

Gut–Liver Axis and Non-Alcoholic Fatty Liver Disease: A Vicious Circle of Dysfunctions Orchestrated by the Gut Microbiome
 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
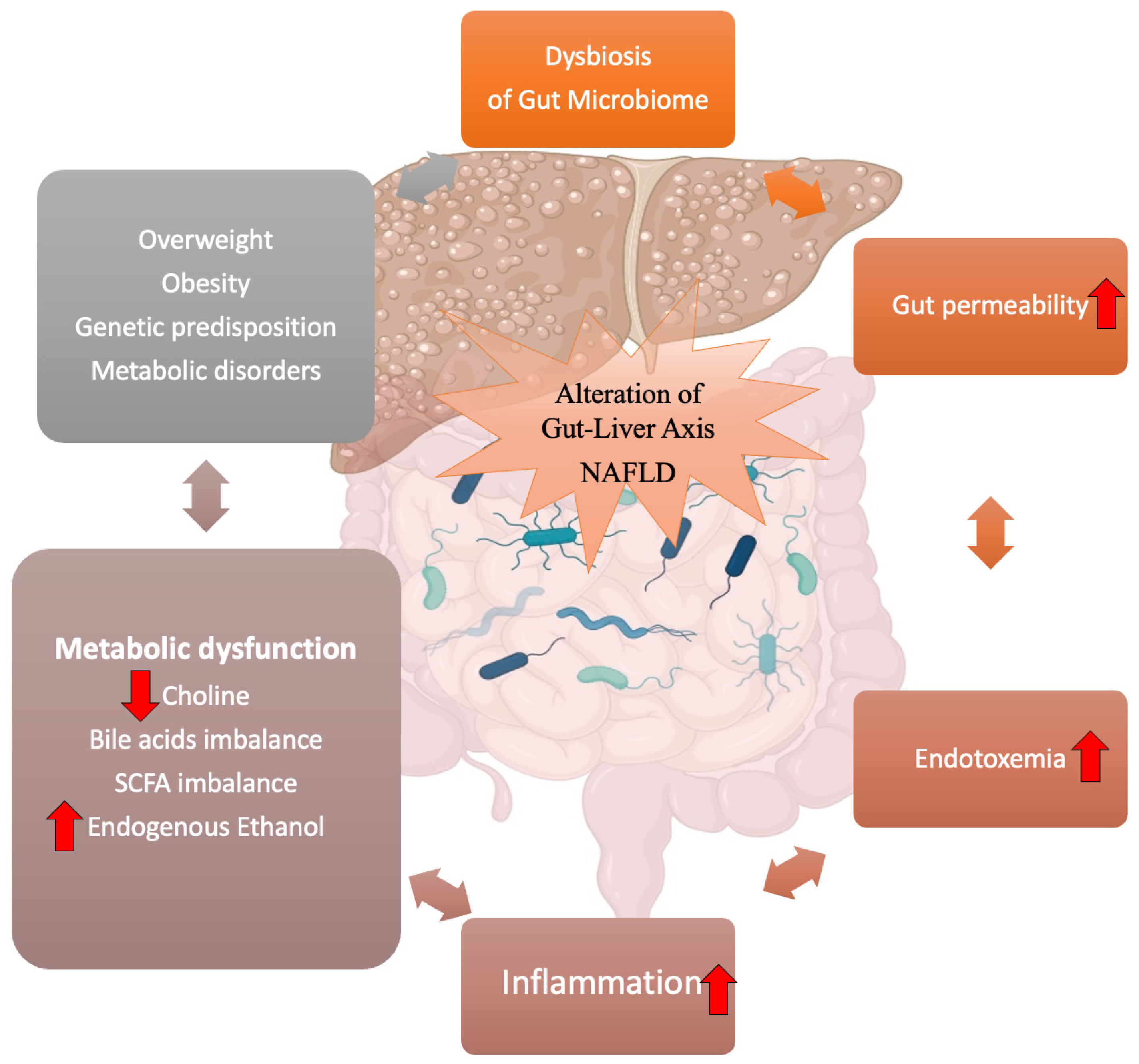
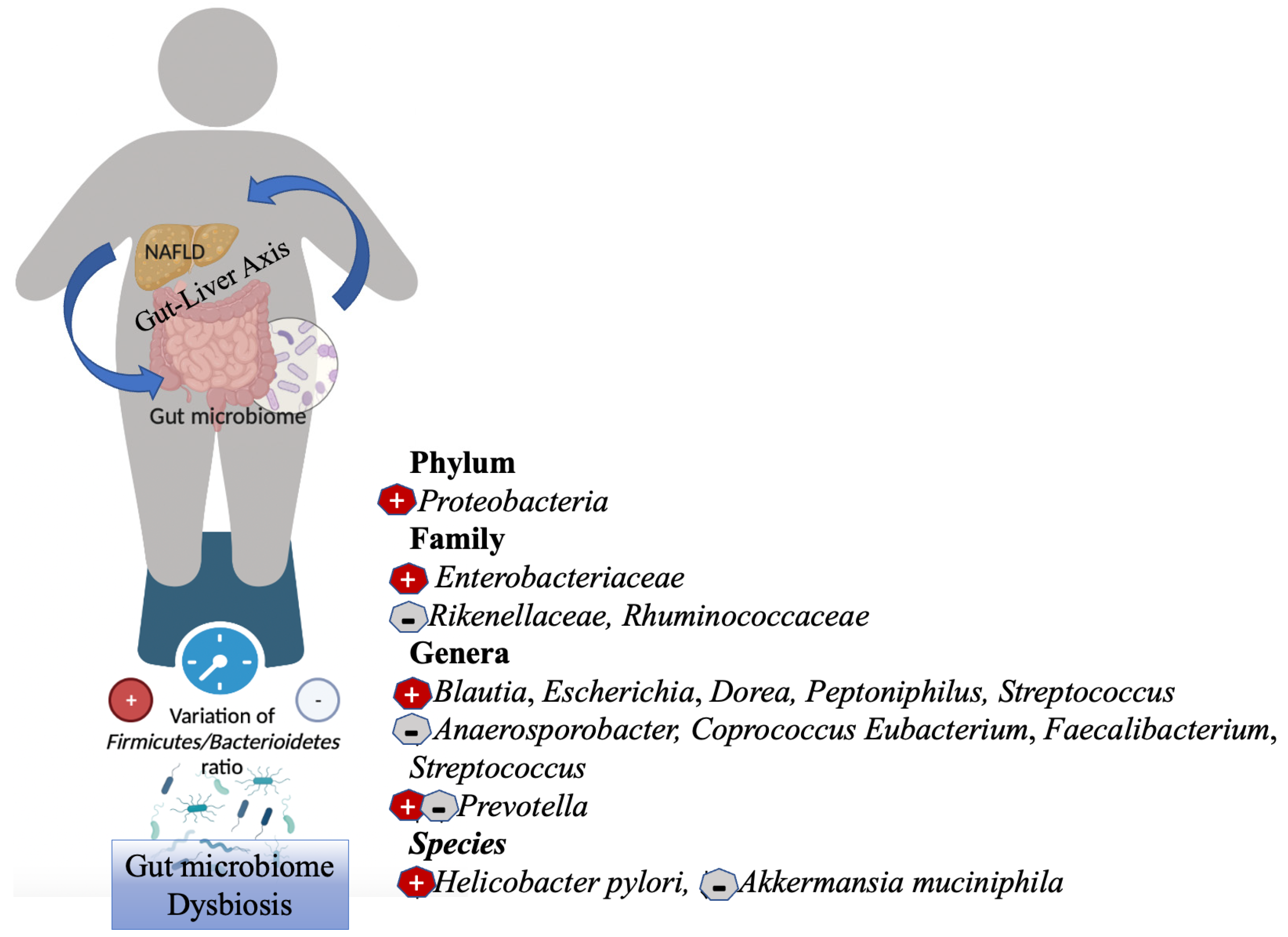
2. Gut Microbiome Dysbiosis in NAFLD and Obesity
3. Gut–Liver Axis and NAFLD
3.1. Intestinal Permeability: The Leaky Gut
3.2. Endotoxins and Inflammation
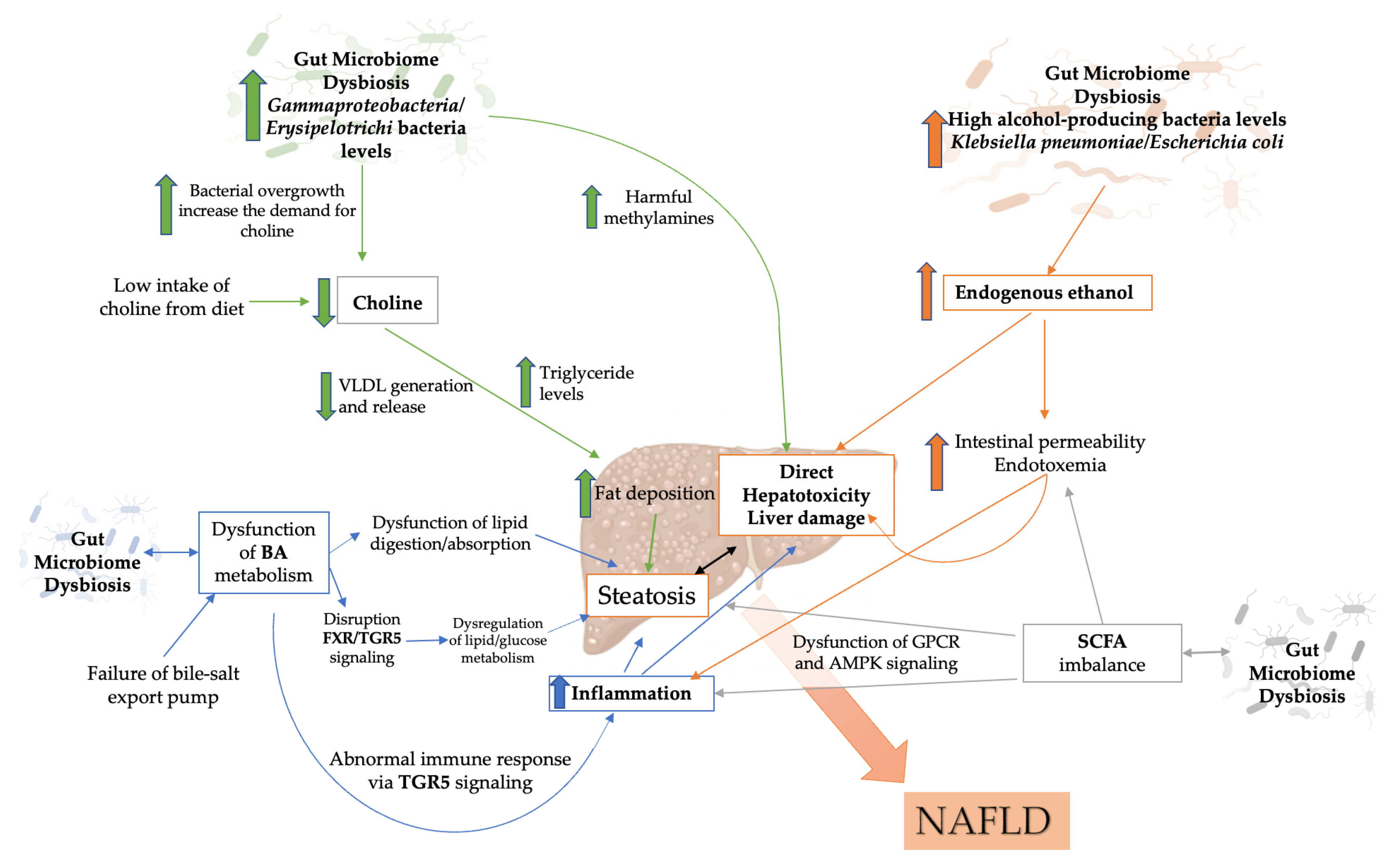
3.3. Metabolites Produced by the Gut Microbiome
3.3.1. Endogenous Ethanol
3.3.2. Choline
3.3.3. Bile Acids
3.3.4. Short-Chain Fatty Acids
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thursby, E.; Juge, N. Introduction to the Human Gut Microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.-M.; et al. Enterotypes of the Human Gut Microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S.; et al. Human Microbiome Project Consortium Structure, Function and Diversity of the Healthy Human Microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Kastl, A.J.; Terry, N.A.; Wu, G.D.; Albenberg, L.G. The Structure and Function of the Human Small Intestinal Microbiota: Current Understanding and Future Directions. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 33–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muscogiuri, G.; Cantone, E.; Cassarano, S.; Tuccinardi, D.; Barrea, L.; Savastano, S.; Colao, A. Gut Microbiota: A New Path to Treat Obesity. Int. J. Obes. Supp. 2019, 9, 10–19. [Google Scholar] [CrossRef]
- Guo, L.; Yang, K.; Zhou, P.; Yong, W. Gut Microbiota in Obesity and Nonalcoholic Fatty Liver Disease. Surg. Pract. Sci. 2021, 5, 100030. [Google Scholar] [CrossRef]
- Pouwels, S.; Sakran, N.; Graham, Y.; Leal, A.; Pintar, T.; Yang, W.; Kassir, R.; Singhal, R.; Mahawar, K.; Ramnarain, D. Non-Alcoholic Fatty Liver Disease (NAFLD): A Review of Pathophysiology, Clinical Management and Effects of Weight Loss. BMC Endocr. Disord. 2022, 22, 63. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between Microbiota and Immunity in Health and Disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Groer, M.; Dutra, S.V.O.; Sarkar, A.; McSkimming, D.I. Gut Microbiota and Immune System Interactions. Microorganisms 2020, 8, 1587. [Google Scholar] [CrossRef]
- Kalra, A.; Yetiskul, E.; Wehrle, C.J.; Tuma, F. Physiology, Liver. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Hornbuckle, W.E.; Simpson, K.W.; Tennant, B.C. Gastrointestinal Function. Clin. Biochem. Domest. Anim. 2008, 14, 413–457. [Google Scholar] [CrossRef]
- Vajro, P.; Paolella, G.; Fasano, A. Microbiota and gut-liver axis: A mini-review on their influences on obesity and obesity related liver disease. J. Pediatr. Gastroenterol. Nutr. 2013, 56, 461–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Targher, G.; Tilg, H.; Byrne, C.D. Non-Alcoholic Fatty Liver Disease: A Multisystem Disease Requiring a Multidisciplinary and Holistic Approach. Lancet Gastroenterol. Hepatol. 2021, 6, 578–588. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD: A Multisystem Disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cholongitas, E.; Pavlopoulou, I.; Papatheodoridi, M.; Markakis, G.E.; Bouras, E.; Haidich, A.-B.; Papatheodoridis, G. Epidemiology of Nonalcoholic Fatty Liver Disease in Europe: A Systematic Review and Meta-Analysis. Ann. Gastroenterol. 2021, 34, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Divella, R.; Mazzocca, A.; Daniele, A.; Sabbà, C.; Paradiso, A. Obesity, Nonalcoholic Fatty Liver Disease and Adipocytokines Network in Promotion of Cancer. Int. J. Biol. Sci. 2019, 15, 610–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellentani, S. The Epidemiology of Non-Alcoholic Fatty Liver Disease. Liver Int. 2017, 37 (Suppl. S1), 81–84. [Google Scholar] [CrossRef] [Green Version]
- Marjot, T.; Moolla, A.; Cobbold, J.F.; Hodson, L.; Tomlinson, J.W. Nonalcoholic Fatty Liver Disease in Adults: Current Concepts in Etiology, Outcomes, and Management. Endocr. Rev. 2020, 41, 66–117. [Google Scholar] [CrossRef]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The Gut-Liver Axis in Liver Disease: Pathophysiological Basis for Therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef] [Green Version]
- Song, Q.; Zhang, X. The Role of Gut–Liver Axis in Gut Microbiome Dysbiosis Associated NAFLD and NAFLD-HCC. Biomedicines 2022, 10, 524. [Google Scholar] [CrossRef]
- Chakraborti, C.K. New-Found Link between Microbiota and Obesity. World J. Gastrointest. Pathophysiol. 2015, 6, 110–119. [Google Scholar] [CrossRef]
- Jahn, D.; Kircher, S.; Hermanns, H.M.; Geier, A. Animal Models of NAFLD from a Hepatologist’s Point of View. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Coker, O.O.; Chu, E.S.; Fu, K.; Lau, H.C.H.; Wang, Y.-X.; Chan, A.W.H.; Wei, H.; Yang, X.; Sung, J.J.Y.; et al. Dietary Cholesterol Drives Fatty Liver-Associated Liver Cancer by Modulating Gut Microbiota and Metabolites. Gut 2021, 70, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.I.; Ijaz, M.U.; Hussain, M.; Haq, I.U.; Zhao, D.; Li, C. High-Fat Proteins Drive Dynamic Changes in Gut Microbiota, Hepatic Metabolome, and Endotoxemia-TLR-4-NFκB-Mediated Inflammation in Mice. J. Agric. Food Chem. 2020, 68, 11710–11725. [Google Scholar] [CrossRef] [PubMed]
- Petrov, P.D.; García-Mediavilla, M.V.; Guzmán, C.; Porras, D.; Nistal, E.; Martínez-Flórez, S.; Castell, J.V.; González-Gallego, J.; Sánchez-Campos, S.; Jover, R. A Network Involving Gut Microbiota, Circulating Bile Acids, and Hepatic Metabolism Genes That Protects against Non-Alcoholic Fatty Liver Disease. Mol. Nutr. Food Res. 2019, 63, 1900487. [Google Scholar] [CrossRef]
- Gart, E.; Souto Lima, E.; Schuren, F.; de Ruiter, C.G.F.; Attema, J.; Verschuren, L.; Keijer, J.; Salic, K.; Morrison, M.C.; Kleemann, R. Diet-Independent Correlations between Bacteria and Dysfunction of Gut, Adipose Tissue, and Liver: A Comprehensive Microbiota Analysis in Feces and Mucosa of the Ileum and Colon in Obese Mice with NAFLD. Int. J. Mol. Sci. 2018, 20, 1. [Google Scholar] [CrossRef] [Green Version]
