Portoles, M.; et al. Intercalated disc in failing hearts from patients with dilated cardiomyopathy: Its role in the depressed left ventricular function. PLoS ONE 2017, 12, e0185062. [Google Scholar] [CrossRef] [Green Version]
- Desplantez, T. Cardiac Cx43, Cx40 and Cx45 co-assembling: Involvement of connexins epitopes in formation of hemichannels and Gap junction channels. BMC Cell. Biol. 2017, 18, 3. [Google Scholar] [CrossRef] [Green Version]
- van der Velden, H.M.; Jongsma, H.J. Cardiac gap junctions and connexins: Their role in atrial fibrillation and potential as therapeutic targets. Cardiovasc. Res. 2002, 54, 270–279. [Google Scholar] [CrossRef]
- Severs, N.J.; Coppen, S.R.; Dupont, E.; Yeh, H.I.; Ko, Y.S.; Matsushita, T. Gap junction alterations in human cardiac disease. Cardiovasc. Res. 2004, 62, 368–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.H.; Shakespeare, T.; Mui, R.; White, T.W.; Valdimarsson, G. Connexin 48.5 is required for normal cardiovascular function and lens development in zebrafish embryos. J. Biol. Chem. 2004, 279, 36993–37003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reaume, A.G.; Desousa, P.A.; Kulkarni, S.; Langille, B.L.; Zhu, D.G.; Davies, T.C.; Juneja, S.C.; Kidder, G.M.; Rossant, J. Cardiac Malformation in Neonatal Mice Lacking Connexin43. Science 1995, 267, 1831–1834. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Smith, F.C.; Taffet, S.M.; Delmar, M. High incidence of cardiac malformations in connexin40-deficient mice. Circ. Res. 2003, 93, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Seki, A.; Ishikawa, T.; Daumy, X.; Mishima, H.; Barc, J.; Sasaki, R.; Nishii, K.; Saito, K.; Urano, M.; Ohno, S.; et al. Progressive Atrial Conduction Defects Associated With Bone Malformation Caused by a Connexin-45 Mutation. J. Am. Coll. Cardiol. 2017, 70, 358–370. [Google Scholar] [CrossRef]
- Munger, S.J.; Kanady, J.D.; Simon, A.M. Absence of venous valves in mice lacking Connexin37. Dev. Biol. 2013, 373, 338–348. [Google Scholar] [CrossRef]
- Kanady, J.D.; Munger, S.J.; Witte, M.H.; Simon, A.M. Combining Foxc2 and Connexin37 deletions in mice leads to severe defects in lymphatic vascular growth and remodeling. Dev. Biol. 2015, 405, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Martins-Marques, T.; Catarino, S.; Goncalves, A.; Miranda-Silva, D.; Goncalves, L.; Antunes, P.; Coutinho, G.; Moreira, A.L.; Pires, I.F.; Girao, H. EHD1 Modulates Cx43 Gap Junction Remodeling Associated With Cardiac Diseases. Circ. Res. 2020, 126, E97–E113. [Google Scholar] [CrossRef]
- Tribulova, N.; Knezl, V.; Okruhlicova, L.; Slezak, J. Myocardial Gap Junctions: Targets for Novel Approaches in the Prevention of Life-Threatening Cardiac Arrhythmias. Physiol Res 2008, 57, S1–S13. [Google Scholar] [CrossRef]
- Spach, M.S.; Starmer, C.F. Altering the Topology of Gap-Junctions—A Major Therapeutic Target for Atrial-Fibrillation. Cardiovasc. Res. 1995, 30, 337–344. [Google Scholar] [CrossRef]
