
Age Estimate of GJB2-p.(Arg143Trp) Founder Variant in Hearing Impairment in Ghana, Suggests Multiple Independent Origins across Populations

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. The Study Participants
2.2. Whole-Exome Sequencing (WES)
2.3. Whole-Exome Sequence Variant Calling
2.4. Variant Filtration and Data Quality Control
2.5. Haplotype Estimation (Phasing) and Genotype Imputation
2.6. Sample Selection for Age Estimation
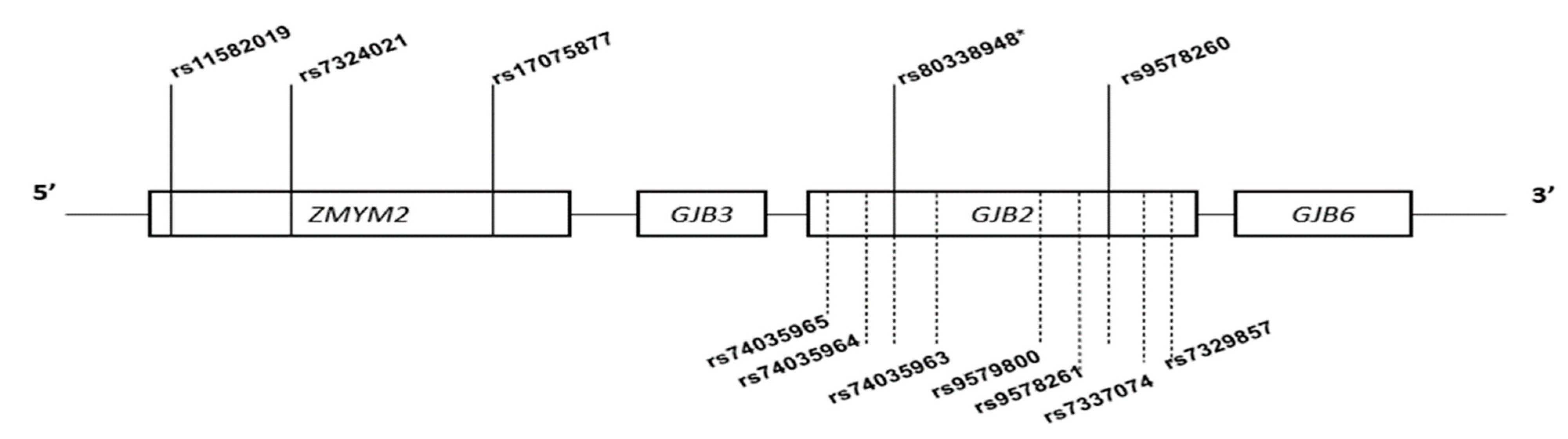
2.7. Markers Selection