- Llorente, C.; Jepsen, P.; Inamine, T.; Wang, L.; Bluemel, S.; Wang, H.J.; Loomba, R.; Bajaj, J.S.; Schubert, M.L.; Sikaroodi, M.; et al. Gastric Acid Suppression Promotes Alcoholic Liver Disease by Inducing Overgrowth of Intestinal Enterococcus. Nat. Commun. 2017, 8, 837. [Google Scholar] [CrossRef] [Green Version]
- Pierantonelli, I.; Rychlicki, C.; Agostinelli, L.; Giordano, D.M.; Gaggini, M.; Fraumene, C.; Saponaro, C.; Manghina, V.; Sartini, L.; Mingarelli, E.; et al. Lack of NLRP3-Inflammasome Leads to Gut-Liver Axis Derangement, Gut Dysbiosis and a Worsened Phenotype in a Mouse Model of NAFLD. Sci. Rep. 2017, 7, 12200. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An Obesity-Associated Gut Microbiome with Increased Capacity for Energy Harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Le Roy, T.; Llopis, M.; Lepage, P.; Bruneau, A.; Rabot, S.; Bevilacqua, C.; Martin, P.; Philippe, C.; Walker, F.; Bado, A.; et al. Intestinal Microbiota Determines Development of Non-Alcoholic Fatty Liver Disease in Mice. Gut 2013, 62, 1787–1794. [Google Scholar] [CrossRef]
- Mazagova, M.; Wang, L.; Anfora, A.T.; Wissmueller, M.; Lesley, S.A.; Miyamoto, Y.; Eckmann, L.; Dhungana, S.; Pathmasiri, W.; Sumner, S.; et al. Commensal Microbiota Is Hepatoprotective and Prevents Liver Fibrosis in Mice. FASEB J. 2015, 29, 1043–1055. [Google Scholar] [CrossRef]
- Cano, P.G.; Santacruz, A.; Trejo, F.M.; Sanz, Y. Bifidobacterium CECT 7765 Improves Metabolic and Immunological Alterations Associated with Obesity in High-Fat Diet-Fed Mice. Obesity (Silver Spring) 2013, 21, 2310–2321. [Google Scholar] [CrossRef]
- Shen, F.; Zheng, R.-D.; Sun, X.-Q.; Ding, W.-J.; Wang, X.-Y.; Fan, J.-G. Gut Microbiota Dysbiosis in Patients with Non-Alcoholic Fatty Liver Disease. Hepatobiliary Pancreat. Dis. Int. 2017, 16, 375–381. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut Microbiota and Human NAFLD: Disentangling Microbial Signatures from Metabolic Disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef]
- Astbury, S.; Atallah, E.; Vijay, A.; Aithal, G.P.; Grove, J.I.; Valdes, A.M. Lower Gut Microbiome Diversity and Higher Abundance of Proinflammatory Genus Collinsella Are Associated with Biopsy-Proven Nonalcoholic Steatohepatitis. Gut Microbes 2020, 11, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Lanthier, N.; Rodriguez, J.; Nachit, M.; Hiel, S.; Trefois, P.; Neyrinck, A.M.; Cani, P.D.; Bindels, L.B.; Thissen, J.-P.; Delzenne, N.M. Microbiota Analysis and Transient Elastography Reveal New Extra-Hepatic Components of Liver Steatosis and Fibrosis in Obese Patients. Sci. Rep. 2021, 11, 659. [Google Scholar] [CrossRef] [PubMed]
- Vallianou, N.; Christodoulatos, G.S.; Karampela, I.; Tsilingiris, D.; Magkos, F.; Stratigou, T.; Kounatidis, D.; Dalamaga, M. Understanding the Role of the Gut Microbiome and Microbial Metabolites in Non-Alcoholic Fatty Liver Disease: Current Evidence and Perspectives. Biomolecules 2021, 12, 56. [Google Scholar] [CrossRef]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The Severity of Nonalcoholic Fatty Liver Disease Is Associated with Gut Dysbiosis and Shift in the Metabolic Function of the Gut Microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.-C.; Liu, Y.-Y.; Lin, C.-C.; Wang, C.-C.; Wu, Y.-J.; Yong, C.-C.; Chen, K.-D.; Chuah, S.-K.; Yao, C.-C.; Huang, P.-Y.; et al. Gut Microbiota Dysbiosis in Patients with Biopsy-Proven Nonalcoholic Fatty Liver Disease: A Cross-Sectional Study in Taiwan. Nutrients 2020, 12, 820. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.W.-S.; Tse, C.-H.; Lam, T.T.-Y.; Wong, G.L.-H.; Chim, A.M.-L.; Chu, W.C.-W.; Yeung, D.K.-W.; Law, P.T.-W.; Kwan, H.-S.; Yu, J.; et al. Molecular Characterization of the Fecal Microbiota in Patients with Nonalcoholic Steatohepatitis—A Longitudinal Study. PLoS ONE 2013, 8, e62885. [Google Scholar] [CrossRef] [Green Version]
- Raman, M.; Ahmed, I.; Gillevet, P.M.; Probert, C.S.; Ratcliffe, N.M.; Smith, S.; Greenwood, R.; Sikaroodi, M.; Lam, V.; Crotty, P.; et al. Fecal Microbiome and Volatile Organic Compound Metabolome in Obese Humans with Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2013, 11, 868–875.e3. [Google Scholar] [CrossRef]
- Mouzaki, M.; Comelli, E.M.; Arendt, B.M.; Bonengel, J.; Fung, S.K.; Fischer, S.E.; McGilvray, I.D.; Allard, J.P. Intestinal Microbiota in Patients with Nonalcoholic Fatty Liver Disease. Hepatology 2013, 58, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Ye, J.; Shao, C.; Zhong, B. Compositional Alterations of Gut Microbiota in Nonalcoholic Fatty Liver Disease Patients: A Systematic Review and Meta-Analysis. Lipids Health Dis. 2021, 20, 22. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, N.; Tan, H.-Y.; Li, S.; Zhang, C.; Feng, Y. Function of Akkermansia Muciniphila in Obesity: Interactions with Lipid Metabolism, Immune Response and Gut Systems. Front. Microbiol. 2020, 11, 219. [Google Scholar] [CrossRef] [Green Version]
- Cover, T.L.; Blaser, M.J. Helicobacter Pylori in Health and Disease. Gastroenterology 2009, 136, 1863–1873. [Google Scholar] [CrossRef] [Green Version]
- Abo-Amer, Y.E.-E.; Sabal, A.; Ahmed, R.; Hasan, N.F.E.; Refaie, R.; Mostafa, S.M.; Mohamed, A.A.; Khalil, M.; Elagawy, W.; Abd-Elsalam, S. Relationship between Helicobacter Pylori Infection and Nonalcoholic Fatty Liver Disease (NAFLD) in a Developing Country: A Cross-Sectional Study. Diabetes Metab. Syndr. Obes. 2020, 13, 619–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijarnpreecha, K.; Thongprayoon, C.; Panjawatanan, P.; Manatsathit, W.; Jaruvongvanich, V.; Ungprasert, P. Helicobacter Pylori and Risk of Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. J. Clin. Gastroenterol. 2018, 52, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Matos, A.F.; Silva Júnior, W.S.; Valerio, C.M. NAFLD as a Continuum: From Obesity to Metabolic Syndrome and Diabetes. Diabetol. Metab. Syndr. 2020, 12, 60. [Google Scholar] [CrossRef]
- Wang, B.; Jiang, X.; Cao, M.; Ge, J.; Bao, Q.; Tang, L.; Chen, Y.; Li, L. Altered Fecal Microbiota Correlates with Liver Biochemistry in Nonobese Patients with Non-Alcoholic Fatty Liver Disease. Sci. Rep. 2016, 6, 32002. [Google Scholar] [CrossRef]