- Kato, T.; Iwasaki, Y.K.; Nattel, S. Connexins and Atrial Fibrillation Filling in the Gaps. Circulation 2012, 125, 203–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wetzel, U.; Boldt, A.; Lauschke, J.; Weigl, J.; Schirdewahn, P.; Dorszewski, A.; Doll, N.; Hindricks, G.; Dhein, S.; Kottkamp, H. Expression of connexins 40 and 43 in human left atrium in atrial fibrillation of different aetiologies. Heart 2005, 91, 166–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polontchouk, L.; Haefliger, J.A.; Ebelt, B.; Schaefer, T.; Stuhlmann, D.; Mehlhorn, U.; Kuhn-Regnier, F.; De Vivie, E.R.; Dhein, S. Effects of chronic atrial fibrillation on gap junction distribution in human and rat atria. J. Am. Coll. Cardiol. 2001, 38, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Kanagaratnam, P.; Cherian, A.; Stanbridge, R.D.L.; Glenville, B.; Severs, N.J.; Peters, N.S. Relationship between connexins and atrial activation during human atrial fibrillation. J. Cardiovasc. Electr. 2004, 15, 206–213. [Google Scholar] [CrossRef]
- Kostin, S.; Klein, G.; Szalay, Z.; Hein, S.; Bauer, E.P.; Schaper, J. Structural correlate of atrial fibrillation in human patients. Cardiovasc. Res. 2002, 54, 361–379. [Google Scholar] [CrossRef] [Green Version]
- Dhein, S.; Rothe, S.; Busch, A.; Rojas Gomez, D.M.; Boldt, A.; Reutemann, A.; Seidel, T.; Salameh, A.; Pfannmuller, B.; Rastan, A.; et al. Effects of metoprolol therapy on cardiac gap junction remodelling and conduction in human chronic atrial fibrillation. Br. J. Pharmacol. 2011, 164, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Rucker-Martin, C.; Milliez, P.; Tan, S.; Decrouy, X.; Recouvreur, M.; Vranckx, R.; Delcayre, C.; Renaud, J.F.; Dunia, I.; Segretain, D.; et al. Chronic hemodynamic overload of the atria is an important factor for gap junction remodeling in human and rat hearts. Cardiovasc. Res. 2006, 72, 69–79. [Google Scholar] [CrossRef]
- Fakuade, F.E.; Tomsits, P.; Voigt, N. Connexin hemichannels in atrial fibrillation: Orphaned and irrelevant? Cardiovasc. Res. 2021, 117, 4–6. [Google Scholar] [CrossRef]
- Dhein, S.; Salameh, A. Remodeling of Cardiac Gap Junctional Cell-Cell Coupling. Cells 2021, 10, 2422. [Google Scholar] [CrossRef]
- Jungk, L.; Franke, H.; Salameh, A.; Dhein, S. Golgi Fragmentation in Human Patients with Chronic Atrial Fibrillation: A New Aspect of Remodeling. Thorac. Cardiov. Surg. 2019, 67, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Oyamada, M.; Takebe, K.; Oyamada, Y. Regulation of connexin expression by transcription factors and epigenetic mechanisms. Biochim. Biophys. Acta 2013, 1828, 118–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.F.; Yang, F.; Mahida, S.N.; Zhao, L.; Chen, X.; Zhang, M.L.; Sun, Z.; Yao, Y.; Zhang, Y.X.; Zheng, G.Y.; et al. TBX5 mutations contribute to early-onset atrial fibrillation in Chinese and Caucasians. Cardiovasc. Res. 2016, 109, 442–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mechakra, A.; Footz, T.; Walter, M.; Aranega, A.; Hernandez-Torres, F.; Morel, E.; Millat, G.; Yang, Y.Q.; Chahine, M.; Chevalier, P.; et al. A Novel PITX2c Gain-of-Function Mutation, p.Met207Val, in Patients With Familial Atrial Fibrillation. Am. J. Cardiol. 2019, 123, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Kong, W.; Zhang, Q.; Beyer, E.C.; Walcott, G.; Fast, V.G.; Ai, X. c-Jun N-terminal kinase activation contributes to reduced connexin43 and development of atrial arrhythmias. Cardiovasc. Res. 2013, 97, 589–597. [Google Scholar] [CrossRef] [Green Version]