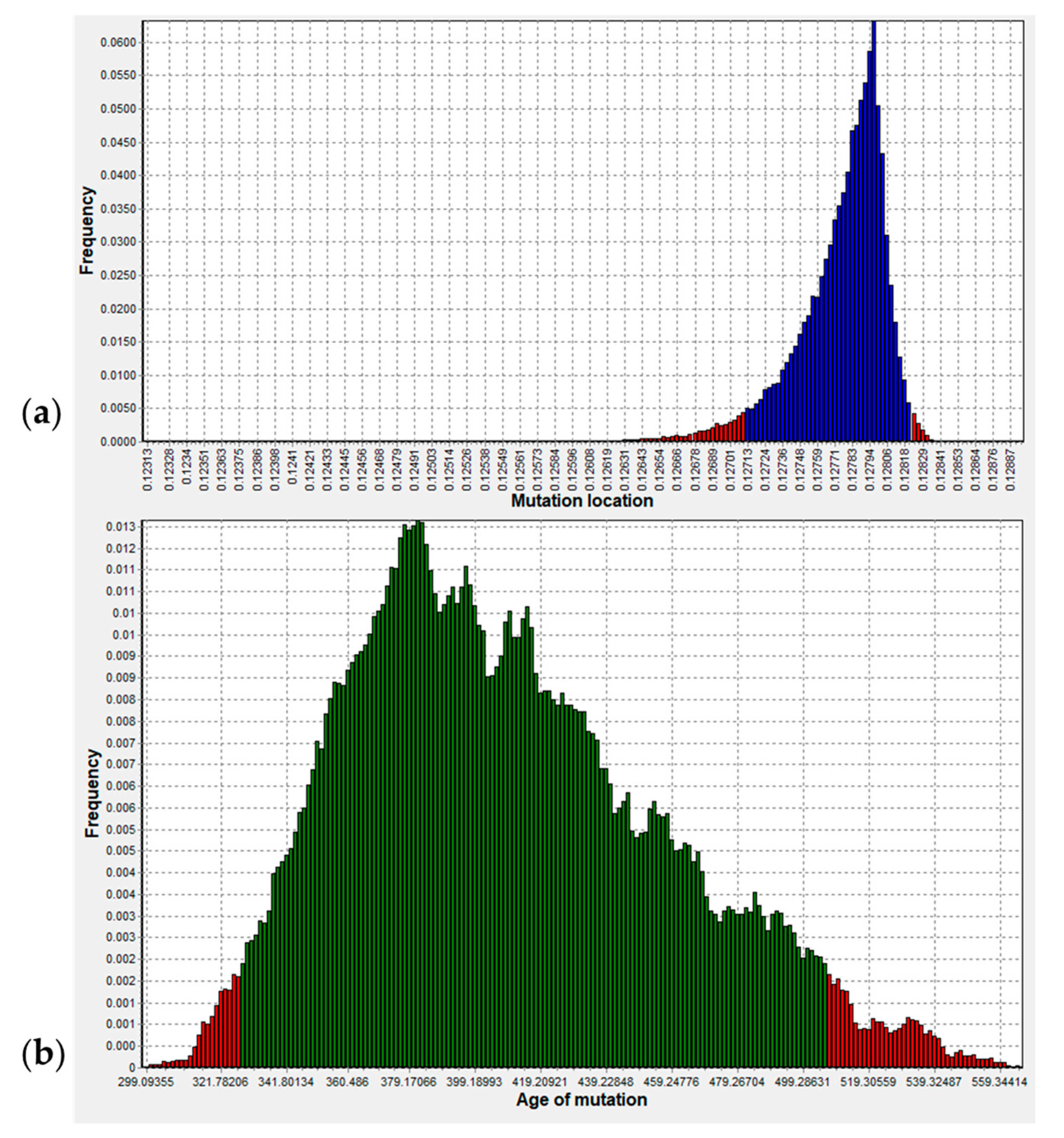
2.8. GJB2: p. (Arg143Trp) (c.427C>T) Variant Age Estimation
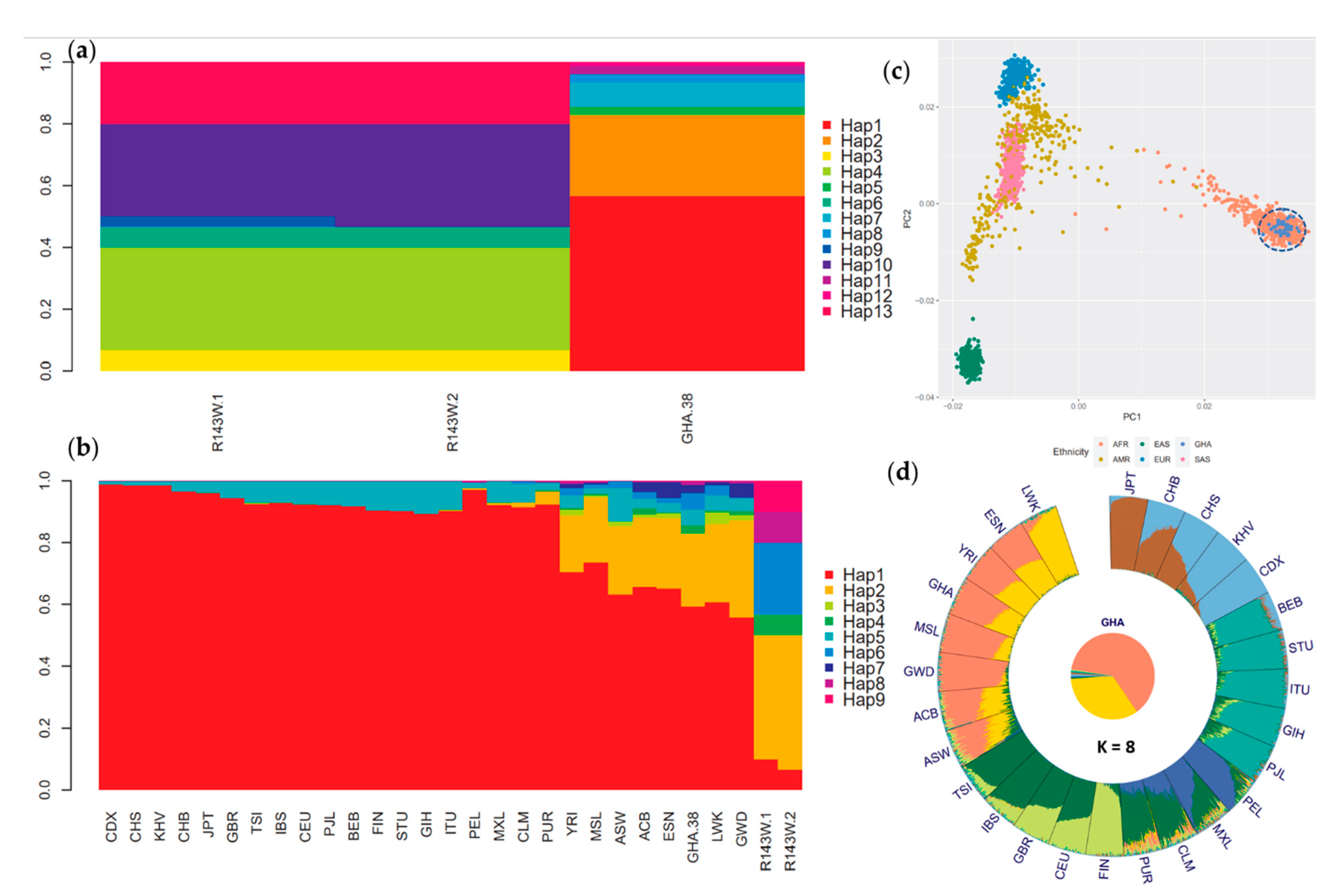
2.9. Principal Component and Ancestry Analyses
2.10. Haplotype Analysis
3. Results
3.1. Characteristics of Study Participants
3.2. Features of Markers Selected for Age Estimation and Haplotype Analysis
3.3. Origin of p.R143W in Ghana
3.4. Haplotype and Ancestry Analyses Confirm Independent Origin of p.(Arg143Trp) in Ghana
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stephenson, J. WHO Report Predicts Hearing Loss for 1 in 4 People Worldwide by 2050. JAMA Health Forum 2021, 2, e210357. [Google Scholar] [CrossRef]
- Morton, C.C.; Nance, W.E. Newborn hearing screening—A silent revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.J.; Bale, J.F., Jr.; White, K.R. Sensorineural hearing loss in children. Lancet 2005, 365, 879–890. [Google Scholar] [CrossRef]
- Van Camp, G.; Smith, R.J. Hereditary Hearing Loss Homepage. 2006. Available online: https://hereditaryhearingloss.org (accessed on 21 November 2021).