- Gensthaler, L.; Felsenreich, D.M.; Jedamzik, J.; Eichelter, J.; Nixdorf, L.; Bichler, C.; Krebs, M.; Itariu, B.; Langer, F.B.; Prager, G. Trends of Overweight and Obesity in Male Adolescents: Prevalence, Socioeconomic Status, and Impact on Cardiovascular Risk in a Central European Country. Obes. Surg. 2022, 32, 1024–1033. [Google Scholar] [CrossRef]
- Hruby, A.; Hu, F.B. The Epidemiology of Obesity: A Big Picture. PharmacoEconomics 2015, 33, 673–689. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, H. Obesity: Epidemiology, Pathophysiology, and Therapeutics. Front. Endocrinol. 2021, 12, 1070. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Moon, J.H.; Kim, H.J.; Kong, M.H.; Oh, Y.H. Sedentary Lifestyle: Overview of Updated Evidence of Potential Health Risks. Korean J. Fam. Med. 2020, 41, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity Alters Gut Microbial Ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, E.F.; Cotter, P.D.; Healy, S.; Marques, T.M.; O’Sullivan, O.; Fouhy, F.; Clarke, S.F.; O’Toole, P.W.; Quigley, E.M.; Stanton, C.; et al. Composition and Energy Harvesting Capacity of the Gut Microbiota: Relationship to Diet, Obesity and Time in Mouse Models. Gut 2010, 59, 1635–1642. [Google Scholar] [CrossRef]
- Kashani, A.; Brejnrod, A.D.; Jin, C.; Kern, T.; Madsen, A.N.; Holm, L.A.; Gerber, G.K.; Holm, J.-C.; Hansen, T.; Holst, B.; et al. Impaired Glucose Metabolism and Altered Gut Microbiome despite Calorie Restriction of Ob/Ob Mice. Anim. Microbiome 2019, 1, 11. [Google Scholar] [CrossRef] [Green Version]
- Kasai, C.; Sugimoto, K.; Moritani, I.; Tanaka, J.; Oya, Y.; Inoue, H.; Tameda, M.; Shiraki, K.; Ito, M.; Takei, Y.; et al. Comparison of the Gut Microbiota Composition between Obese and Non-Obese Individuals in a Japanese Population, as Analyzed by Terminal Restriction Fragment Length Polymorphism and next-Generation Sequencing. BMC Gastroenterol. 2015, 15, 100. [Google Scholar] [CrossRef] [Green Version]
- Indiani, C.M.D.S.P.; Rizzardi, K.F.; Castelo, P.M.; Ferraz, L.F.C.; Darrieux, M.; Parisotto, T.M. Childhood Obesity and Firmicutes/Bacteroidetes Ratio in the Gut Microbiota: A Systematic Review. Child. Obes. 2018, 14, 501–509. [Google Scholar] [CrossRef]
- Gomes, A.C.; Hoffmann, C.; Mota, J.F. The Human Gut Microbiota: Metabolism and Perspective in Obesity. Gut Microbes 2018, 9, 308–325. [Google Scholar] [CrossRef] [Green Version]
- Crovesy, L.; Masterson, D.; Rosado, E.L. Profile of the Gut Microbiota of Adults with Obesity: A Systematic Review. Eur. J. Clin. Nutr. 2020, 74, 1251–1262. [Google Scholar] [CrossRef]
- Palmas, V.; Pisanu, S.; Madau, V.; Casula, E.; Deledda, A.; Cusano, R.; Uva, P.; Vascellari, S.; Loviselli, A.; Manzin, A.; et al. Gut Microbiota Markers Associated with Obesity and Overweight in Italian Adults. Sci. Rep. 2021, 11, 5532. [Google Scholar] [CrossRef] [PubMed]
- Schwiertz, A.; Taras, D.; Schäfer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in Lean and Overweight Healthy Subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.H.; Lobley, G.E.; Holtrop, G.; Ince, J.; Johnstone, A.M.; Louis, P.; Flint, H.J. Human Colonic Microbiota Associated with Diet, Obesity and Weight Loss. Int. J. Obes. 2008, 32, 1720–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andoh, A.; Nishida, A.; Takahashi, K.; Inatomi, O.; Imaeda, H.; Bamba, S.; Kito, K.; Sugimoto, M.; Kobayashi, T. Comparison of the Gut Microbial Community between Obese and Lean Peoples Using 16S Gene Sequencing in a Japanese Population. J. Clin. Biochem. Nutr. 2016, 59, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Payne, A.N.; Chassard, C.; Zimmermann, M.; Müller, P.; Stinca, S.; Lacroix, C. The Metabolic Activity of Gut Microbiota in Obese Children Is Increased Compared with Normal-Weight Children and Exhibits More Exhaustive Substrate Utilization. Nutr. Diabetes 2011, 1, e12. [Google Scholar] [CrossRef] [Green Version]
- Pinart, M.; Dötsch, A.; Schlicht, K.; Laudes, M.; Bouwman, J.; Forslund, S.K.; Pischon, T.; Nimptsch, K. Gut Microbiome Composition in Obese and Non-Obese Persons: A Systematic Review and Meta-Analysis. Nutrients 2021, 14, 12. [Google Scholar] [CrossRef]
- Buckley, A.; Turner, J.R. Cell Biology of Tight Junction Barrier Regulation and Mucosal Disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a029314. [Google Scholar] [CrossRef]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased Intestinal Permeability and Tight Junction Alterations in Nonalcoholic Fatty Liver Disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef]
- Giorgio, V.; Miele, L.; Principessa, L.; Ferretti, F.; Villa, M.P.; Negro, V.; Grieco, A.; Alisi, A.; Nobili, V. Intestinal Permeability Is Increased in Children with Non-Alcoholic Fatty Liver Disease, and Correlates with Liver Disease Severity. Dig. Liver Dis. 2014, 46, 556–560. [Google Scholar] [CrossRef]
- Cani, P.D.; Osto, M.; Geurts, L.; Everard, A. Involvement of Gut Microbiota in the Development of Low-Grade Inflammation and Type 2 Diabetes Associated with Obesity. Gut Microbes 2012, 3, 279–288. [Google Scholar] [CrossRef]
- Brun, P.; Castagliuolo, I.; Di Leo, V.; Buda, A.; Pinzani, M.; Palù, G.; Martines, D. Increased Intestinal Permeability in Obese Mice: New Evidence in the Pathogenesis of Nonalcoholic Steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G518–G525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laukoetter, M.G.; Nava, P.; Lee, W.Y.; Severson, E.A.; Capaldo, C.T.; Babbin, B.A.; Williams, I.R.; Koval, M.; Peatman, E.; Campbell, J.A.; et al. JAM-A Regulates Permeability and Inflammation in the Intestine in Vivo. J. Exp. Med. 2007, 204, 3067–3076. [Google Scholar] [CrossRef] [PubMed]
- Vetrano, S.; Rescigno, M.; Cera, M.R.; Correale, C.; Rumio, C.; Doni, A.; Fantini, M.; Sturm, A.; Borroni, E.; Repici, A.; et al. Unique Role of Junctional Adhesion Molecule-A in Maintaining Mucosal Homeostasis in Inflammatory Bowel Disease. Gastroenterology 2008, 135, 173–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ménard, S.; Cerf-Bensussan, N.; Heyman, M. Multiple Facets of Intestinal Permeability and Epithelial Handling of Dietary Antigens. Mucosal Immunol. 2010, 3, 247–259. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, A.C.; Sumagin, R.; Rankin, C.R.; Leoni, G.; Mina, M.J.; Reiter, D.M.; Stehle, T.; Dermody, T.S.; Schaefer, S.A.; Hall, R.A.; et al. JAM-A Associates with ZO-2, Afadin, and PDZ-GEF1 to Activate Rap2c and Regulate Epithelial Barrier Function. MBoC 2013, 24, 2849–2860. [Google Scholar] [CrossRef]