- Wijffels, M.C.E.F.; Kirchhof, C.J.H.J.; Dorland, R.; Allessie, M.A. Atrial-Fibrillation Begets Atrial-Fibrillation—A Study in Awake Chronically Instrumented Goats. Circulation 1995, 92, 1954–1968. [Google Scholar] [CrossRef]
- Rohr, S. Role of gap junctions in the propagation of the cardiac action potential. Cardiovasc. Res. 2004, 62, 309–322. [Google Scholar] [CrossRef]
- Kleber, A.G.; Rudy, Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol. Rev. 2004, 84, 431–488. [Google Scholar] [CrossRef] [Green Version]
- Molica, F.; Merlijn, J.P.; Morel, S.; Kwak, B.R. Mutations in cardiovascular connexin genes. Biol. Cell. 2014, 106, 269–293. [Google Scholar] [CrossRef]
- Kanagaratnam, P.; Rothery, S.; Patel, P.; Severs, N.J.; Peters, N.S. Relative expression of immunolocalized connexins 40 and 43 correlates with human atrial conduction properties. J. Am. Coll. Cardiol. 2002, 39, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Valiunas, V.; Gemel, J.; Brink, P.R.; Beyer, E.C. Gap junction channels formed by coexpressed connexin40 and connexin43. Am. J. Physiol.-Heart Circ. Physiol. 2001, 281, H1675–H1689. [Google Scholar] [CrossRef] [PubMed]
- Gemel, J.; Levy, A.E.; Simon, A.R.; Bennett, K.B.; Ai, X.; Akhter, S.; Beyer, E.C. Connexin40 abnormalities and atrial fibrillation in the human heart. J. Mol. Cell. Cardiol. 2014, 76, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Gutstein, D.E.; Morley, G.E.; Vaidya, D.; Liu, F.Y.; Chen, F.L.; Stuhlmann, H.; Fishman, G.I. Heterogeneous expression of gap junction channels in the heart leads to conduction defects and ventricular dysfunction. Circulation 2001, 104, 1194–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firouzi, M.; Ramanna, H.; Kok, B.; Jongsma, H.J.; Koeleman, B.P.C.; Doevendans, P.A.; Groenewegen, W.A.; Hauer, R.N.W. Association of human connexin40 gene polymorphisms with atrial vulnerability as a risk factor for idiopathic atrial fibrillation. Circ. Res. 2004, 95, 29–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juang, J.M.; Chern, Y.R.; Tsai, C.T.; Chiang, F.T.; Lin, J.L.; Hwang, J.J.; Hsu, K.L.; Tseng, C.D.; Tseng, Y.Z.; Lai, L.P. The association of human connexin 40 genetic polymorphisms with atrial fibrillation. Int. J. Cardiol. 2007, 116, 107–112. [Google Scholar] [CrossRef]
- Wirka, R.C.; Gore, S.; Van Wagoner, D.R.; Arking, D.E.; Lubitz, S.A.; Lunetta, K.L.; Benjamin, E.J.; Alonso, A.; Ellinor, P.T.; Barnard, J.; et al. A Common Connexin-40 Gene Promoter Variant Affects Connexin-40 Expression in Human Atria and Is Associated With Atrial Fibrillation. Circ. Arrhythmia Electrophysiol. 2011, 4, 87–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christophersen, I.E.; Holmegard, H.N.; Jabbari, J.; Haunso, S.; Tveit, A.; Svendsen, J.H.; Olesen, M.S. Rare Variants in GJA5 Are Associated With Early-Onset Lone Atrial Fibrillation. Can. J. Cardiol. 2013, 29, 111–116. [Google Scholar] [CrossRef]
- Carballo, S.; Pfenniger, A.; Carballo, D.; Garin, N.; James, R.W.; Mach, F.; Shah, D.; Kwak, B.R. Differential Association of Cx37 and Cx40 Genetic Variants in Atrial Fibrillation with and without Underlying Structural Heart Disease. Int. J. Mol. Sci. 2018, 19, 295. [Google Scholar] [CrossRef] [Green Version]