- Kenneson, A.; Braun, K.V.N.; Boyle, C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: A HuGE review. Genet. Med. 2002, 4, 258–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bliznetz, E.A.; Lalayants, M.R.; Markova, T.G.; Balanovsky, O.P.; Balanovska, E.V.; Skhalyakho, R.A.; Pocheshkhova, E.A.; Nikitina, N.V.; Voronin, S.V.; Kudryashova, E.K. Update of the GJB2/DFNB1 mutation spectrum in Russia: A founder Ingush mutation del (GJB2-D13S175) is the most frequent among other large deletions. J. Hum. Genet. 2017, 62, 789–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Lu, Y.; Wei, Q.; Cao, X.; Xing, G. A systematic review and meta-analysis of 235delC mutation of GJB2 gene. J. Transl. Med. 2012, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Park, G.; Han, K.-H.; Kim, A.; Koo, J.-W.; Chang, S.O.; Oh, S.H.; Park, W.-Y.; Choi, B.Y. Prevalence of p. V37I variant of GJB2 in mild or moderate hearing loss in a pediatric population and the interpretation of its pathogenicity. PLoS ONE 2013, 8, e61592. [Google Scholar]
- Bouwer, S.; Angelicheva, D.; Chandler, D.; Seeman, P.; Tournev, I.; Kalaydjieva, L. Carrier rates of the ancestral Indian W24X mutation in GJB2 in the general Gypsy population and individual subisolates. Genet. Test. 2007, 11, 455–458. [Google Scholar] [CrossRef]
- Sobe, T.; Vreugde, S.; Shahin, H.; Berlin, M.; Davis, N.; Kanaan, M.; Yaron, Y.; Orr-Urtreger, A.; Frydman, M.; Shohat, M. The prevalence and expression of inherited connexin 26 mutations associated with nonsyndromic hearing loss in the Israeli population. Hum. Genet. 2000, 106, 50–57. [Google Scholar] [CrossRef]
- Brobby, G.W.; Müller-Myhsok, B.; Horstmann, R.D. Connexin 26 R143W mutation associated with recessive nonsyndromic sensorineural deafness in Africa. N. Engl. J. Med. 1998, 338, 548–550. [Google Scholar] [CrossRef]
- Hamelmann, C.; Amedofu, G.K.; Albrecht, K.; Muntau, B.; Gelhaus, A.; Brobby, G.W.; Horstmann, R.D. Pattern of connexin 26 (GJB2) mutations causing sensorineural hearing impairment in Ghana. Hum. Mutat. 2001, 18, 84–85. [Google Scholar] [CrossRef] [PubMed]
- Adadey, S.M.; Manyisa, N.; Mnika, K.; De Kock, C.; Nembaware, V.; Quaye, O.; Amedofu, G.K.; Awandare, G.A.; Wonkam, A. GJB2 and GJB6 mutations in non-syndromic childhood hearing impairment in Ghana. Front. Genet. 2019, 10, 841. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, K.; Nishio, S.; Hattori, M.; Usami, S. Ethnic-specific spectrum of GJB2 and SLC26A4 mutations: Their origin and a literature review. Ann. Otol. Rhinol. Laryngol. 2015, 124, 61S–76S. [Google Scholar] [CrossRef] [PubMed]
- Common, J.E.; Di, W.-L.; Davies, D.; Kelsell, D.P. Further evidence for heterozygote advantage of GJB2 deafness mutations: A link with cell survival. J. Med. Genet. 2004, 41, 573–575. [Google Scholar] [CrossRef] [Green Version]
- Shinagawa, J.; Moteki, H.; Nishio, S.; Noguchi, Y.; Usami, S. Haplotype Analysis of GJB2 Mutations: Founder Effect or Mutational Hot Spot? Genes 2020, 11, 250. [Google Scholar] [CrossRef] [Green Version]
- Nance, W.E. The genetics of deafness. Ment. Retard. Dev. Disabil. Res. Rev. 2003, 9, 109–119. [Google Scholar] [CrossRef]
- Deafness and Hearing Loss. Available online: https://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss (accessed on 23 June 2021).