- Rahman, K.; Desai, C.; Iyer, S.S.; Thorn, N.E.; Kumar, P.; Liu, Y.; Smith, T.; Neish, A.S.; Li, H.; Tan, S.; et al. Loss of Junctional Adhesion Molecule A Promotes Severe Steatohepatitis in Mice on a Diet High in Saturated Fat, Fructose, and Cholesterol. Gastroenterology 2016, 151, 733–746.e12. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Tan, J.; Qian, W.; Zhang, L.; Hou, X. Gut Inflammation Exacerbates Hepatic Injury in the High-Fat Diet Induced NAFLD Mouse: Attention to the Gut-Vascular Barrier Dysfunction. Life Sci. 2018, 209, 157–166. [Google Scholar] [CrossRef]
- Shen, B.; Wang, J.; Guo, Y.; Gu, T.; Shen, Z.; Zhou, C.; Li, B.; Xu, X.; Li, F.; Zhang, Q.; et al. Dextran Sulfate Sodium Salt-Induced Colitis Aggravates Gut Microbiota Dysbiosis and Liver Injury in Mice With Non-Alcoholic Steatohepatitis. Front. Microbiol. 2021, 12, 756299. [Google Scholar] [CrossRef]
- Nicoletti, A.; Ponziani, F.R.; Biolato, M.; Valenza, V.; Marrone, G.; Sganga, G.; Gasbarrini, A.; Miele, L.; Grieco, A. Intestinal Permeability in the Pathogenesis of Liver Damage: From Non-Alcoholic Fatty Liver Disease to Liver Transplantation. World J. Gastroenterol. 2019, 25, 4814–4834. [Google Scholar] [CrossRef]
- Jiang, W.; Wu, N.; Wang, X.; Chi, Y.; Zhang, Y.; Qiu, X.; Hu, Y.; Li, J.; Liu, Y. Dysbiosis Gut Microbiota Associated with Inflammation and Impaired Mucosal Immune Function in Intestine of Humans with Non-Alcoholic Fatty Liver Disease. Sci. Rep. 2015, 5, 8096. [Google Scholar] [CrossRef] [Green Version]
- De Munck, T.J.I.; Xu, P.; Verwijs, H.J.A.; Masclee, A.A.M.; Jonkers, D.; Verbeek, J.; Koek, G.H. Intestinal Permeability in Human Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-analysis. Liver Int. 2020, 40, 2906–2916. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Nighot, M.; Al-Sadi, R.; Alhmoud, T.; Nighot, P.; Ma, T.Y. Lipopolysaccharide Regulation of Intestinal Tight Junction Permeability Is Mediated by TLR4 Signal Transduction Pathway Activation of FAK and MyD88. J. Immunol. 2015, 195, 4999–5010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Semba, S.; Khan, R.I.; Bochimoto, H.; Watanabe, T.; Fujiya, M.; Kohgo, Y.; Liu, Y.; Taniguchi, T. Focal Adhesion Kinase Regulates Intestinal Epithelial Barrier Function via Redistribution of Tight Junction. Biochim. Biophys. Acta 2013, 1832, 151–159. [Google Scholar] [CrossRef] [Green Version]
- Sturgeon, C.; Fasano, A. Zonulin, a Regulator of Epithelial and Endothelial Barrier Functions, and Its Involvement in Chronic Inflammatory Diseases. Tissue Barriers 2016, 4, e1251384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zak-Gołąb, A.; Kocełak, P.; Aptekorz, M.; Zientara, M.; Juszczyk, L.; Martirosian, G.; Chudek, J.; Olszanecka-Glinianowicz, M. Gut Microbiota, Microinflammation, Metabolic Profile, and Zonulin Concentration in Obese and Normal Weight Subjects. Int. J. Endocrinol. 2013, 2013, 674106. [Google Scholar] [CrossRef] [Green Version]
- Pacifico, L.; Bonci, E.; Marandola, L.; Romaggioli, S.; Bascetta, S.; Chiesa, C. Increased Circulating Zonulin in Children with Biopsy-Proven Nonalcoholic Fatty Liver Disease. World J. Gastroenterol. 2014, 20, 17107–17114. [Google Scholar] [CrossRef]
- Çakır, M.; Aksel İşbilen, A.; Eyüpoğlu, İ.; Sağ, E.; Örem, A.; Mazlum Şen, T.; Kaklıkkaya, N.; Kaya, G. Effects of Long-Term Synbiotic Supplementation in Addition to Lifestyle Changes in Children with Obesity-Related Non-Alcoholic Fatty Liver Disease. Turk. J. Gastroenterol. 2017, 28, 377–383. [Google Scholar] [CrossRef] [Green Version]
- Loffredo, L.; Zicari, A.M.; Perri, L.; Carnevale, R.; Nocella, C.; Angelico, F.; Del Ben, M.; Mosca, A.; Zaffina, S.; Panera, N.; et al. Does Nox2 Overactivate in Children with Nonalcoholic Fatty Liver Disease? Antioxid. Redox Signal. 2019, 30, 1325–1330. [Google Scholar] [CrossRef]
- Chwist, A.; Hartleb, M.; Lekstan, A.; Kukla, M.; Gutkowski, K.; Kajor, M. A Composite Model Including Visfatin, Tissue Polypeptide-Specific Antigen, Hyaluronic Acid, and Hematological Variables for the Diagnosis of Moderate-to-Severe Fibrosis in Nonalcoholic Fatty Liver Disease: A Preliminary Study. Pol. Arch. Intern. Med. 2014, 124, 704–712. [Google Scholar] [CrossRef]
- Naryzny, S.N.; Legina, O.K. Haptoglobin as a Biomarker. Biochem. Moscow Suppl. Ser. B 2021, 15, 184–198. [Google Scholar] [CrossRef]
- Kamada, Y.; Akita, M.; Takeda, Y.; Yamada, S.; Fujii, H.; Sawai, Y.; Doi, Y.; Asazawa, H.; Nakayama, K.; Mizutani, K.; et al. Serum Fucosylated Haptoglobin as a Novel Diagnostic Biomarker for Predicting Hepatocyte Ballooning and Nonalcoholic Steatohepatitis. PLoS ONE 2013, 8, e66328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperandeo, P.; Martorana, A.M.; Polissi, A. Lipopolysaccharide Biogenesis and Transport at the Outer Membrane of Gram-Negative Bacteria. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2017, 1862, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Lindeman, J.H.; Menke, A.L.; Koonen, D.P.; Morrison, M.; Havekes, L.M.; van den Hoek, A.M.; Kleemann, R. Metabolically Induced Liver Inflammation Leads to NASH and Differs from LPS- or IL-1β-Induced Chronic Inflammation. Lab. Investig. 2014, 94, 491–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccarelli, S.; Panera, N.; Mina, M.; Gnani, D.; De Stefanis, C.; Crudele, A.; Rychlicki, C.; Petrini, S.; Bruscalupi, G.; Agostinelli, L.; et al. LPS-Induced TNF-α Factor Mediates pro-Inflammatory and pro-Fibrogenic Pattern in Non-Alcoholic Fatty Liver Disease. Oncotarget 2015, 6, 41434–41452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Diao, N.; Yuan, R.; Chen, K.; Geng, S.; Li, M.; Li, L. Subclinical Dose Endotoxin Sustains Low-Grade Inflammation and Exacerbates Steatohepatitis in High-Fat Diet Fed Mice. J. Immunol. 2016, 196, 2300–2308. [Google Scholar] [CrossRef] [Green Version]