- Gollob, M.H.; Jones, D.L.; Krahn, A.D.; Danis, L.; Gong, X.Q.; Shao, Q.; Liu, X.Q.; Veinot, J.P.; Tang, A.S.L.; Stewart, A.F.R.; et al. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N. Engl. J. Med. 2006, 354, 2677–2688. [Google Scholar] [CrossRef]
- Lubkemeier, I.; Andrie, R.; Lickfett, L.; Bosen, F.; Stockigt, F.; Dobrowolski, R.; Draffehn, A.M.; Fregeac, J.; Schultze, J.L.; Bukauskas, F.F.; et al. The Connexin40A96S mutation from a patient with atrial fibrillation causes decreased atrial conduction velocities and sustained episodes of induced atrial fibrillation in mice. J. Mol. Cell. Cardiol. 2013, 65, 19–32. [Google Scholar] [CrossRef]
- Yang, Y.Q.; Zhang, X.L.; Wang, X.H.; Tan, H.W.; Shi, H.F.; Jiang, W.F.; Fang, W.Y.; Liu, X. Connexin40 nonsense mutation in familial atrial fibrillation. Int. J. Mol. Med. 2010, 26, 605–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.G.; Tong, X.L.; Chen, H.H.; Huang, T.; Shao, Q.; Huang, W.X.; Laird, D.W.; Bai, D.L. An atrial-fibrillation-linked connexin40 mutant is retained in the endoplasmic reticulum and impairs the function of atrial gap-junction channels. Dis. Model. Mech. 2014, 7, 561–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.Q.; Liu, X.; Zhang, X.L.; Wang, X.H.; Tan, H.W.; Shi, H.F.; Jiang, W.F.; Fang, W.Y. Novel connexin40 missense mutations in patients with familial atrial fibrillation. Europace 2010, 12, 1421–1427. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.G.; Yang, Y.Q.; Gong, X.Q.; Wang, X.H.; Li, R.G.; Tan, H.W.; Liu, X.; Fang, W.Y.; Bai, D.L. Novel Germline GJA5/Connexin40 Mutations Associated with Lone Atrial Fibrillation Impair Gap Junctional Intercellular Communication. Hum. Mutat. 2013, 34, 603–609. [Google Scholar] [PubMed]
- Sun, Y.G.; Hills, M.D.; Ye, W.G.; Tong, X.L.; Bai, D.L. Atrial Fibrillation-Linked Germline GJA5/Connexin40 Mutants Showed an Increased Hemichannel Function. PLoS ONE 2014, 9, e95125. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.F.; Yang, J.F.; Wang, Q.; Li, R.G.; Xu, Y.J.; Qu, X.K.; Fang, W.Y.; Liu, X.; Yang, Y.Q. Prevalence and spectrum of GJA5 mutations associated with lone atrial fibrillation. Mol. Med. Rep. 2013, 7, 767–774. [Google Scholar] [CrossRef] [Green Version]
- Noureldin, M.; Chen, H.H.; Bai, D.L. Functional Characterization of Novel Atrial Fibrillation-Linked GJA5 (Cx40) Mutants. Int. J. Mol. Sci. 2018, 19, 977. [Google Scholar] [CrossRef] [Green Version]
- Rudy, Y. Molecular basis of cardiac action potential repolarization. Ann. N. Y. Acad. Sci. 2008, 1123, 113–118. [Google Scholar] [CrossRef]
- Dale, N. Dynamic ATP signalling and neural development. J. Physiol. 2008, 586, 2429–2436. [Google Scholar] [CrossRef]
- Paznekas, W.A.; Boyadjiev, S.A.; Shapiro, R.E.; Daniels, O.; Wollnik, B.; Keegan, C.E.; Innis, J.W.; Dinulos, M.B.; Christian, C.; Hannibal, M.C.; et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am. J. Hum. Genet. 2003, 72, 408–418. [Google Scholar] [CrossRef] [Green Version]
