
Gap Junctions and Hemichannels Composed of Connexins and Pannexins Mediate the Secondary Brain Injury Following Intracerebral Hemorrhage

 , and
, and 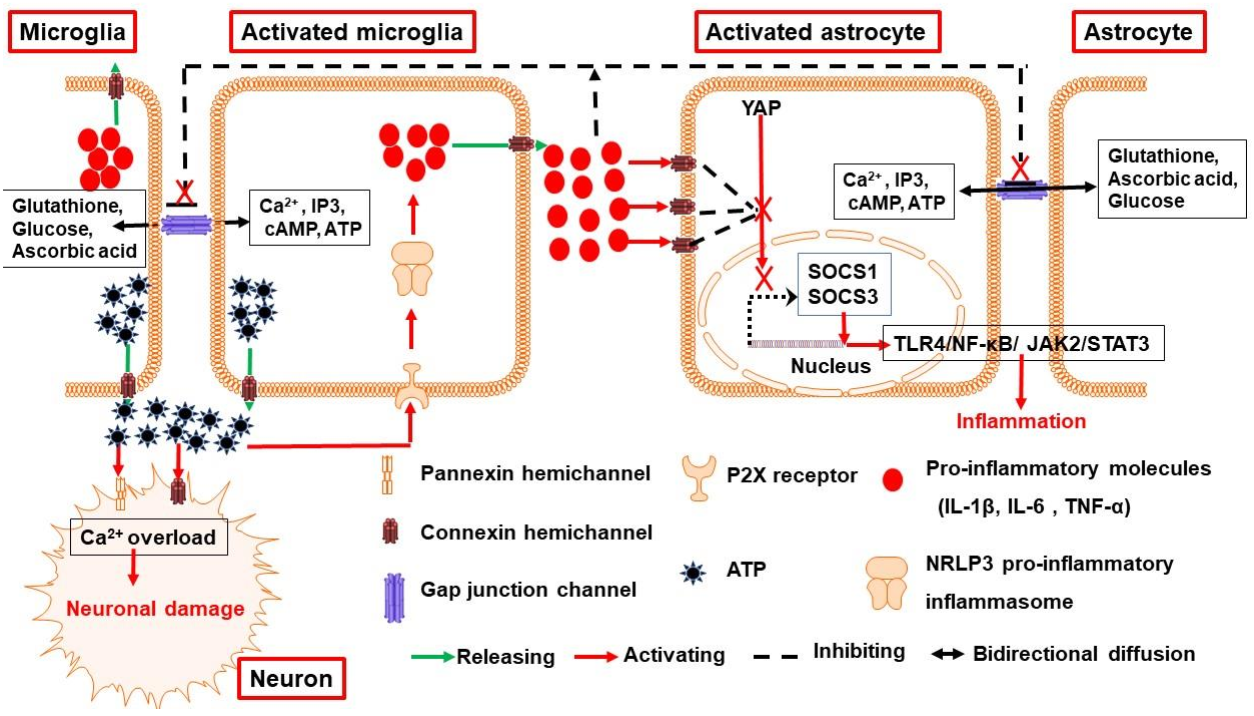{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. The Expression and Functions of Connexins and Pannexins in the Brain
2.1. The Expression and Function of Gap Junction Channels in the Brain
2.2. The Expression and Function of Connexin and Pannexin Hemichannels in the Brain
3. Connexin and Pannexin Channels in ICH
3.1. Connexin and Pannexin Channels Are Implicated in Neuroinflammation Following ICH
3.2. Connexin Channels Mediate the Permeability of BBB Following ICH
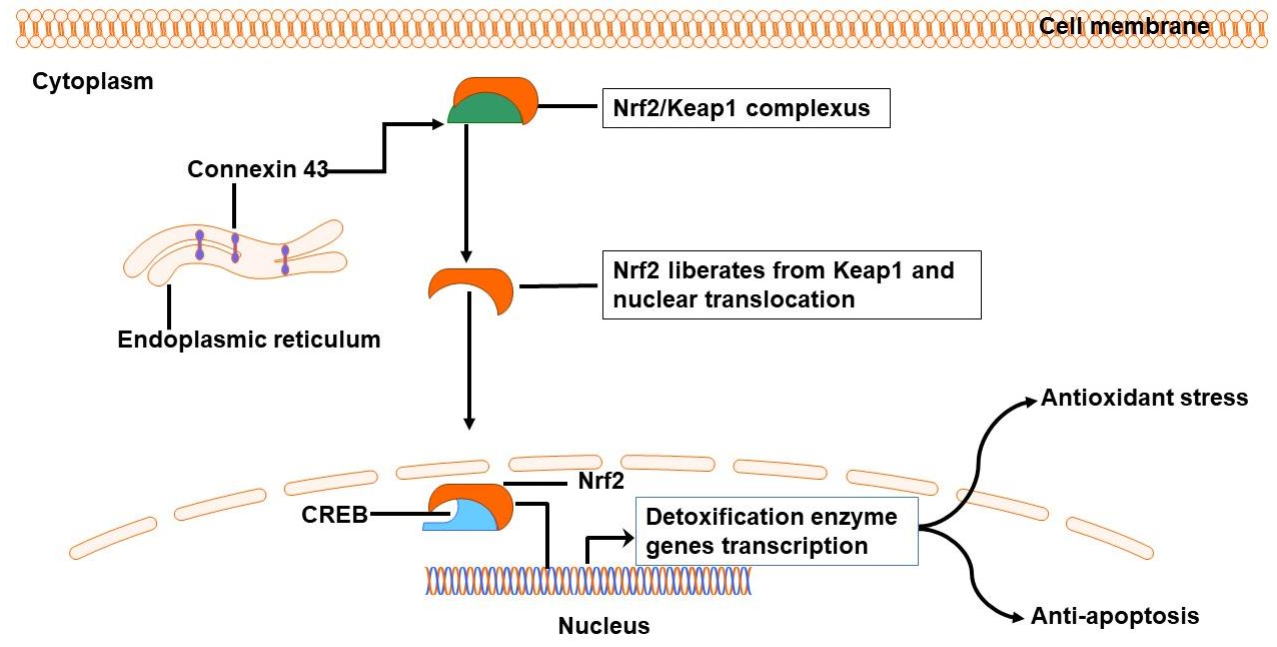
3.3. Connexin Channels Are Involved in Oxidative Stress Following ICH
3.4. Connexin Channels Take Part in Regulating Cerebral Vasospasm Following ICH
3.5. Hemichannels Are Involved in Cell Death Following ICH
4. Strategies Target Gap Junctions and Hemichannels Following ICH
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gross, B.A.; Jankowitz, B.T.; Friedlander, R.M. Cerebral Intraparenchymal Hemorrhage: A Review. JAMA 2019, 321, 1295–1303. [Google Scholar] [CrossRef]
- Chen, L.; Chen, T.; Mao, G.; Chen, B.; Li, M.; Zhang, H.; Xi, H.; She, X.; Tang, Z.; Zhang, P.; et al. Clinical Neurorestorative Therapeutic Guideline for Brainstem Hemorrhage (2020 China Version). J. Neurorestoratology 2020, 8, 232–240. [Google Scholar] [CrossRef]
- Xue, M.; Yong, V.W. Neuroinflammation in Intracerebral Haemorrhage: Immunotherapies with Potential for Translation. Lancet Neurol. 2020, 19, 1023–1032. [Google Scholar] [CrossRef]
- Yao, Z.; Bai, Q.; Wang, G. Mechanisms of Oxidative Stress and Therapeutic Targets Following Intracerebral Hemorrhage. Oxid. Med. Cell Longev. 2021, 2021, 8815441. [Google Scholar] [CrossRef] [PubMed]
- Athiraman, U.; Zipfel, G.J. Role of Anesthetics and their Adjuvants in Neurovascular Protection in Secondary Brain Injury after Aneurysmal Subarachnoid Hemorrhage. Int. J. Mol. Sci. 2021, 22, 6550. [Google Scholar] [CrossRef]
- Keep, R.F.; Andjelkovic, A.V.; Xiang, J.; Stamatovic, S.M.; Antonetti, D.A.; Hua, Y.; Xi, G. Brain Endothelial Cell Junctions After Cerebral Hemorrhage: Changes, Mechanisms and Therapeutic Targets. J. Cereb. Blood Flow Metab. 2018, 38, 1255–1275. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Sheng, Z.; Liu, Y.; Zhang, R.; Yong, V.W.; Xue, M. Intracerebral Haemorrhage: From Clinical Settings to Animal Models. Stroke Vasc. Neurol. 2020, 5, 388–395. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Xue, Q.; Zhao, J.; Yang, Y.; Yu, Y.; Liu, D.; Liu, J.; Yang, W.; Mu, L.; Zhang, P.; et al. Clinical Diagnostic and Therapeutic Guidelines of Stroke Neurorestoration (2020 China Version). J. Neurorestoratology 2020, 8, 241–251. [Google Scholar] [CrossRef]