- Gäbele, E.; Dostert, K.; Hofmann, C.; Wiest, R.; Schölmerich, J.; Hellerbrand, C.; Obermeier, F. DSS Induced Colitis Increases Portal LPS Levels and Enhances Hepatic Inflammation and Fibrogenesis in Experimental NASH. J. Hepatol. 2011, 55, 1391–1399. [Google Scholar] [CrossRef]
- Harte, A.L.; da Silva, N.F.; Creely, S.J.; McGee, K.C.; Billyard, T.; Youssef-Elabd, E.M.; Tripathi, G.; Ashour, E.; Abdalla, M.S.; Sharada, H.M.; et al. Elevated Endotoxin Levels in Non-Alcoholic Fatty Liver Disease. J. Inflamm. 2010, 7, 15. [Google Scholar] [CrossRef] [Green Version]
- Kessoku, T.; Kobayashi, T.; Imajo, K.; Tanaka, K.; Yamamoto, A.; Takahashi, K.; Kasai, Y.; Ozaki, A.; Iwaki, M.; Nogami, A.; et al. Endotoxins and Non-Alcoholic Fatty Liver Disease. Front. Endocrinol. 2021, 12, 770986. [Google Scholar] [CrossRef]
- Dornas, W.; Lagente, V. Intestinally Derived Bacterial Products Stimulate Development of Nonalcoholic Steatohepatitis. Pharmacol. Res. 2019, 141, 418–428. [Google Scholar] [CrossRef]
- Fukunishi, S.; Sujishi, T.; Takeshita, A.; Ohama, H.; Tsuchimoto, Y.; Asai, A.; Tsuda, Y.; Higuchi, K. Lipopolysaccharides Accelerate Hepatic Steatosis in the Development of Nonalcoholic Fatty Liver Disease in Zucker Rats. J. Clin. Biochem. Nutr. 2014, 54, 39–44. [Google Scholar] [CrossRef]
- An, L.; Wirth, U.; Koch, D.; Schirren, M.; Drefs, M.; Koliogiannis, D.; Nieß, H.; Andrassy, J.; Guba, M.; Bazhin, A.V.; et al. The Role of Gut-Derived Lipopolysaccharides and the Intestinal Barrier in Fatty Liver Diseases. J. Gastrointest. Surg. 2022, 26, 671–683. [Google Scholar] [CrossRef] [PubMed]
- De Roza, M.A.; Lamba, M.; Goh, G.B.-B.; Lum, J.H.-M.; Cheah, M.C.-C.; Ngu, J.H.J. Immunoglobulin G in Non-Alcoholic Steatohepatitis Predicts Clinical Outcome: A Prospective Multi-Centre Cohort Study. World J. Gastroenterol. 2021, 27, 7563–7571. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Janeway, C.A. Innate Immunity: Impact on the Adaptive Immune Response. Curr. Opin. Immunol. 1997, 9, 4–9. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The Roles of TLRs, RLRs and NLRs in Pathogen Recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, K.; Ohnishi, H. Role of Gut Microbiota and Toll-like Receptors in Nonalcoholic Fatty Liver Disease. World J. Gastroenterol. 2014, 20, 7381–7391. [Google Scholar] [CrossRef]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like Receptor-4 Signaling and Kupffer Cells Play Pivotal Roles in the Pathogenesis of Non-Alcoholic Steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef] [Green Version]
- Alisi, A.; Manco, M.; Devito, R.; Piemonte, F.; Nobili, V. Endotoxin and Plasminogen Activator Inhibitor-1 Serum Levels Associated with Nonalcoholic Steatohepatitis in Children. J. Pediatr. Gastroenterol. Nutr. 2010, 50, 645–649. [Google Scholar] [CrossRef]
- Kapil, S.; Duseja, A.; Sharma, B.K.; Singla, B.; Chakraborti, A.; Das, A.; Ray, P.; Dhiman, R.K.; Chawla, Y. Small Intestinal Bacterial Overgrowth and Toll-like Receptor Signaling in Patients with Non-Alcoholic Fatty Liver Disease. J. Gastroenterol. Hepatol. 2016, 31, 213–221. [Google Scholar] [CrossRef]
- Sharifnia, T.; Antoun, J.; Verriere, T.G.C.; Suarez, G.; Wattacheril, J.; Wilson, K.T.; Peek, R.M.; Abumrad, N.N.; Flynn, C.R. Hepatic TLR4 Signaling in Obese NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G270–G278. [Google Scholar] [CrossRef]
- Jia, L.; Chang, X.; Qian, S.; Liu, C.; Lord, C.C.; Ahmed, N.; Lee, C.E.; Lee, S.; Gautron, L.; Mitchell, M.C.; et al. Hepatocyte Toll-like Receptor 4 Deficiency Protects against Alcohol-Induced Fatty Liver Disease. Mol. Metab. 2018, 14, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Mencin, A.; Kluwe, J.; Schwabe, R.F. Toll-like Receptors as Targets in Chronic Liver Diseases. Gut 2009, 58, 704–720. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Friedman, S.L. Toll-like Receptor 4 Signaling in Liver Injury and Hepatic Fibrogenesis. Fibrogenesis Tissue Repair 2010, 3, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, J.-B.; Pimentel-Nunes, P.; Roncon-Albuquerque, R.; Leite-Moreira, A. The Role of Lipopolysaccharide/Toll-like Receptor 4 Signaling in Chronic Liver Diseases. Hepatol. Int. 2010, 4, 659–672. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhang, X.; Yang, W.; Yu, H.; He, Q.; Xu, H.; Li, S.; Shang, Z.; Gao, X.; Wang, Y.; et al. Gut Microbiota in Adipose Tissue Dysfunction Induced Cardiovascular Disease: Role as a Metabolic Organ. Front. Endocrinol. 2021, 12, 749125. [Google Scholar] [CrossRef]
- Caesar, R.; Tremaroli, V.; Kovatcheva-Datchary, P.; Cani, P.D.; Bäckhed, F. Crosstalk between Gut Microbiota and Dietary Lipids Aggravates WAT Inflammation through TLR Signaling. Cell Metab. 2015, 22, 658–668. [Google Scholar] [CrossRef] [Green Version]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Hong, W.; Lu, S.; Li, Y.; Guan, Y.; Weng, X.; Feng, Z. The NLRP3 Inflammasome in Non-Alcoholic Fatty Liver Disease and Steatohepatitis: Therapeutic Targets and Treatment. Front. Pharmacol. 2022, 13, 682. [Google Scholar] [CrossRef]
- Semin, I.; Ninnemann, J.; Bondareva, M.; Gimaev, I.; Kruglov, A.A. Interplay Between Microbiota, Toll-Like Receptors and Cytokines for the Maintenance of Epithelial Barrier Integrity. Front. Med. 2021, 8, 644333. [Google Scholar] [CrossRef]
- Wree, A.; McGeough, M.D.; Peña, C.A.; Schlattjan, M.; Li, H.; Inzaugarat, M.E.; Messer, K.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 Inflammasome Activation Is Required for Fibrosis Development in NAFLD. J. Mol. Med. 2014, 92, 1069–1082. [Google Scholar] [CrossRef]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.-H.; Savard, C.; Ioannou, G.N.; et al. NLRP3 Inflammasome Blockade Reduces Liver Inflammation and Fibrosis in Experimental NASH in Mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Idalsoaga, F.; Kulkarni, A.V.; Mousa, O.Y.; Arrese, M.; Arab, J.P. Non-Alcoholic Fatty Liver Disease and Alcohol-Related Liver Disease: Two Intertwined Entities. Front. Med. 2020, 7, 448. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Hou, H.; Zhang, W.; Liu, T.; Li, Y.; Wang, S.; Wang, B.; Cao, H. Microbial Metabolites: Critical Regulators in NAFLD. Front. Microbiol. 2020, 11, 567654. [Google Scholar] [CrossRef] [PubMed]