- Díaz-de Usera, A.; Lorenzo-Salazar, J.M.; Rubio-Rodríguez, L.A.; Muñoz-Barrera, A.; Guillen-Guio, B.; Marcelino-Rodríguez, I.; García-Olivares, V.; Mendoza-Alvarez, A.; Corrales, A.; Íñigo-Campos, A. Evaluation of Whole-Exome Enrichment Solutions: Lessons from the High-End of the Short-Read Sequencing Scale. J. Clin. Med. 2020, 9, 3656. [Google Scholar] [CrossRef]
- Buchan, D.W.; Jones, D.T. The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, s13742-015. [Google Scholar] [CrossRef]
- Manichaikul, A.; Mychaleckyj, J.C.; Rich, S.S.; Daly, K.; Sale, M.; Chen, W.-M. Robust relationship inference in genome-wide association studies. Bioinformatics 2010, 26, 2867–2873. [Google Scholar] [CrossRef] [Green Version]
- Release 1.11·Samtools/Bcftools. Available online: https://github.com/samtools/bcftools/releases/tag/1.11 (accessed on 24 November 2021).
- Browning, S.R.; Browning, B.L. Rapid and Accurate Haplotype Phasing and Missing-Data Inference for Whole-Genome Association Studies By Use of Localized Haplotype Clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A haplotype map of the human genome. Nature 2005, 437, 1299–1320. [CrossRef] [PubMed] [Green Version]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef]
- DMLE+ Disease Mapping Using Linkage Disequilibrium. Available online: http://www.dmle.org/ (accessed on 17 August 2021).
- Ghana Demographics 2020 (Population, Age, Sex, Trends)—Worldometer. Available online: https://www.worldometers.info/demographics/ghana-demographics/#pop (accessed on 25 June 2021).
- Population Growth (Annual%)—Ghana|Data. Available online: https://data.worldbank.org/indicator/SP.POP.GROW?locations=GH (accessed on 17 August 2021).
- Ghana Population Growth Rate, 1960–2020—knoema.com. Available online: https://knoema.com//atlas/Ghana/Population-growth-rate (accessed on 17 August 2021).
- Zhou, H.; Alexander, D.; Lange, K. A quasi-Newton acceleration for high-dimensional optimization algorithms. Stat. Comput. 2011, 21, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Lu, D.; Xu, S. AncestryPainter: A Graphic Program for Displaying Ancestry Composition of Populations and Individuals. Genom. Proteom. Bioinform. 2018, 16, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Rabionet, R.; Zelante, L.; López-Bigas, N.; D’Agruma, L.; Melchionda, S.; Restagno, G.; Arbonés, M.L.; Gasparini, P.; Estivill, X. Molecular basis of childhood deafness resulting from mutations in the GJB2 (connexin 26) gene. Hum. Genet. 2000, 106, 40–44. [Google Scholar] [PubMed]
- Dahl, H.-H.M.; Ching, T.Y.; Hutchison, W.; Hou, S.; Seeto, M.; Sjahalam-King, J. Etiology and audiological outcomes at 3 years for 364 children in Australia. PLoS ONE 2013, 8, e59624. [Google Scholar] [CrossRef] [Green Version]
- Danilenko, N.; Merkulava, E.; Siniauskaya, M.; Olejnik, O.; Levaya-Smaliak, A.; Kushniarevich, A.; Shymkevich, A.; Davydenko, O. Spectrum of genetic changes in patients with non-syndromic hearing impairment and extremely high carrier frequency of 35delG GJB2 mutation in Belarus. PLoS ONE 2012, 7, e36354. [Google Scholar] [CrossRef]