- Zhang, R.; Xue, M.; Yong, V.W. Central Nervous System Tissue Regeneration after Intracerebral Hemorrhage: The Next Frontier. Cells 2021, 10, 2513. [Google Scholar] [CrossRef] [PubMed]
- Li, X. Seeing is Believing: Gap Junctions in Motion. Biology 2021, 10, 494. [Google Scholar] [CrossRef]
- Orellana, J.A.; Martinez, A.D.; Retamal, M.A. Gap Junction Channels and Hemichannels in the Cns: Regulation by Signaling Molecules. Neuropharmacology 2013, 75, 567–582. [Google Scholar] [CrossRef] [PubMed]
- Decrock, E.; Vinken, M.; De Vuyst, E.; Krysko, D.V.; D’Herde, K.; Vanhaecke, T.; Vandenabeele, P.; Rogiers, V.; Ley-baert, L. Connexin-Related Signaling in Cell Death: To Live Or Let Die? Cell Death Differ. 2009, 16, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Shestopalov, V.I.; Slepak, V.Z. Molecular Pathways of Pannexin1-Mediated Neurotoxicity. Front Physiol. 2014, 5, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Davidson, J.O.; Green, C.R.; Nicholson, L.; O’Carroll, S.J.; Zhang, J. Connexins and Pannexins in Cerebral Ischemia. Biochim. Biophys. Acta Biomembr. 2018, 1860, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Bargiotas, P.; Krenz, A.; Hormuzdi, S.G.; Ridder, D.A.; Herb, A.; Barakat, W.; Penuela, S.; von Engelhardt, J.; Monyer, H.; Schwaninger, M. Pannexins in Ischemia-Induced Neurodegeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 20772–20777. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, H.; Suzumura, A. Gap Junctions and Hemichannels Composed of Connexins: Potential Therapeutic Targets for Neurodegenerative Diseases. Front. Cell Neurosci. 2014, 8, 189. [Google Scholar] [CrossRef] [Green Version]
- Lan, S.H.; Lai, W.T.; Zheng, S.Y.; Yang, L.; Fang, L.C.; Zhou, L.; Tang, B.; Duan, J.; Hong, T. Upregulation of Connexin 40 Mediated by Nitric Oxide Attenuates Cerebral Vasospasm After Subarachnoid Hemorrhage Via the Nitric Oxide-Cyclic Guanosine Monophosphate-Protein Kinase G Pathway. World Neurosurg. 2020, 136, e476–e486. [Google Scholar] [CrossRef]
- Lynn, B.D.; Li, X.; Hormuzdi, S.G.; Griffiths, E.K.; McGlade, C.J.; Nagy, J.I. E3 Ubiquitin Ligases Lnx1 and Lnx2 Localize at Neuronal Gap Junctions Formed by Connexin36 in Rodent Brain and Molecularly Interact with Connexin36. Eur. J. Neurosci. 2018, 48, 3062–3081. [Google Scholar] [CrossRef] [Green Version]
- Molchanova, S.M.; Huupponen, J.; Lauri, S.E.; Taira, T. Gap Junctions Between Ca3 Pyramidal Cells Contribute to Network Synchronization in Neonatal Hippocampus. Neuropharmacology 2016, 107, 9–17. [Google Scholar] [CrossRef]