- Elshaghabee, F.M.F.; Bockelmann, W.; Meske, D.; de Vrese, M.; Walte, H.-G.; Schrezenmeir, J.; Heller, K.J. Ethanol Production by Selected Intestinal Microorganisms and Lactic Acid Bacteria Growing under Different Nutritional Conditions. Front. Microbiol. 2016, 7, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, S.S.; Baker, R.D.; Liu, W.; Nowak, N.J.; Zhu, L. Role of Alcohol Metabolism in Non-Alcoholic Steatohepatitis. PLoS ONE 2010, 5, e9570. [Google Scholar] [CrossRef] [Green Version]
- Park, E.; Jeong, J.-J.; Won, S.-M.; Sharma, S.P.; Gebru, Y.A.; Ganesan, R.; Gupta, H.; Suk, K.T.; Kim, D.J. Gut Microbiota-Related Cellular and Molecular Mechanisms in the Progression of Nonalcoholic Fatty Liver Disease. Cells 2021, 10, 2634. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.; Li, S.; Xue, G.; et al. Fatty Liver Disease Caused by High-Alcohol-Producing Klebsiella Pneumoniae. Cell Metab. 2019, 30, 675–688. [Google Scholar] [CrossRef]
- Sanders, L.M.; Zeisel, S.H. Choline: Dietary Requirements and Role in Brain Development. Nutr. Today 2007, 42, 181–186. [Google Scholar] [CrossRef]
- Cole, L.K.; Vance, J.E.; Vance, D.E. Phosphatidylcholine Biosynthesis and Lipoprotein Metabolism. Biochim. Biophys. Acta 2012, 1821, 754–761. [Google Scholar] [CrossRef]
- van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The Critical Role of Phosphatidylcholine and Phosphatidylethanolamine Metabolism in Health and Disease. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef]
- Vance, D.E. Role of Phosphatidylcholine Biosynthesis in the Regulation of Lipoprotein Homeostasis. Curr. Opin. Lipidol. 2008, 19, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Van Herck, M.A.; Vonghia, L.; Francque, S.M. Animal Models of Nonalcoholic Fatty Liver Disease—A Starter’s Guide. Nutrients 2017, 9, 1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchman, A.L.; Ament, M.E.; Sohel, M.; Dubin, M.; Jenden, D.J.; Roch, M.; Pownall, H.; Farley, W.; Awal, M.; Ahn, C. Choline Deficiency Causes Reversible Hepatic Abnormalities in Patients Receiving Parenteral Nutrition: Proof of a Human Choline Requirement: A Placebo-Controlled Trial. JPEN J. Parenter. Enteral. Nutr. 2001, 25, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Shu, X.-O.; Xiang, Y.-B.; Li, H.; Yang, G.; Gao, Y.-T.; Zheng, W.; Zhang, X. Higher Dietary Choline Intake Is Associated with Lower Risk of Nonalcoholic Fatty Liver in Normal-Weight Chinese Women12. J. Nutr. 2014, 144, 2034–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.M.; Vance, D.E. The Active Synthesis of Phosphatidylcholine Is Required for Very Low Density Lipoprotein Secretion from Rat Hepatocytes. J. Biol. Chem. 1988, 263, 2998–3004. [Google Scholar] [CrossRef]
- Huang, J.-K.; Lee, H.-C. Emerging Evidence of Pathological Roles of Very-Low-Density Lipoprotein (VLDL). Int. J. Mol. Sci. 2022, 23, 4300. [Google Scholar] [CrossRef]
- Yao, Z.M.; Vance, D.E. Reduction in VLDL, but Not HDL, in Plasma of Rats Deficient in Choline. Biochem. Cell Biol. 1990, 68, 552–558. [Google Scholar] [CrossRef]
- Stephenson, K.; Kennedy, L.; Hargrove, L.; Demieville, J.; Thomson, J.; Alpini, G.; Francis, H. Updates on Dietary Models of Nonalcoholic Fatty Liver Disease: Current Studies and Insights. Gene Expr. 2018, 18, 5–17. [Google Scholar] [CrossRef]
- Soret, P.-A.; Magusto, J.; Housset, C.; Gautheron, J. In Vitro and In Vivo Models of Non-Alcoholic Fatty Liver Disease: A Critical Appraisal. J. Clin. Med. 2021, 10, 36. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; DuGar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.-M.; et al. Gut Flora Metabolism of Phosphatidylcholine Promotes Cardiovascular Disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef]
- Spencer, M.D.; Hamp, T.J.; Reid, R.W.; Fischer, L.M.; Zeisel, S.H.; Fodor, A.A. Association Between Composition of the Human Gastrointestinal Microbiome and Development of Fatty Liver with Choline Deficiency. Gastroenterology 2011, 140, 976–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumas, M.-E.; Barton, R.H.; Toye, A.; Cloarec, O.; Blancher, C.; Rothwell, A.; Fearnside, J.; Tatoud, R.; Blanc, V.; Lindon, J.C.; et al. Metabolic Profiling Reveals a Contribution of Gut Microbiota to Fatty Liver Phenotype in Insulin-Resistant Mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12511–12516. [Google Scholar] [CrossRef] [Green Version]
- Guerrerio, A.L.; Colvin, R.M.; Schwartz, A.K.; Molleston, J.P.; Murray, K.F.; Diehl, A.; Mohan, P.; Schwimmer, J.B.; Lavine, J.E.; Torbenson, M.S.; et al. Choline Intake in a Large Cohort of Patients with Nonalcoholic Fatty Liver Disease123. Am. J. Clin. Nutr. 2012, 95, 892–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.-Y.; Wu, C.-H.; Chang, C.-Y.; Lee, F.-J.; Wang, B.-W.; Doong, J.-Y.; Lin, Y.-S.; Kuo, C.-S.; Huang, R.-F.S. Optimal Dietary Intake Composition of Choline and Betaine Is Associated with Minimized Visceral Obesity-Related Hepatic Steatosis in a Case-Control Study. Nutrients 2022, 14, 261. [Google Scholar] [CrossRef]
- Geiger, O.; López-Lara, I.M.; Sohlenkamp, C. Phosphatidylcholine Biosynthesis and Function in Bacteria. Biochim. Biophys. Acta 2013, 1831, 503–513. [Google Scholar] [CrossRef]
- Sohlenkamp, C.; López-Lara, I.M.; Geiger, O. Biosynthesis of Phosphatidylcholine in Bacteria. Prog. Lipid Res. 2003, 42, 115–162. [Google Scholar] [CrossRef]
- Jha, P.; Knopf, A.; Koefeler, H.; Mueller, M.; Lackner, C.; Hoefler, G.; Claudel, T.; Trauner, M. Role of Adipose Tissue in Methionine–Choline-Deficient Model of Non-Alcoholic Steatohepatitis (NASH). Biochim. Biophys. Acta 2014, 1842, 959–970. [Google Scholar] [CrossRef] [Green Version]
- Arias, N.; Arboleya, S.; Allison, J.; Kaliszewska, A.; Higarza, S.G.; Gueimonde, M.; Arias, J.L. The Relationship between Choline Bioavailability from Diet, Intestinal Microbiota Composition, and Its Modulation of Human Diseases. Nutrients 2020, 12, 2340. [Google Scholar] [CrossRef]