- Rothrock, C.R.; Murgia, A.; Sartorato, E.L.; Leonardi, E.; Wei, S.; Lebeis, S.L.; Laura, E.Y.; Elfenbein, J.L.; Fisher, R.A.; Friderici, K.H. Connexin 26 35delG does not represent a mutational hotspot. Hum. Genet. 2003, 113, 18–23. [Google Scholar] [CrossRef]
- Adadey, S.M.; Wonkam-Tingang, E.; Twumasi Aboagye, E.; Nayo-Gyan, D.W.; Boatemaa Ansong, M.; Quaye, O.; Awandare, G.A.; Wonkam, A. Connexin Genes Variants Associated with Non-Syndromic Hearing Impairment: A Systematic Review of the Global Burden. Life 2020, 10, 258. [Google Scholar] [CrossRef]
- de Oliveira, C.A.; Alexandrino, F.; Christiani, T.V.; Steiner, C.E.; Cunha, J.L.R.; Guerra, A.T.M.; Sartorato, E.L. Molecular genetics study of deafness in Brazil: 8-year experience. Am. J. Med. Genet. A 2007, 143A, 1574–1579. [Google Scholar] [CrossRef] [PubMed]
- Nance, W.E.; Liu, X.-Z.; Pandya, A. Relation between choice of partner and high frequency of connexin-26 deafness. Lancet 2000, 356, 500–501. [Google Scholar] [CrossRef]
- Rose, S.P. Genetic Studies of Profound Prelingual Deafness; Indiana University: Bloomington, IN, USA, 1975. [Google Scholar]
- Vona, B.; Nanda, I.; Hofrichter, M.A.; Shehata-Dieler, W.; Haaf, T. Non-syndromic hearing loss gene identification: A brief history and glimpse into the future. Mol. Cell. Probes 2015, 29, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Pigeon, C.; Ilyin, G.; Courselaud, B.; Leroyer, P.; Turlin, B.; Brissot, P.; Loréal, O. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J. Biol. Chem. 2001, 276, 7811–7819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolas, G.; Bennoun, M.; Devaux, I.; Beaumont, C.; Grandchamp, B.; Kahn, A.; Vaulont, S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc. Natl. Acad. Sci. USA 2001, 98, 8780–8785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganz, T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003, 102, 783–788. [Google Scholar] [CrossRef] [Green Version]
- Aidoo, M.; Terlouw, D.J.; Kolczak, M.S.; McElroy, P.D.; ter Kuile, F.O.; Kariuki, S.; Nahlen, B.L.; Lal, A.A.; Udhayakumar, V. Protective effects of the sickle cell gene against malaria morbidity and mortality. Lancet 2002, 359, 1311–1312. [Google Scholar] [CrossRef]
- Lindqvist, P.G.; Dahlback, B. Carriership of Factor V Leiden and Evolutionary Selection Advantage. Curr. Med. Chem. 2008, 15, 1541–1544. [Google Scholar] [CrossRef]
- Laer, L.V.; Coucke, P.; Mueller, R.F.; Caethoven, G.; Flothmann, K.; Prasad, S.D.; Chamberlin, G.P.; Houseman, M.; Taylor, G.R.; de Heyning, C.M.V.; et al. A common founder for the 35delG GJB2 gene mutation in connexin 26 hearing impairment. J. Med. Genet. 2001, 38, 515–518. [Google Scholar] [CrossRef] [Green Version]
- Gerido, D.A.; DeRosa, A.M.; Richard, G.; White, T.W. Aberrant hemichannel properties of Cx26 mutations causing skin disease and deafness. Am. J. Physiol.-Cell Physiol. 2007, 293, C337–C345. [Google Scholar] [CrossRef] [Green Version]
- Jousse, H. A new contribution to the history of pastoralism in West Africa. J. Afr. Archaeol. 2004, 2, 187–201. [Google Scholar] [CrossRef]
- Coluzzi, M. The clay feet of the malaria giant and its African roots: Hypotheses and inferences about origin, spread and control of Plasmodium falciparum. Parassitologia 1999, 41, 277–283. [Google Scholar] [PubMed]