- Del, C.C.; Iglesias, R.; Zoidl, G.; Dermietzel, R.; Spray, D.C. Calmodulin Dependent Protein Kinase Increases Conductance at Gap Junctions Formed by the Neuronal Gap Junction Protein Connexin36. Brain Res. 2012, 1487, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Lanciotti, A.; Brignone, M.S.; Belfiore, M.; Columba-Cabezas, S.; Mallozzi, C.; Vincentini, O.; Molinari, P.; Petrucci, T.C.; Visentin, S.; Ambrosini, E. Megalencephalic Leukoencephalopathy with Subcortical Cysts Disease-Linked Mlc1 Protein Favors Gap-Junction Intercellular Communication by Regulating Connexin 43 Trafficking in Astrocytes. Cells 2020, 9, 1425. [Google Scholar] [CrossRef] [PubMed]
- Giaume, C.; Naus, C.C.; Saez, J.C.; Leybaert, L. Glial Connexins and Pannexins in the Healthy and Diseased Brain. Physiol. Rev. 2021, 101, 93–145. [Google Scholar] [CrossRef] [PubMed]
- Krutovskikh, V.A.; Piccoli, C.; Yamasaki, H. Gap Junction Intercellular Communication Propagates Cell Death in Cancerous Cells. Oncogene 2002, 21, 1989–1999. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.H.; Weigel, H.; Cotrina, M.L.; Liu, S.; Bueno, E.; Hansen, A.J.; Hansen, T.W.; Goldman, S.; Nedergaard, M. Gap-Junction-Mediated Propagation and Amplification of Cell Injury. Nat. Neurosci. 1998, 1, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Fontes, J.D.; Ramsey, J.; Polk, J.M.; Koop, A.; Denisova, J.V.; Belousov, A.B. Death of Neurons Following Injury Requires Conductive Neuronal Gap Junction Channels but Not a Specific Connexin. PLoS ONE 2015, 10, e125395. [Google Scholar] [CrossRef]
- Ma, D.; Feng, L.; Cheng, Y.; Xin, M.; You, J.; Yin, X.; Hao, Y.; Cui, L.; Feng, J. Astrocytic Gap Junction Inhibition by Carbenoxolone Enhances the Protective Effects of Ischemic Preconditioning Following Cerebral Ischemia. J. Neuroinflammation 2018, 15, 198. [Google Scholar] [CrossRef]
- Manjarrez-Marmolejo, J.; Franco-Perez, J. Gap Junction Blockers: An Overview of their Effects On Induced Seizures in Animal Models. Curr. Neuropharmacol. 2016, 14, 759–771. [Google Scholar] [CrossRef]
- Zhou, S.; Fang, Z.; Wang, G.; Wu, S. Gap Junctional Intercellular Communication Dysfunction Mediates the Cognitive Impairment Induced by Cerebral Ischemia-Reperfusion Injury: Pi3K/Akt Pathway Involved. Am. J. Transl. Res. 2017, 9, 5442–5451. [Google Scholar]
- Bruzzone, R.; Dermietzel, R. Structure and Function of Gap Junctions in the Developing Brain. Cell Tissue Res. 2006, 326, 239–248. [Google Scholar] [CrossRef]