- Kalcheva, N.; Qu, J.X.; Sandeep, N.; Garcia, L.; Zhang, J.; Wang, Z.Y.; Lampe, P.D.; Suadicani, S.O.; Spray, D.C.; Fishman, G.I. Gap junction remodeling and cardiac arrhythmogenesis in a murine model of oculodentodigital dysplasia. Proc. Natl. Acad. Sci. USA 2007, 104, 20512–20516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrowolski, R.; Sasse, P.; Schrickel, J.W.; Watkins, M.; Kim, J.S.; Rackauskas, M.; Troatz, C.; Ghanem, A.; Tiemann, K.; Degen, J.; et al. The conditional connexin43G138R mouse mutant represents a new model of hereditary oculodentodigital dysplasia in humans. Hum. Mol. Genet. 2008, 17, 539–554. [Google Scholar] [CrossRef] [PubMed]
- Delmar, M.; Makita, N. Cardiac connexins, mutations and arrhythmias. Curr. Opin. Cardiol. 2012, 27, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Thibodeau, I.L.; Xu, J.; Li, Q.J.; Liu, G.L.; Lam, K.; Veinot, J.P.; Birnie, D.H.; Jones, D.L.; Krahn, A.D.; Lemery, R.; et al. Paradigm of Genetic Mosaicism and Lone Atrial Fibrillation Physiological Characterization of a Connexin 43-Deletion Mutant Identified From Atrial Tissue. Circulation 2010, 122, 236–244. [Google Scholar] [CrossRef] [Green Version]
- Kruger, O.; Plum, A.; Kim, J.S.; Winterhager, E.; Maxeiner, S.; Hallas, G.; Kirchhoff, S.; Traub, O.; Lamers, W.H.; Willecke, K. Defective vascular development in connexin 45-deficient mice. Development 2000, 127, 4179–4193. [Google Scholar] [CrossRef]
- Kumai, M.; Nishii, K.; Nakamura, K.; Takeda, N.; Suzuki, M.; Shibata, Y. Loss of connexin45 causes a cushion defect in early cardiogenesis. Development 2000, 127, 3501–3512. [Google Scholar] [CrossRef]
- Frank, M.; Wirth, A.; Andrie, R.P.; Kreuzberg, M.M.; Dobrowolski, R.; Seifert, G.; Offermanns, S.; Nickenig, G.; Willecke, K.; Schrickel, J.W. Connexin45 Provides Optimal Atrioventricular Nodal Conduction in the Adult Mouse Heart. Circ. Res. 2012, 111, 1528–1538. [Google Scholar] [CrossRef] [Green Version]
- Rackauskas, M.; Kreuzberg, M.M.; Pranevicius, M.; Willecke, K.; Verselis, V.K.; Bukauskas, F.F. Gating properties of heterotypic gap junction channels formed of connexins 40, 43, 45. Biophys. J. 2007, 92, 1952–1965. [Google Scholar] [CrossRef] [Green Version]
- Li, R.G.; Xu, Y.J.; Ye, W.G.; Li, Y.J.; Chen, H.H.; Qiu, X.B.; Yang, Y.Q.; Bai, D.L. Connexin45 (GJC1) loss-of-function mutation contributes to familial atrial fibrillation and conduction disease. Heart Rhythm. 2021, 18, 684–693. [Google Scholar] [CrossRef]
- Richard, G.; Lin, J.P.; Smith, L.; Whyte, Y.M.; Itin, P.; Wollina, U.; Epstein, E.; Hohl, D.; Giroux, J.M.; Charnas, L.; et al. Linkage studies in erythrokeratodermias: Fine mapping, genetic heterogeneity, and analysis of candidate genes. J. Investig. Dermatol. 1997, 109, 666–671. [Google Scholar] [CrossRef] [Green Version]
- Derouette, J.P.; Desplantez, T.; Wong, C.W.; Roth, I.; Kwak, B.R.; Weingart, R. Functional differences between human Cx37 polymorphic hemichannels. J. Mol. Cell. Cardiol. 2009, 46, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.S.; Varadaraj, K.; Valiunas, V.; Ramanan, S.V.; Christensen, E.A.; Beyer, E.C.; Brink, P.R. Functional expression and biophysical properties of polymorphic variants of the human gap junction protein connexin37. Biochem. Bioph. Res. Commun. 