
Heterozygosity of the Complex Corfu δ0β+ Thalassemic Allele (HBD Deletion and HBB:c.92+5G>A) Revisited

 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Patients and Methods
2.1. Patients
2.2. Methods
- (a)
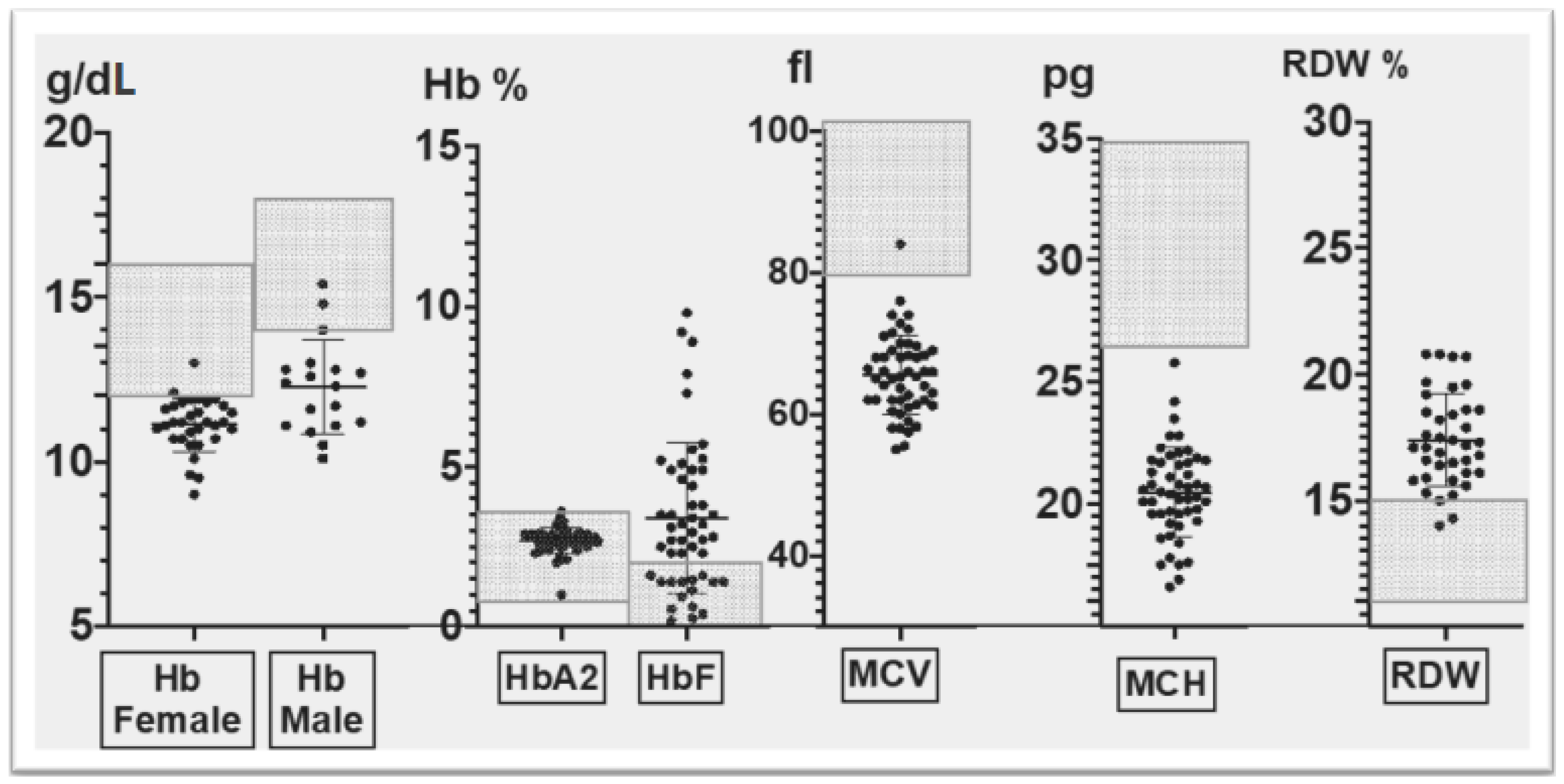
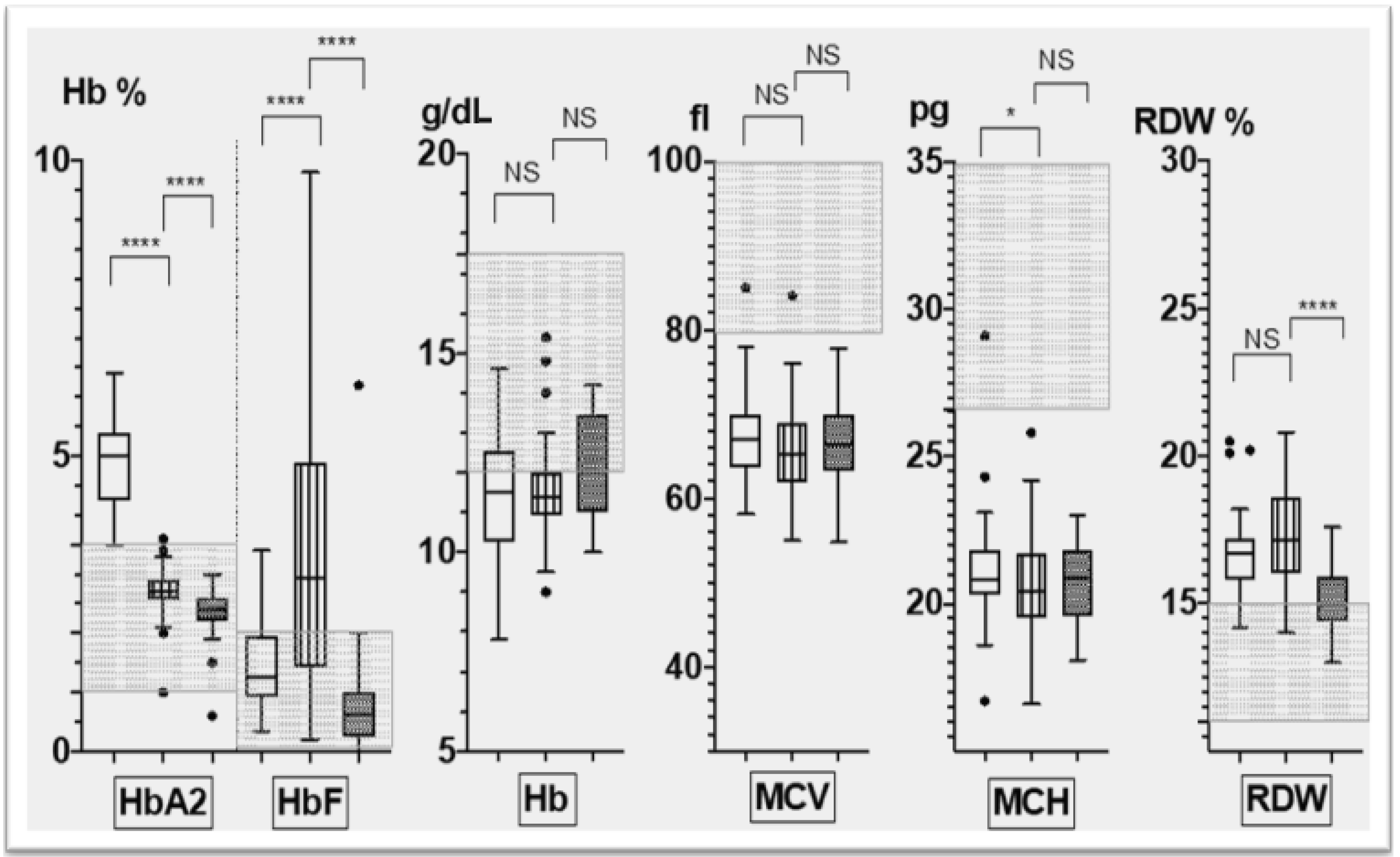
- The hematological phenotype based on relevant red cell parameters, including Hb (g/dL), MCV (Mean Corpuscular Volume) (fl), MCH (Mean Corpuscular Hemoglobin)(pg), RDW (Red cell Distribution Width)(%), HbA2 (%) and HbF (%),as measured by standard hematological and biochemical methods; and
- (b)
2.3. Statistical Analysis
3. Results
3.1. Types and Prevalence of HBB Variants
3.2. Corfu δ0β+ Hematological Phenotype
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Phylipsen, M.; Gallivan, M.V.; Arkesteijn, S.G.; Harteveld, C.L.; Giordano, P.C. Occurrence of common and rare delta-globin gene defects in two multiethnic populations: Thirteen new mutations and the significance of delta-globin gene defects in beta-thalassemia diagnostics. Int. J. Lab. Hematol. 2011, 33, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Metaxotou-Mavromati, A.; Kattamis, C.; Matathia, L.; Tzetis, M.; Kanavakis, E. Clinical, haematological, and genetic studies of type 2 normal Hb A2 beta thalassaemia. J. Med. Genet. 1988, 25, 195–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kattamis, C.; Metaxotou-Mavromati, A.; Wood, W.G.; Nash, J.R.; Weatherall, D.J. The heterogeneity of normal Hb A2-beta thalassaemia in Greece. Br. J. Haematol. 1979, 42, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Wainscoat, J.S.; Thein, S.L.; Wood, W.G.; Weatherall, D.J.; Metaxotou-Mavromati, A.; Tzotos, S.; Kanavakis, E.; Kattamis, C. A novel deletion in the beta-globin gene complex. Ann. N. Y. Acad. Sci. 1985, 445, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Kulozik, A.E.; Yarwood, N.; Jones, R.W. The Corfu delta beta zero thalassemia: A small