- Orellana, J.A.; Saez, P.J.; Shoji, K.F.; Schalper, K.A.; Palacios-Prado, N.; Velarde, V.; Giaume, C.; Bennett, M.V.; Saez, J.C. Modulation of Brain Hemichannels and Gap Junction Channels by Pro-Inflammatory Agents and their Possible Role in Neurodegeneration. Antioxid. Redox Signal 2009, 11, 369–399. [Google Scholar] [CrossRef]
- Bennett, M.V.; Garre, J.M.; Orellana, J.A.; Bukauskas, F.F.; Nedergaard, M.; Saez, J.C. Connexin and Pannexin Hemichannels in Inflammatory Responses of Glia and Neurons. Brain Res. 2012, 1487, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Acosta, M.L.; Mat, N.M.; Guo, C.X.; Mugisho, O.O.; Coutinho, F.P.; Rupenthal, I.D.; Green, C.R. Connexin Therapeutics: Blocking Connexin Hemichannel Pores Is Distinct from Blocking Pannexin Channels or Gap Junctions. Neural Regen. Res. 2021, 16, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; von Bernhardi, R.; Giaume, C.; Saez, J.C. Glial Hemichannels and their Involvement in Aging and Neurodegenerative Diseases. Rev. Neurosci. 2012, 23, 163–177. [Google Scholar] [CrossRef]
- Gangoso, E.; Talaveron, R.; Jaraiz-Rodriguez, M.; Dominguez-Prieto, M.; Ezan, P.; Koulakoff, A.; Medina, J.M.; Giaume, C.; Tabernero, A. A C-Src Inhibitor Peptide Based On Connexin43 Exerts Neuroprotective Effects through the Inhibition of Glial Hemichannel Activity. Front. Mol. Neurosci. 2017, 10, 418. [Google Scholar] [CrossRef] [Green Version]
- Orellana, J.A.; Froger, N.; Ezan, P.; Jiang, J.X.; Bennett, M.V.; Naus, C.C.; Giaume, C.; Saez, J.C. Atp and Glutamate Released Via Astroglial Connexin 43 Hemichannels Mediate Neuronal Death through Activation of Pannexin 1 Hemichannels. J. Neurochem. 2011, 118, 826–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Z.; Diao, H.; Yu, M.; Ji, X.; Liu, Q.; Chang, X.; Wu, Q. Connexin 43 Mediates Changes in Protein Phosphorylation in Hk-2 Cells During Chronic Cadmium Exposure. Environ. Toxicol. Pharmacol. 2017, 53, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Shen, J.; Zhu, K.; Zhou, H.; Tian, H.; Yu, G. Efficacy of Statins in Cerebral Vasospasm, Mortality, and Delayed Cerebral Ischemia in Patients with Aneurysmal Subarachnoid Hemorrhage: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. World Neurosurg. 2019, 131, e65–e73. [Google Scholar] [CrossRef]
- Bai, Q.; Xue, M.; Yong, V.W. Microglia and Macrophage Phenotypes in Intracerebral Haemorrhage Injury: Therapeutic Opportunities. Brain 2020, 143, 1297–1314. [Google Scholar] [CrossRef]
- Yong, H.; Rawji, K.S.; Ghorbani, S.; Xue, M.; Yong, V.W. The Benefits of Neuroinflammation for the Repair of the Injured Central Nervous System. Cell Mol. Immunol. 2019, 16, 540–546. [Google Scholar] [CrossRef]