- Liu, Y.; Dai, M. Trimethylamine N-Oxide Generated by the Gut Microbiota Is Associated with Vascular Inflammation: New Insights into Atherosclerosis. Mediat. Inflamm. 2020, 2020, 4634172. [Google Scholar] [CrossRef] [Green Version]
- Parséus, A.; Sommer, N.; Sommer, F.; Caesar, R.; Molinaro, A.; Ståhlman, M.; Greiner, T.U.; Perkins, R.; Bäckhed, F. Microbiota-Induced Obesity Requires Farnesoid X Receptor. Gut 2017, 66, 429–437. [Google Scholar] [CrossRef]
- Krueger, E.S.; Lloyd, T.S.; Tessem, J.S. The Accumulation and Molecular Effects of Trimethylamine N-Oxide on Metabolic Tissues: It’s Not All Bad. Nutrients 2021, 13, 2873. [Google Scholar] [CrossRef] [PubMed]
- Flores-Guerrero, J.L.; Post, A.; van Dijk, P.R.; Connelly, M.A.; Garcia, E.; Navis, G.; Bakker, S.J.L.; Dullaart, R.P.F. Circulating Trimethylamine-N-Oxide Is Associated with All-Cause Mortality in Subjects with Nonalcoholic Fatty Liver Disease. Liver Int. 2021, 41, 2371–2382. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L. Bile Acid Metabolism and Signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [Green Version]
- Di Ciaula, A.; Garruti, G.; Lunardi Baccetto, R.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.-H.; Portincasa, P. Bile Acid Physiology. Ann. Hepatol. 2017, 16, s4–s14. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Hu, J.; Yan, X. Cross-Talk between Bile Acids and Intestinal Microbiota in Host Metabolism and Health. J. Zhejiang Univ. Sci. B 2015, 16, 436–446. [Google Scholar] [CrossRef] [Green Version]
- Kiriyama, Y.; Nochi, H. Physiological Role of Bile Acids Modified by the Gut Microbiome. Microorganisms 2022, 10, 68. [Google Scholar] [CrossRef]
- Trauner, M.; Claudel, T.; Fickert, P.; Moustafa, T.; Wagner, M. Bile Acids as Regulators of Hepatic Lipid and Glucose Metabolism. Dig. Dis. 2010, 28, 220–224. [Google Scholar] [CrossRef]
- Vítek, L.; Haluzík, M. The Role of Bile Acids in Metabolic Regulation. J. Endocrinol. 2016, 228, R85–R96. [Google Scholar] [CrossRef]
- Watanabe, M.; Fukiya, S.; Yokota, A. Comprehensive Evaluation of the Bactericidal Activities of Free Bile Acids in the Large Intestine of Humans and Rodents. J. Lipid Res. 2017, 58, 1143–1152. [Google Scholar] [CrossRef] [Green Version]
- Guzior, D.V.; Quinn, R.A. Review: Microbial Transformations of Human Bile Acids. Microbiome 2021, 9, 140. [Google Scholar] [CrossRef]
- Nimer, N.; Choucair, I.; Wang, Z.; Nemet, I.; Li, L.; Gukasyan, J.; Weeks, T.L.; Alkhouri, N.; Zein, N.; Tang, W.H.W.; et al. Bile Acids Profile, Histopathological Indices and Genetic Variants for Non-Alcoholic Fatty Liver Disease Progression. Metabolism 2021, 116, 154457. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Doi, H.; Ko, T.; Fukuma, T.; Kadono, T.; Asaeda, K.; Kobayashi, R.; Nakano, T.; Doi, T.; Nakatsugawa, Y.; et al. Frequently Abnormal Serum Gamma-Glutamyl Transferase Activity Is Associated with Future Development of Fatty Liver: A Retrospective Cohort Study. BMC Gastroenterol. 2020, 20, 217. [Google Scholar] [CrossRef] [PubMed]
- Caussy, C.; Hsu, C.; Singh, S.; Bassirian, S.; Kolar, J.; Faulkner, C.; Sinha, N.; Bettencourt, R.; Gara, N.; Valasek, M.A.; et al. Serum Bile Acid Patterns Are Associated with the Presence of NAFLD in Twins, and Dose-Dependent Changes with Increase in Fibrosis Stage in Patients with Biopsy-Proven NAFLD. Aliment. Pharmacol. Ther. 2019, 49, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Yang, M.; Xu, H.; Wang, L.; Wei, H.; Ji, G. Serum Bile Acid Profiles Improve Clinical Prediction of Nonalcoholic Fatty Liver in T2DM Patients. J. Proteome Res. 2021, 20, 3814–3825. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Ferrell, J.M. Bile Acid Metabolism in Liver Pathobiology. Gene Expr. 2018, 18, 71–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soroka, C.J.; Boyer, J.L. Biosynthesis and Trafficking of the Bile Salt Export Pump, BSEP: Therapeutic Implications of BSEP Mutations. Mol. Asp. Med. 2014, 37, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Jackson, K.G.; Way, G.W.; Zhou, H. Bile Acids and Sphingolipids in Non-Alcoholic Fatty Liver Disease. Chin. Med. J. 2022, 135, 1163–1171. [Google Scholar] [CrossRef]
- Alberto González-Regueiro, J.; Moreno-Castañeda, L.; Uribe, M.; Carlos Chávez-Tapia, N. The Role of Bile Acids in Glucose Metabolism and Their Relation with Diabetes. Ann. Hepatol. 2017, 16, S15–S20. [Google Scholar] [CrossRef]
- Chávez-Talavera, O.; Tailleux, A.; Lefebvre, P.; Staels, B. Bile Acid Control of Metabolism and Inflammation in Obesity, Type 2 Diabetes, Dyslipidemia, and Nonalcoholic Fatty Liver Disease. Gastroenterology 2017, 152, 1679–1694.e3. [Google Scholar] [CrossRef]
- Mori, H.; Svegliati Baroni, G.; Marzioni, M.; Di Nicola, F.; Santori, P.; Maroni, L.; Abenavoli, L.; Scarpellini, E. Farnesoid X Receptor, Bile Acid Metabolism, and Gut Microbiota. Metabolites 2022, 12, 647. [Google Scholar] [CrossRef]
- Ferrell, J.M.; Chiang, J.Y.L. Understanding Bile Acid Signaling in Diabetes: From Pathophysiology to Therapeutic Targets. Diabetes Metab. J. 2019, 43, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, G.; Renga, B.; Mencarelli, A.; Pellicciari, R.; Fiorucci, S. Role of FXR in Regulating Bile Acid Homeostasis and Relevance for Human Diseases. Curr. Drug Targets Immune Endocr. Metab. Disord. 2005, 5, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L.; Ferrell, J.M. Up to Date on Cholesterol 7 Alpha-Hydroxylase (CYP7A1) in Bile Acid Synthesis. Liver Res. 2020, 4, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Lu, Y.; Li, X. Farnesoid X Receptor: A Master Regulator of Hepatic Triglyceride and Glucose Homeostasis. Acta Pharmacol. Sin. 2015, 36, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Pols, T.W.H.; Noriega, L.G.; Nomura, M.; Auwerx, J.; Schoonjans, K. The Bile Acid Membrane Receptor TGR5: A Valuable Metabolic Target. Dig. Dis. 2011, 29, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Lou, G.; Ma, X.; Fu, X.; Meng, Z.; Zhang, W.; Wang, Y.-D.; Van Ness, C.; Yu, D.; Xu, R.; Huang, W. GPBAR1/TGR5 Mediates Bile Acid-Induced Cytokine Expression in Murine Kupffer Cells. PLoS ONE 2014, 9, e93567. [Google Scholar] [CrossRef] [Green Version]
