
Bcl-xL Is Required by Primary Hippocampal Neurons during Development to Support Local Energy Metabolism at Neurites

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Culture of Primary Hippocampal Neurons
2.2. Live Imaging and Mitochondrial Motility Analysis
2.3. Analysis of Neurite Branching
2.4. Measurement of the ATP/ADP Ratio
2.5. Measurement of Mitochondrial Potential (Δψ)
2.6. Immunoblots
2.7. Calcein-AM, and Propidium Iodide (PI) Staining
2.8. Fluo-4 Staining
2.9. Immunocytochemistry
2.10. Statistical Analysis
3. Results
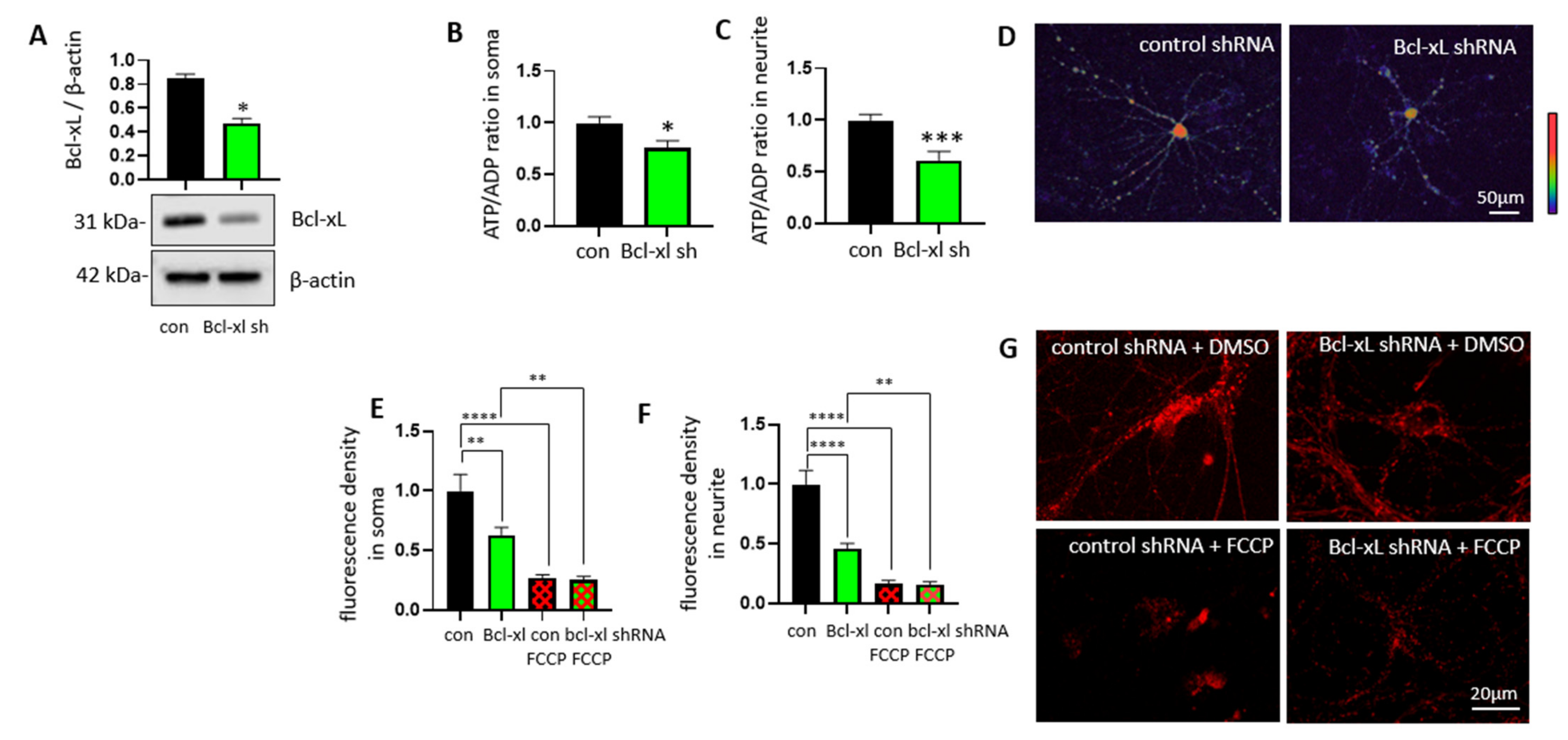
3.1. Bcl-xL Depletion Lowers ATP/ADP Ratios at Neurites of Primary Hippocampal Neurons