- Yang, Y.; Xi, Z.; Xue, Y.; Ren, J.; Sun, Y.; Wang, B.; Zhong, Z.; Yang, G.Y.; Sun, Q.; Bian, L. Hemoglobin Pretreatment Endows Rat Cortical Astrocytes Resistance to Hemin-Induced Toxicity Via Nrf2/Ho-1 Pathway. Exp. Cell Res. 2017, 361, 217–224. [Google Scholar] [CrossRef]
- Munakata, M.; Shirakawa, H.; Nagayasu, K.; Miyanohara, J.; Miyake, T.; Nakagawa, T.; Katsuki, H.; Kaneko, S. Transient Receptor Potential Canonical 3 Inhibitor Pyr3 Improves Outcomes and Attenuates Astrogliosis After Intracerebral Hemorrhage in Mice. Stroke 2013, 44, 1981–1987. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Davidson, J.O.; Gunn, K.C.; Phillips, A.R.; Green, C.R.; Gunn, A.J. Role of Hemichannels in Cns Inflammation and the Inflammasome Pathway. Adv. Protein Chem. Struct. Biol. 2016, 104, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Cao, X.; Li, W.; Liu, P.; Zhao, Y.; Song, L.; Chen, J.; Chen, B.; Yu, W.; Xu, Y. Targeting Connexin 43 Provides Anti-Inflammatory Effects after Intracerebral Hemorrhage Injury by Regulating Yap Signaling. J. Neuroinflammation 2020, 17, 322. [Google Scholar] [CrossRef]
- Yang, Y.; Ren, J.; Sun, Y.; Xue, Y.; Zhang, Z.; Gong, A.; Wang, B.; Zhong, Z.; Cui, Z.; Xi, Z.; et al. A Connexin43/Yap Axis Regulates Astroglial-Mesenchymal Transition in Hemoglobin Induced Astrocyte Activation. Cell Death Differ. 2018, 25, 1870–1884. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Liu, Y.; Wei, R.; Khan, S.; Xue, M.; Yong, V.W. The Combination of Deferoxamine and Minocycline Strengthens Neuroprotective Effect On Acute Intracerebral Hemorrhage in Rats. Neurol. Res. 2021, 43, 854–864. [Google Scholar] [CrossRef]
- Davalos, D.; Ryu, J.K.; Merlini, M.; Baeten, K.M.; Le Moan, N.; Petersen, M.A.; Deerinck, T.J.; Smirnoff, D.S.; Be-dard, C.; Hakozaki, H.; et al. Fibrinogen-Induced Perivascular Microglial Clustering is Required for the Development of Axonal Damage in Neuroinflammation. Nat. Commun. 2012, 3, 1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Retamal, M.A.; Froger, N.; Palacios-Prado, N.; Ezan, P.; Saez, P.J.; Saez, J.C.; Giaume, C. Cx43 Hemichannels and Gap Junction Channels in Astrocytes are Regulated Oppositely by Proinflammatory Cytokines Released From Activated Microglia. J. Neurosci. 2007, 27, 13781–13792. [Google Scholar] [CrossRef]
- Makarenkova, H.P.; Shestopalov, V.I. The Role of Pannexin Hemichannels in Inflammation and Regeneration. Front Physiol. 2014, 5, 63. [Google Scholar] [CrossRef] [Green Version]
- Meme, W.; Ezan, P.; Venance, L.; Glowinski, J.; Giaume, C. Atp-Induced Inhibition of Gap Junctional Communication is Enhanced by Interleukin-1 Beta Treatment in Cultured Astrocytes. Neuroscience 2004, 126, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Abudara, V.; Roux, L.; Dallerac, G.; Matias, I.; Dulong, J.; Mothet, J.P.; Rouach, N.; Giaume, C. Activated Microglia Impairs Neuroglial Interaction by Opening Cx43 Hemichannels in Hippocampal Astrocytes. Glia 2015, 63, 795–811. [Google Scholar] [CrossRef]