- Keitel, V.; Reinehr, R.; Gatsios, P.; Rupprecht, C.; Görg, B.; Selbach, O.; Häussinger, D.; Kubitz, R. The G-Protein Coupled Bile Salt Receptor TGR5 Is Expressed in Liver Sinusoidal Endothelial Cells. Hepatology 2007, 45, 695–704. [Google Scholar] [CrossRef]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-Mediated Bile Acid Sensing Controls Glucose Homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Meijnikman, A.S.; Davids, M.; Herrema, H.; Aydin, O.; Tremaroli, V.; Rios-Morales, M.; Levels, H.; Bruin, S.; de Brauw, M.; Verheij, J.; et al. Microbiome-Derived Ethanol in Nonalcoholic Fatty Liver Disease. Nat. Med. 2022, 28, 2100–2106. [Google Scholar] [CrossRef]
- Tan, J.; McKenzie, C.; Potamitis, M.; Thorburn, A.N.; Mackay, C.R.; Macia, L. The Role of Short-Chain Fatty Acids in Health and Disease. Adv. Immunol. 2014, 121, 91–119. [Google Scholar] [CrossRef]
- Spivak, I.; Fluhr, L.; Elinav, E. Local and Systemic Effects of Microbiome-Derived Metabolites. EMBO Rep. 2022, 23, e55664. [Google Scholar] [CrossRef] [PubMed]
- Baxter, N.T.; Schmidt, A.W.; Venkataraman, A.; Kim, K.S.; Waldron, C.; Schmidt, T.M. Dynamics of Human Gut Microbiota and Short-Chain Fatty Acids in Response to Dietary Interventions with Three Fermentable Fibers. mBio 2019, 10, e02566-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ríos-Covián, D.; Ruas-Madiedo, P.; Margolles, A.; Gueimonde, M.; de los Reyes-Gavilán, C.G.; Salazar, N. Intestinal Short Chain Fatty Acids and Their Link with Diet and Human Health. Front. Microbiol. 2016, 7, 185. [Google Scholar] [CrossRef] [Green Version]
- Schönfeld, P.; Wojtczak, L. Short- and Medium-Chain Fatty Acids in Energy Metabolism: The Cellular Perspective. J. Lipid Res. 2016, 57, 943–954. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Lv, Y.-W.; Long, J.; Chen, J.-M.; He, J.; Ruan, X.-Z.; Zhu, H. Butyrate Improves the Metabolic Disorder and Gut Microbiome Dysbiosis in Mice Induced by a High-Fat Diet. Front. Pharmacol. 2019, 10, 1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.-Q.; An, Y.-X.; Yu, C.-G.; Ke, J.; Zhao, D.; Yu, K. The Association Between Fecal Short-Chain Fatty Acids, Gut Microbiota, and Visceral Fat in Monozygotic Twin Pairs. DMSO 2022, 15, 359–368. [Google Scholar] [CrossRef]
- Rau, M.; Rehman, A.; Dittrich, M.; Groen, A.K.; Hermanns, H.M.; Seyfried, F.; Beyersdorf, N.; Dandekar, T.; Rosenstiel, P.; Geier, A. Fecal SCFAs and SCFA-Producing Bacteria in Gut Microbiome of Human NAFLD as a Putative Link to Systemic T-Cell Activation and Advanced Disease. United Eur. Gastroenterol. J. 2018, 6, 1496–1507. [Google Scholar] [CrossRef]
- Zhou, J.; Tripathi, M.; Sinha, R.A.; Singh, B.K.; Yen, P.M. Gut Microbiota and Their Metabolites in the Progression of Non-Alcoholic Fatty Liver Disease. Hepatoma Res. 2021, 7, 11. [Google Scholar] [CrossRef]
- He, J.; Zhang, P.; Shen, L.; Niu, L.; Tan, Y.; Chen, L.; Zhao, Y.; Bai, L.; Hao, X.; Li, X.; et al. Short-Chain Fatty Acids and Their Association with Signalling Pathways in Inflammation, Glucose and Lipid Metabolism. Int. J. Mol. Sci. 2020, 21, 6356. [Google Scholar] [CrossRef]
- Ang, Z.; Ding, J.L. GPR41 and GPR43 in Obesity and Inflammation – Protective or Causative? Front. Immunol. 2016, 7, 28. [Google Scholar] [CrossRef]
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; Hammer, R.E.; Williams, S.C.; Crowley, J.; Yanagisawa, M.; et al. Effects of the Gut Microbiota on Host Adiposity Are Modulated by the Short-Chain Fatty-Acid Binding G Protein-Coupled Receptor, Gpr41. Proc. Natl. Acad. Sci. USA 2008, 105, 16767–16772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Hong, S.-W.; Rhee, E.-J.; Lee, W.-Y. GLP-1 Receptor Agonist and Non-Alcoholic Fatty Liver Disease. Diabetes Metab. J. 2012, 36, 262–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos-Perez, W.; Martinez-Lopez, E. Effects of Short Chain Fatty Acids on Metabolic and Inflammatory Processes in Human Health. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158900. [Google Scholar] [CrossRef] [PubMed]
- Rangan, P.; Mondino, A. Microbial Short-Chain Fatty Acids: A Strategy to Tune Adoptive T Cell Therapy. J. Immunother. Cancer 2022, 10, e004147. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Chen, X.; Zhao, Z.; Liao, Y.; Zhou, T.; Xiang, Q. A Potential Link between Plasma Short-Chain Fatty Acids, TNF-α Level and Disease Progression in Non-Alcoholic Fatty Liver Disease: A Retrospective Study. Exp. Ther. Med. 2022, 24, 598. [Google Scholar] [CrossRef]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global Epidemiology of NAFLD-Related HCC: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the Epidemic of Nonalcoholic Fatty Liver Disease Demonstrates an Exponential Increase in Burden of Disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.-Y.; Li, L.; Yu, C.-H.; Shen, Z.; Chen, L.-H.; Li, Y.-M. Effects of Probiotics on Nonalcoholic Fatty Liver Disease: A Meta-Analysis. World J. Gastroenterol. 2013, 19, 6911–6918. [Google Scholar] [CrossRef]
- Suk, K.T.; Koh, H. New Perspective on Fecal Microbiota Transplantation in Liver Diseases. J. Gastroenterol. Hepatol. 2022, 37, 24–33. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pezzino, S.; Sofia, M.; Faletra, G.; Mazzone, C.; Litrico, G.; La Greca, G.; Latteri, S. Gut–Liver Axis and Non-Alcoholic Fatty Liver Disease: A Vicious Circle of Dysfunctions Orchestrated by the Gut Microbiome. Biology 2022, 11, 1622. https://doi.org/10.3390/biology11111622
Pezzino S, Sofia M, Faletra G, Mazzone C, Litrico G, La Greca G, Latteri S. Gut–Liver Axis and Non-Alcoholic Fatty Liver Disease: A Vicious Circle of Dysfunctions Orchestrated by the Gut Microbiome. Biology. 2022; 11(11):1622. https://doi.org/10.3390/biology11111622
Chicago/Turabian StylePezzino, Salvatore, Maria Sofia, Gloria Faletra, Chiara Mazzone, Giorgia Litrico, Gaetano La Greca, and Saverio Latteri. 2022. "Gut–Liver Axis and Non-Alcoholic Fatty Liver Disease: A Vicious Circle of Dysfunctions Orchestrated by the Gut Microbiome" Biology 11, no. 11: 1622. https://doi.org/10.3390/biology11111622