deletion acts at a distance to selectively abolish beta globin gene expression. Blood 1988, 71, 457–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traeger-Synodinos, J.; Tzetis, M.; Kanavakis, E.; Metaxotou-Mavromati, A.; Kattamis, C. The Corfu delta beta thalassaemia mutation in Greece: Haematological phenotype and prevalence. Br. J. Haematol. 1991, 79, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Musallam, K.M.; Cappellini, M.D. beta-Thalassemias. N. Engl. J. Med. 2021, 384, 727–743. [Google Scholar] [CrossRef] [PubMed]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and beta-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Kattamis, C.; Metaxotou-Mavromati, A.; Ladis, V.; Tsiarta, H.; Laskari, S.; Kanavakis, E. The clinical phenotype of beta and delta beta thalassemias in Greece. Eur. J. Pediatr. 1982, 139, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Vrettou, C.; Kanavakis, E.; Traeger-Synodinos, J.; Metaxotou-Mavrommati, A.; Basiakos, I.; Maragoudaki, E.; Stamoulakatou, A.; Papassotiriou, I.; Kattamis, C. Molecular studies of beta-thalassemia heterozygotes with raised Hb F levels. Hemoglobin 2000, 24, 203–220. [Google Scholar] [CrossRef]
- Tzetis, M.; Traeger-Synodinos, J.; Kanavakis, E.; Metaxotou-Mavromati, A.; Kattamis, C. The molecular basis of normal HbA2 (type 2) beta-thalassemia in Greece. Hematol. Pathol. 1994, 8, 25–34. [Google Scholar]
- Boussiou, M.; Karababa, P.; Sinopoulou, K.; Tsaftaridis, P.; Plata, E.; Loutradi-Anagnostou, A. The molecular heterogeneity of beta-thalassemia in Greece. Blood Cells Mol. Dis. 2008, 40, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Chakalova, L.; Osborne, C.S.; Dai, Y.F.; Goyenechea, B.; Metaxotou-Mavromati, A.; Kattamis, A.; Kattamis, C.; Fraser, P. The Corfu deltabeta thalassemia deletion disrupts gamma-globin gene silencing and reveals post-transcriptional regulation of HbF expression. Blood 2005, 105, 2154–2160. [Google Scholar] [CrossRef] [PubMed]