- John, G.R.; Scemes, E.; Suadicani, S.O.; Liu, J.S.; Charles, P.C.; Lee, S.C.; Spray, D.C.; Brosnan, C.F. Il-1Beta Differentially Regulates Calcium Wave Propagation Between Primary Human Fetal Astrocytes Via Pathways Involving P2 Receptors and Gap Junction Channels. Proc. Natl. Acad. Sci. USA 1999, 96, 11613–11618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zvalova, D.; Cordier, J.; Mesnil, M.; Junier, M.P.; Chneiweiss, H. P38/Sapk2 Controls Gap Junction Closure in Astrocytes. Glia 2004, 46, 323–333. [Google Scholar] [CrossRef]
- Johnson, A.M.; Roach, J.P.; Hu, A.; Stamatovic, S.M.; Zochowski, M.R.; Keep, R.F.; Andjelkovic, A.V. Connexin 43 Gap Junctions Contribute to Brain Endothelial Barrier Hyperpermeability in Familial Cerebral Cavernous Malformations Type Iii by Modulating Tight Junction Structure. FASEB J. 2018, 32, 2615–2629. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Lu, S.; Nagy, J.I. Direct Association of Connexin36 with Zonula Occludens-2 and Zonula Occludens-3. Neurochem. Int. 2009, 54, 393–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Chu, H.; Tang, Y.; Dong, Q. The Role of Connexin43 in Hemorrhagic Transformation After Thrombolysis in Vivo and in Vitro. Neuroscience 2016, 329, 54–65. [Google Scholar] [CrossRef]
- De Bock, M.; Culot, M.; Wang, N.; Bol, M.; Decrock, E.; De Vuyst, E.; Da, C.A.; Dauwe, I.; Vinken, M.; Simon, A.M.; et al. Connexin Channels Provide a Target to Manipulate Brain Endothelial Calcium Dynamics and Blood-Brain Barrier Permeability. J. Cereb. Blood Flow Metab. 2011, 31, 1942–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danesh-Meyer, H.V.; Kerr, N.M.; Zhang, J.; Eady, E.K.; O’Carroll, S.J.; Nicholson, L.F.; Johnson, C.S.; Green, C.R. Connexin43 Mimetic Peptide Reduces Vascular Leak and Retinal Ganglion Cell Death Following Retinal Ischaemia. Brain 2012, 135, 506–520. [Google Scholar] [CrossRef] [Green Version]
- Rozas-Villanueva, M.F.; Casanello, P.; Retamal, M.A. Role of Ros/Rns in Preeclampsia: Are Connexins the Missing Piece? Int. J. Mol. Sci. 2020, 21, 4698. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Orellana, J.A.; von Bernhardi, R. Understanding Risk Factors for Alzheimer’s Disease: Interplay of Neuroinflammation, Connexin-Based Communication and Oxidative Stress. Arch. Med. Res. 2012, 43, 632–644. [Google Scholar] [CrossRef]