- Soffer, O.; Gamble, C. The World at 18000 BP: Volume 1, High Latitudes; Unwin Hyman: London, UK, 1990. [Google Scholar]
- Livincstone, F.B. Malaria and human polymorphisms. Annu. Rev. Genet. 1971, 5, 33–64. [Google Scholar] [CrossRef] [PubMed]
- Wiesenfeld, S.L. Sickle-cell trait in human biological and cultural evolution. Development of agriculture causing increased malaria is bound to gene-pool changes causing malaria reduction. Science 1967, 157, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.G.; Amedofu, G.K.; Brandner, J.M.; Pohland, D.; Timmann, C.; Horstmann, R.D. Selection for deafness? Nat. Med. 2002, 8, 1332–1333. [Google Scholar] [CrossRef] [PubMed]
- Man, Y.S.; Trolove, C.; Tattersall, D.; Thomas, A.C.; Papakonstantinopoulou, A.; Patel, D.; Scott, C.; Chong, J.; Jagger, D.J.; O’Toole, E.A. A deafness-associated mutant human connexin 26 improves the epithelial barrier in vitro. J. Membr. Biol. 2007, 218, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Guastalla, P.; Guerci, V.I.; Fabretto, A.; Faletra, F.; Grasso, D.L.; Zocconi, E.; Stefanidou, D.; D’Adamo, P.; Ronfani, L.; Montico, M. Detection of epidermal thickening in GJB2 carriers with epidermal US. Radiology 2009, 251, 280–286. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Marker | Chr:Pos | Ref/Alt | Gene | LD (r2) | Genetic Map (cM) | Alternate Allele Frequency (AAF) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R143W-1N | R143W-2N | GHA-38 | AFR | EUR | AMR | Asian | EAS | ||||||
| rs115802719 | 13:20635310 | A/G | ZMYM2 | 0.102 | 0.00000 | 0.1875 | 0.1875 | 0.0375 | 0.0253 | 0.000088 | 0.0256 | 0.0000 | 0.0000 |
| rs7324021 | 13:20637140 | T/G | ZMYM2 | 0.220 | 0.00183 | 0.3438 | 0.3438 | 0.1125 | 0.0705 | 0.05039 | 0.0700 | 0.0100 | 0.0110 |
| rs1705877 | 13:20641422 | A/G | ZMYM2 | 0.105 | 0.00611 | 0.2500 | 0.2500 | 0.0625 | 0.1595 | 0.12264 | 0.1600 | 0.1430 | 0.1600 |
| *rs80338948 | 13:20763294 | G/A | GJB2 | 1.000 | 0.12798 | 1.0000 * | 1.0000 * | 0.0000 * | 0.0002 | 0.000024 | 0.0002 | 0.0002 | 0.0002 |
| rs9578260 | 13:20763754 | G/A | GJB2 | 0.341 | 0.12844 | 0.9062 | 0.9375 | 0.3250 | 0.2167 | 0.00082 | 0.2165 | 0.0000 | 0.0000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aboagye, E.T.; Adadey, S.M.; Esoh, K.; Jonas, M.; de Kock, C.; Amenga-Etego, L.; Awandare, G.A.; Wonkam, A. Age Estimate of GJB2-p.(Arg143Trp) Founder Variant in Hearing Impairment in Ghana, Suggests Multiple Independent Origins across Populations. Biology 2022, 11, 476. https://doi.org/10.3390/biology11030476
Aboagye ET, Adadey SM, Esoh K, Jonas M, de Kock C, Amenga-Etego L, Awandare GA, Wonkam A. Age Estimate of GJB2-p.(Arg143Trp) Founder Variant in Hearing Impairment in Ghana, Suggests Multiple Independent Origins across Populations. Biology. 2022; 11(3):476. https://doi.org/10.3390/biology11030476
Chicago/Turabian StyleAboagye, Elvis Twumasi, Samuel Mawuli Adadey, Kevin Esoh, Mario Jonas, Carmen de Kock, Lucas Amenga-Etego, Gordon A. Awandare, and Ambroise Wonkam. 2022. "Age Estimate of GJB2-p.(Arg143Trp) Founder Variant in Hearing Impairment in Ghana, Suggests Multiple Independent Origins across Populations" Biology 11, no. 3: 476. https://doi.org/10.3390/biology11030476