2000, 274, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.W.; Christen, T.; Pfenniger, A.; James, R.W.; Kwak, B.R. Do allelic variants of the connexin37 1019 gene polymorphism differentially predict for coronary artery disease and myocardial infarction? Atherosclerosis 2007, 191, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Katakami, N.; Sakamoto, K.; Kaneto, H.; Matsuhisa, M.; Shimizu, I.; Ishibashi, F.; Osonoi, T.; Kashiwagi, A.; Kawamori, R.; Hori, M.; et al. Association between the connexin37 polymorphism and peripheral arterial disease in subjects with type 2 diabetes. Diabetes Care 2009, 32, e53–e54. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.X.; Yang, Z.Y.; Wang, R.X.; Yang, Y.; Cao, H.M.; Zhang, T. Association between C1019T polymorphism of the connexin37 gene and coronary heart disease in patients with in-stent restenosis. Exp. Ther. Med. 2013, 5, 539–544. [Google Scholar] [CrossRef]
- Listi, F.; Candore, G.; Balistreri, C.R.; Caruso, M.; Incalcaterra, E.; Hoffmann, E.; Lio, D.; Caruso, C. Connexin37 1019 gene polymorphism in myocardial infarction patients and centenarians. Atherosclerosis 2007, 191, 460–461. [Google Scholar] [CrossRef] [Green Version]
- Leu, H.B.; Chung, C.M.; Chuang, S.Y.; Bai, C.H.; Chen, J.R.; Chen, J.W.; Pan, W.H. Genetic variants of connexin37 are associated with carotid intima-medial thickness and future onset of ischemic stroke. Atherosclerosis 2011, 214, 101–106. [Google Scholar] [CrossRef]
- Wong, C.W.; Christen, T.; Roth, I.; Chadjichristos, C.E.; Derouette, J.P.; Foglia, B.F.; Chanson, M.; Goodenough, D.A.; Kwak, B.R. Connexin37 protects against atherosclerosis by regulating monocyte adhesion. Nat. Med. 2006, 12, 950–954. [Google Scholar] [CrossRef]
- Wen, D.; Du, X.; Nie, S.P.; Dong, J.Z.; Ma, C.S. Association of Connexin37 C1019T with myocardial infarction and coronary artery disease: A meta-analysis. Exp. Gerontol. 2014, 58, 203–207. [Google Scholar] [CrossRef]
- Zhao, L.; Li, Y.; Wu, D.; Ma, T.; Xia, S.Y.; Liu, Z. Cx37 C1019T Polymorphism May Contribute to the Pathogenesis of Coronary Heart Disease. Genet. Test. Mol. Biomark. 2014, 18, 497–504. [Google Scholar] [CrossRef]
- Kjolbye, A.L.; Knudsen, C.B.; Jepsen, T.; Larsen, B.D.; Petersen, J.S. Pharmacological characterization of the new stable antiarrhythmic peptide analog Ac-D-Tyr-D-Pro-D-Hyp-Gly-D-Ala-Gly-NH2 (ZP123): In vivo and in vitro studies. J. Pharmacol. Exp. Ther. 2003, 306, 1191–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiroshita-Takeshita, A.; Sakabe, M.; Haugan, K.; Hennan, J.K.; Nattel, S. Model-dependent effects of the gap junction conduction-enhancing antiarrhythmic peptide rotigaptide (ZP123) on experimental atrial fibrillation in dogs. Circulation 2007, 115, 310–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghazizadeh, Z.; Kiviniemi, T.; Olafsson, S.; Plotnick, D.; Beerens, M.E.; Zhang, K.; Gillon, L.; Steinbaugh, M.J.; Barrera, V.; Sui, S.H.; et al. Metastable Atrial State Underlies the Primary Genetic Substrate for MYL4 Mutation-Associated Atrial Fibrillation. Circulation 2020, 141, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Orr, N.; Arnaout, R.; Gula, L.J.; Spears, D.A.; Leong-Sit, P.; Li, Q.J.; Tarhuni, W.; Reischauer, S.; Chauhan, V.S.; Borkovich, M.; et al. A mutation in the atrial-specific myosin light chain gene (MYL4) causes familial atrial fibrillation. Nat. Commun. 2016, 7, 11303. [Google Scholar] [CrossRef] [PubMed]