- Le, H.T.; Sin, W.C.; Lozinsky, S.; Bechberger, J.; Vega, J.L.; Guo, X.Q.; Saez, J.C.; Naus, C.C. Gap Junction Intercellular Communication Mediated by Connexin43 in Astrocytes is Essential for their Resistance to Oxidative Stress. J. Biol. Chem. 2014, 289, 1345–1354. [Google Scholar] [CrossRef] [Green Version]
- Balboa, E.; Saavedra, F.; Cea, L.A.; Ramirez, V.; Escamilla, R.; Vargas, A.A.; Regueira, T.; Saez, J.C. Vitamin E Blocks Connexin Hemichannels and Prevents Deleterious Effects of Glucocorticoid Treatment On Skeletal Muscles. Int. J. Mol. Sci. 2020, 21, 4094. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.W.; Ji, D.D.; Li, Q.Q.; Zhang, T.; Luo, L. Inhibition of Connexin 43 Attenuates Oxidative Stress and Apoptosis in Human Umbilical Vein Endothelial Cells. BMC Pulm. Med. 2020, 20, 19. [Google Scholar] [CrossRef]
- Chen, X.; Liang, H.; Xi, Z.; Yang, Y.; Shan, H.; Wang, B.; Zhong, Z.; Xu, C.; Yang, G.Y.; Sun, Q.; et al. Bm-Msc Transplantation Alleviates Intracerebral Hemorrhage-Induced Brain Injury, Promotes Astrocytes Vimentin Expression, and Enhances Astrocytes Antioxidation Via the Cx43/Nrf2/Ho-1 Axis. Front Cell Dev. Biol. 2020, 8, 302. [Google Scholar] [CrossRef]
- Smetana, K.S.; Buschur, P.L.; Owusu-Guha, J.; May, C.C. Pharmacologic Management of Cerebral Vasospasm in Aneurysmal Subarachnoid Hemorrhage. Crit. Care Nurs. Q. 2020, 43, 138–156. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yan, J.; Zhang, J.A.; Zhou, X.H.; Fang, C.; Zeng, E.M.; Tang, B.; Duan, J.; Lu, G.H.; Hong, T. The Important Role of Connexin 43 in Subarachnoid Hemorrhage-Induced Cerebral Vasospasm. J. Transl. Med. 2019, 17, 433. [Google Scholar] [CrossRef] [Green Version]
- Lei, C.; Ruan, Y.; Cai, C.; He, B.; Zhao, D. Role of P38 Mitogen-Activated Protein Kinase On Cx43 Phosphorylation in Cerebral Vasospasm After Subarachnoid Hemorrhage. Int. J. Neurosci. 2019, 129, 461–469. [Google Scholar] [CrossRef]
- Hong, T.; Wang, H.; Wang, Y.; Wang, H. Effects of Gap Junctional Blockers On Cerebral Vasospasm After Subarachnoid Hemorrhage in Rabbits. Neurol. Res. 2009, 31, 238–244. [Google Scholar] [CrossRef]
- Li, H.; Liu, S.; Sun, X.; Yang, J.; Yang, Z.; Shen, H.; Li, X.; Liu, Y.; Chen, G. Critical Role for Annexin a7 in Secondary Brain Injury Mediated by its Phosphorylation After Experimental Intracerebral Hemorrhage in Rats. Neurobiol. Dis. 2018, 110, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Weilinger, N.L.; Lohman, A.W.; Rakai, B.D.; Ma, E.M.; Bialecki, J.; Maslieieva, V.; Rilea, T.; Bandet, M.V.; Ikuta, N.T.; Scott, L.; et al. Metabotropic Nmda Receptor Signaling Couples Src Family Kinases to Pannexin-1 During Excitotoxicity. Nat. Neurosci. 2016, 19, 432–442. [Google Scholar] [CrossRef]