- Antoniani, C.; Meneghini, V.; Lattanzi, A.; Felix, T.; Romano, O.; Magrin, E.; Weber, L.; Pavani, G.; El Hoss, S.; Kurita, R.; et al. Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human beta-globin locus. Blood 2018, 131, 1960–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankaran, V.G.; Xu, J.; Byron, R.; Greisman, H.A.; Fisher, C.; Weatherall, D.J.; Sabath, D.E.; Groudine, M.; Orkin, S.H.; Premawardhena, A.; et al. A functional element necessary for fetal hemoglobin silencing. N. Engl. J. Med. 2011, 365, 807–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.E.; Magis, W.; Vu, J.; Heo, S.J.; Wartiovaara, K.; Walters, M.C.; Kurita, R.; Nakamura, Y.; Boffelli, D.; Martin, D.I.K.; et al. CRISPR-Cas9 interrogation of a putative fetal globin repressor in human erythroid cells. PLoS ONE 2019, 14, e0208237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basak, A.; Sankaran, V.G. Regulation of the fetal hemoglobin silencing factor BCL11A. Ann. N. Y. Acad. Sci. 2016, 1368, 25–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galanello, R.; Melis, M.A.; Podda, A.; Monne, M.; Perseu, L.; Loudianos, G.; Cao, A.; Pirastu, M.; Piga, A. Deletion delta-thalassemia: The 7.2 kb deletion of Corfu delta beta-thalassemia in a non-beta-thalassemia chromosome. Blood 1990, 75, 1747–1749. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Verboon, J.M.; Zhang, Y.; Liu, N.; Kim, Y.J.; Marglous, S.; Nandakumar, S.K.; Voit, R.A.; Fiorini, C.; Ejaz, A.; et al. A unified model of human hemoglobin switching through single-cell genome editing. Nat. Commun. 2021, 12, 4991. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Ν | Thalassemia Variant | Hematologic Phenotype * | Number of Cases | Frequency (%) | |
|---|---|---|---|---|---|
| NM_000518.5 | Known as | ||||
| 1 | c.93-21G>A | IVSI-110 G>A | β+ | 1034 | 40.42 |
| 2 | c.118C>T | CD39 C>T | β0 | 452 | 17.67 |