- O’Quinn, M.P.; Palatinus, J.A.; Harris, B.S.; Hewett, K.W.; Gourdie, R.G. A Peptide Mimetic of the Connexin43 Carboxyl Terminus Reduces Gap Junction Remodeling and Induced Arrhythmia Following Ventricular Injury. Circ. Res. 2011, 108, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Salameh, A.; Apel, D.; Casanova, J.G.; von Salisch, S.; Mohr, F.W.; Daehnert, I.; Dhein, S. On the different roles of AT(1) and AT(2) receptors in stretch-induced changes of connexin43 expression and localisation. Pflügers Arch. Eur. J. Physiol. 2012, 464, 535–547. [Google Scholar] [CrossRef]
- Yamazaki, T.; Yazaki, Y. Role of tissue angiotensin II in myocardial remodelling induced by mechanical stress. J. Hum. Hypertens. 1999, 13, S43–S47. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Hou, M.C.; Li, J.J.; Qi, Y.; Zhang, Y.; She, G.; Ren, Y.J.; Wu, W.; Pang, Z.D.; Xie, W.; et al. Cardiac beta-adrenergic receptor activation mediates distinct and cell type-dependent changes in the expression and distribution of connexin 43. J. Cell. Mol. Med. 2020, 24, 8505–8517. [Google Scholar] [CrossRef]
- Lissoni, A.; Hulpiau, P.; Martins-Marques, T.; Wang, N.; Bultynck, G.; Schulz, R.; Witschas, K.; Girao, H.; De Smet, M.; Leybaert, L. RyR2 regulates Cx43 hemichannel intracellular Ca2+-dependent activation in cardiomyocytes. Cardiovasc. Res. 2021, 117, 123–136. [Google Scholar] [CrossRef]
- Igarashi, T.; Finet, J.E.; Takeuchi, A.; Fujino, Y.; Strom, M.; Greener, I.D.; Rosenbaum, D.S.; Donahue, J.K. Connexin gene transfer preserves conduction velocity and prevents atrial fibrillation. Circulation 2012, 125, 216–225. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| AF-Linked Mutations | Gap Junction Function | Hemichannel Function | |||
|---|---|---|---|---|---|
| Mutant Alone | Mutant on Wild-Type Cx40 | Mutant on Wild-Type Cx43 | |||
| Cx43 | c.932delC | eliminated | reduced | reduced | not tested |
| Cx40 | G38D | reduced | not tested | not tested | not tested |
| P88S | eliminated | reduced | reduced | not tested | |
| A96S | reduced | reduced | reduced | not tested | |
| M163V | normal | not tested | not tested | not tested | |
| Q49X | eliminated | reduced | reduced | normal | |
| V85I | normal | not tested | normal | enhanced | |
| L221I | normal | not tested | normal | enhanced | |
| L229M | normal | normal | reduced | normal | |
| I75F | eliminated | reduced | reduced | normal | |
| K107R | normal | normal | normal | normal | |
| L223M | normal | normal | normal | normal | |
| Q236H | reduced | not tested | reduced | not tested | |
| I257L | normal | normal | normal | normal | |
| Cx45 | M235L | reduced | reduced | reduced | not tested |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.-H.; Yang, Y.-Q. Atrial Fibrillation: Focus on Myocardial Connexins and Gap Junctions. Biology 2022, 11, 489. https://doi.org/10.3390/biology11040489
Guo Y-H, Yang Y-Q. Atrial Fibrillation: Focus on Myocardial Connexins and Gap Junctions. Biology. 2022; 11(4):489. https://doi.org/10.3390/biology11040489
Chicago/Turabian StyleGuo, Yu-Han, and Yi-Qing Yang. 2022. "Atrial Fibrillation: Focus on Myocardial Connexins and Gap Junctions" Biology 11, no. 4: 489. https://doi.org/10.3390/biology11040489