- Huang, Y.; Lin, J.; Chen, X.; Lin, J. Pannexin-1 Contributes to the Apoptosis of Spinal Neurocytes in Spinal Cord Injury. Front. Physiol. 2021, 12, 656647. [Google Scholar] [CrossRef]
- Imamura, H.; Sakamoto, S.; Yoshida, T.; Matsui, Y.; Penuela, S.; Laird, D.W.; Mizukami, S.; Kikuchi, K.; Kakizuka, A. Single-Cell Dynamics of Pannexin-1-Facilitated Programmed Atp Loss During Apoptosis. Elife 2020, 9, e61960. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, C.; Wang, Z.; Shen, H.; Wen, Z.; Chen, D.; Sun, Q.; Chen, G. Pannexin-1 is Involved in Neuronal Apoptosis and Degeneration in Experimental Intracerebral Hemorrhage in Rats. Mol. Med. Rep. 2018, 17, 5684–5691. [Google Scholar] [CrossRef]
- Chu, H.; Gao, Z.; Huang, C.; Dong, J.; Tang, Y.; Dong, Q. Relationship Between Hematoma Expansion Induced by Hypertension and Hyperglycemia and Blood-Brain Barrier Disruption in Mice and its Possible Mechanism: Role of Aquaporin-4 and Connexin43. Neurosci. Bull. 2020, 36, 1369–1380. [Google Scholar] [CrossRef] [PubMed]
- de Pina-Benabou, M.H.; Szostak, V.; Kyrozis, A.; Rempe, D.; Uziel, D.; Urban-Maldonado, M.; Benabou, S.; Spray, D.C.; Federoff, H.J.; Stanton, P.K.; et al. Blockade of Gap Junctions in Vivo Provides Neuroprotection After Perinatal Global Ischemia. Stroke 2005, 36, 2232–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayer, R.; Chen, W.; Sugawara, T.; Suzuki, H.; Zhang, J.H. Role of Gap Junctions in Early Brain Injury Following Subarachnoid Hemorrhage. Brain Res. 2010, 1315, 150–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manaenko, A.; Lekic, T.; Sozen, T.; Tsuchiyama, R.; Zhang, J.H.; Tang, J. Effect of Gap Junction Inhibition On Intracerebral Hemorrhage-Induced Brain Injury in Mice. Neurol. Res. 2009, 31, 173–178. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Khan, S.; Liu, Y.; Siddique, R.; Zhang, R.; Yong, V.W.; Xue, M. Gap Junctions and Hemichannels Composed of Connexins and Pannexins Mediate the Secondary Brain Injury Following Intracerebral Hemorrhage. Biology 2022, 11, 27. https://doi.org/10.3390/biology11010027
Zhang Y, Khan S, Liu Y, Siddique R, Zhang R, Yong VW, Xue M. Gap Junctions and Hemichannels Composed of Connexins and Pannexins Mediate the Secondary Brain Injury Following Intracerebral Hemorrhage. Biology. 2022; 11(1):27. https://doi.org/10.3390/biology11010027
Chicago/Turabian StyleZhang, Yan, Suliman Khan, Yang Liu, Rabeea Siddique, Ruiyi Zhang, Voon Wee Yong, and Mengzhou Xue. 2022. "Gap Junctions and Hemichannels Composed of Connexins and Pannexins Mediate the Secondary Brain Injury Following Intracerebral Hemorrhage" Biology 11, no. 1: 27. https://doi.org/10.3390/biology11010027