| 3 | c.92+1G>A | IVSI-1 G>A | β0 | 306 | 11.96 |
| 4 | c.92+6T>C | IVSI-6 T>C | β++ | 267 | 10.44 |
| 5 | c.316−106C>G | IVSII-745 C>G | β+ | 126 | 4.93 |
| 6 | c.315+1G>A | IVSII-1 G>A | β0 | 74 | 2.89 |
| 7 | c.-137C>G | −87 C>G | β++ | 67 | 2.62 |
| 8 | c.20delA | Cd6 del A | β0 | 61 | 2.38 |
| 9 | c.-151C>T | −101 C>T | βsil | 45 | 1.76 |
| 10 | c.92+5G>A | IVSI-5G>A plus Corfu delta | δ0β+ | 40 | 1.56 |
| 11 | c.25_26delAA | Cd8 del AA | β0 | 21 | 0.82 |
| 12 | c.17_18delCT | Cd5 del CT | β0 | 18 | 0.70 |
| 13 | c.*6C>G | +1480 C>G | βsil | 16 | 0.63 |
| 14 | c.*111A>G | PolyA A>G | β++ | 14 | 0.55 |
| 15 | c.76_92+27del | 44bp del | β0 | 4 | 0.16 |
| 16 | c.-78A>C | −28 A>C | β++ | 3 | 0.12 |
| 17 | c.92G>C | CD30 AGG>ACG | β0 | 3 | 0.12 |
| 18 | c.316-3C>A | IVSII-848 C>A | β+ | 2 | 0.08 |
| 19 | c.−80T>A | −30 T>A | β++ | 2 | 0.08 |
| 20 | c.*96T>C | +1570 T>C | βsil | 1 | 0.04 |
| 21 | c.114G>A | CD37 TGG>TGA | β0 | 1 | 0.04 |
| 22 | c.135delC | CD44 del C | β0 | 1 | 0.04 |
| TOTAL | 2558 | 100.00 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kattamis, C.; Skafida, M.; Delaporta, P.; Vrettou, C.; Traeger-Synodinos, J.; Sofocleous, C.; Kattamis, A. Heterozygosity of the Complex Corfu δ0β+ Thalassemic Allele (HBD Deletion and HBB:c.92+5G>A) Revisited. Biology 2022, 11, 432. https://doi.org/10.3390/biology11030432
Kattamis C, Skafida M, Delaporta P, Vrettou C, Traeger-Synodinos J, Sofocleous C, Kattamis A. Heterozygosity of the Complex Corfu δ0β+ Thalassemic Allele (HBD Deletion and HBB:c.92+5G>A) Revisited. Biology. 2022; 11(3):432. https://doi.org/10.3390/biology11030432
Chicago/Turabian StyleKattamis, Christos, Myrto Skafida, Polyxeni Delaporta, Christina Vrettou, Joanne Traeger-Synodinos, Christalena Sofocleous, and Antonis Kattamis. 2022. "Heterozygosity of the Complex Corfu δ0β+ Thalassemic Allele (HBD Deletion and HBB:c.92+5G>A) Revisited" Biology 11, no. 3: 432. https://doi.org/10.3390/biology11030432