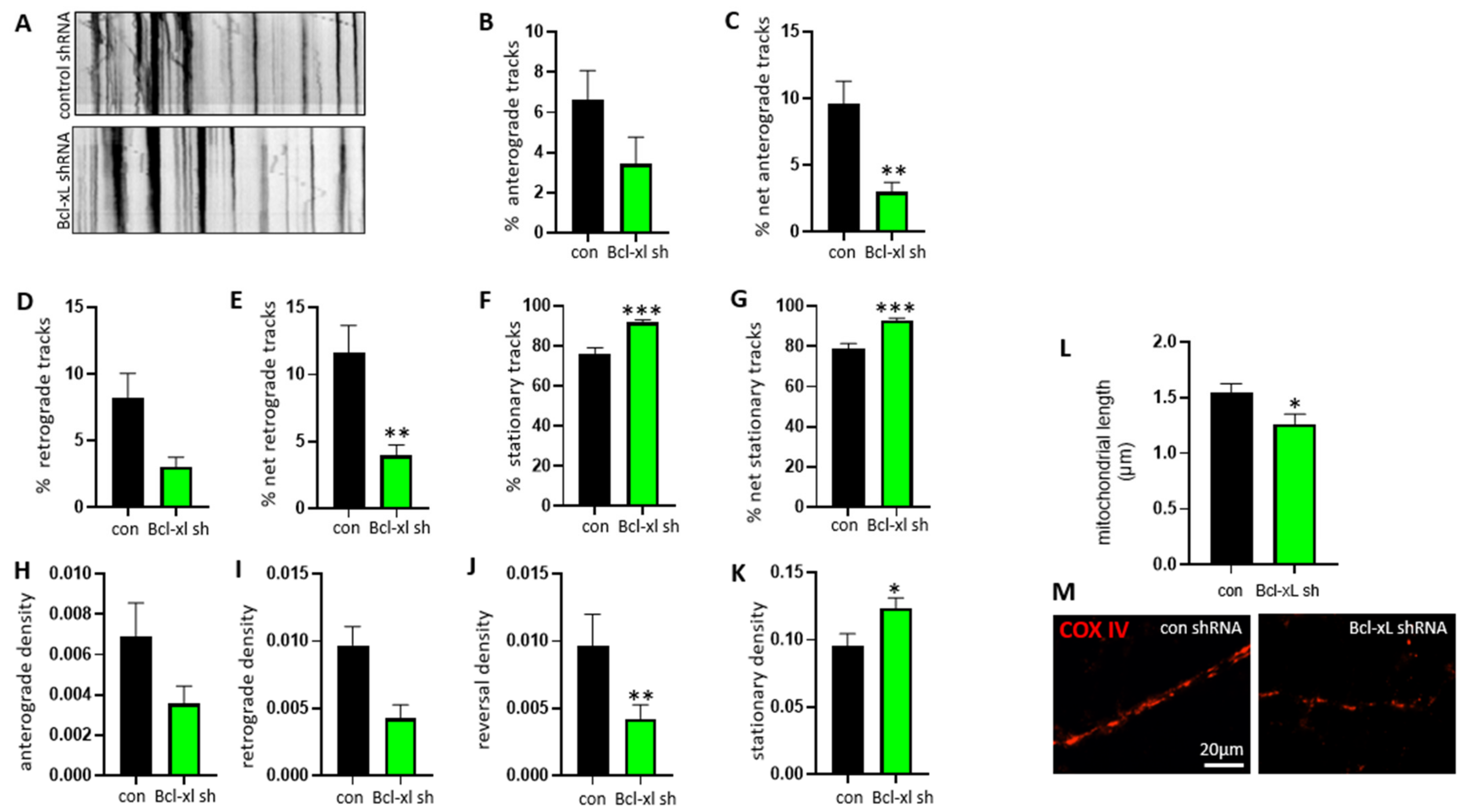
3.2. Bcl-xL Depletion Impairs Normal Mitochondrial Motility Patterns in Neurites of Primary Hippocampal Neurons
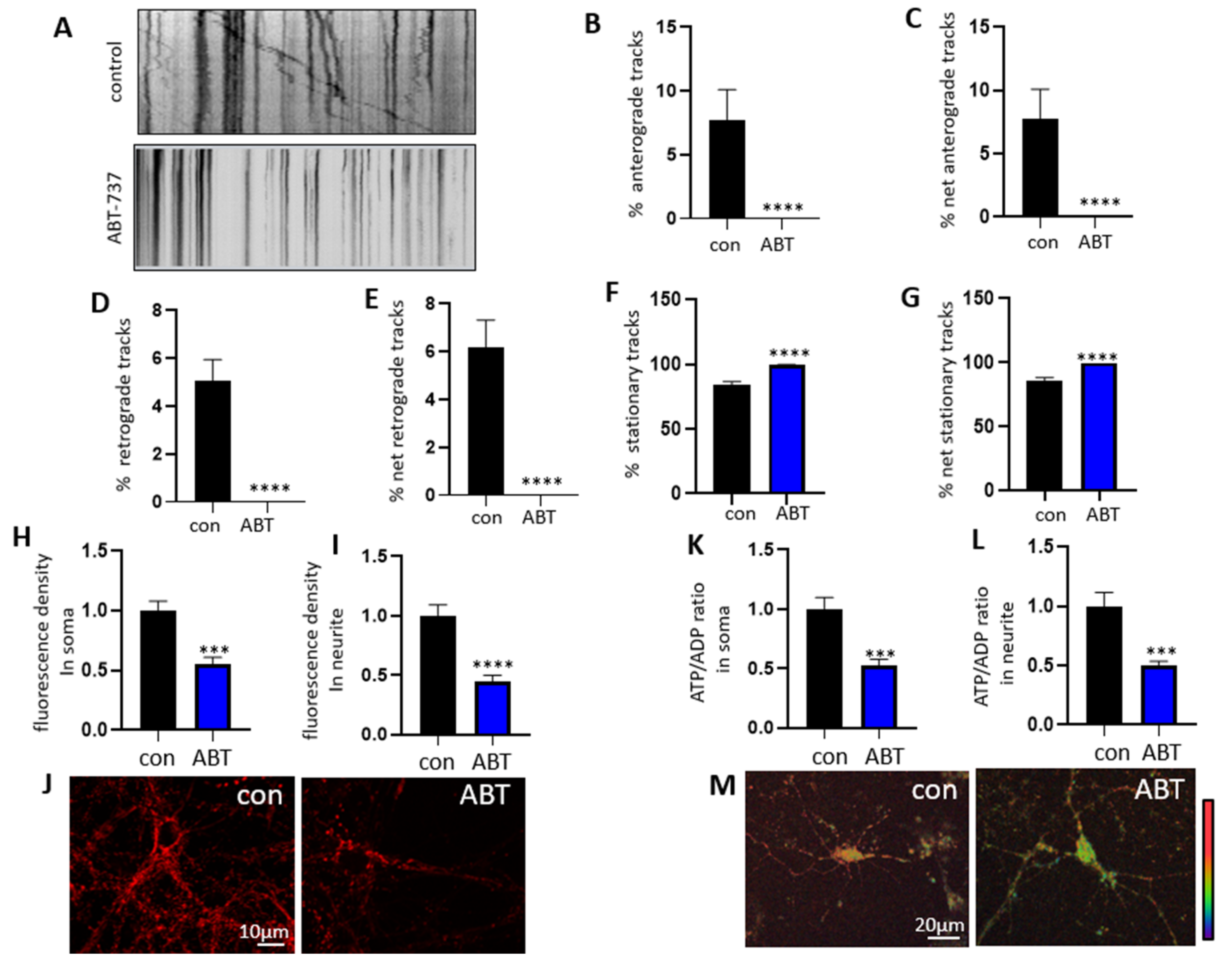
3.3. Treatment with ABT-263 Impairs Mitochondrial Motility in Primary Hippocampal Neurons
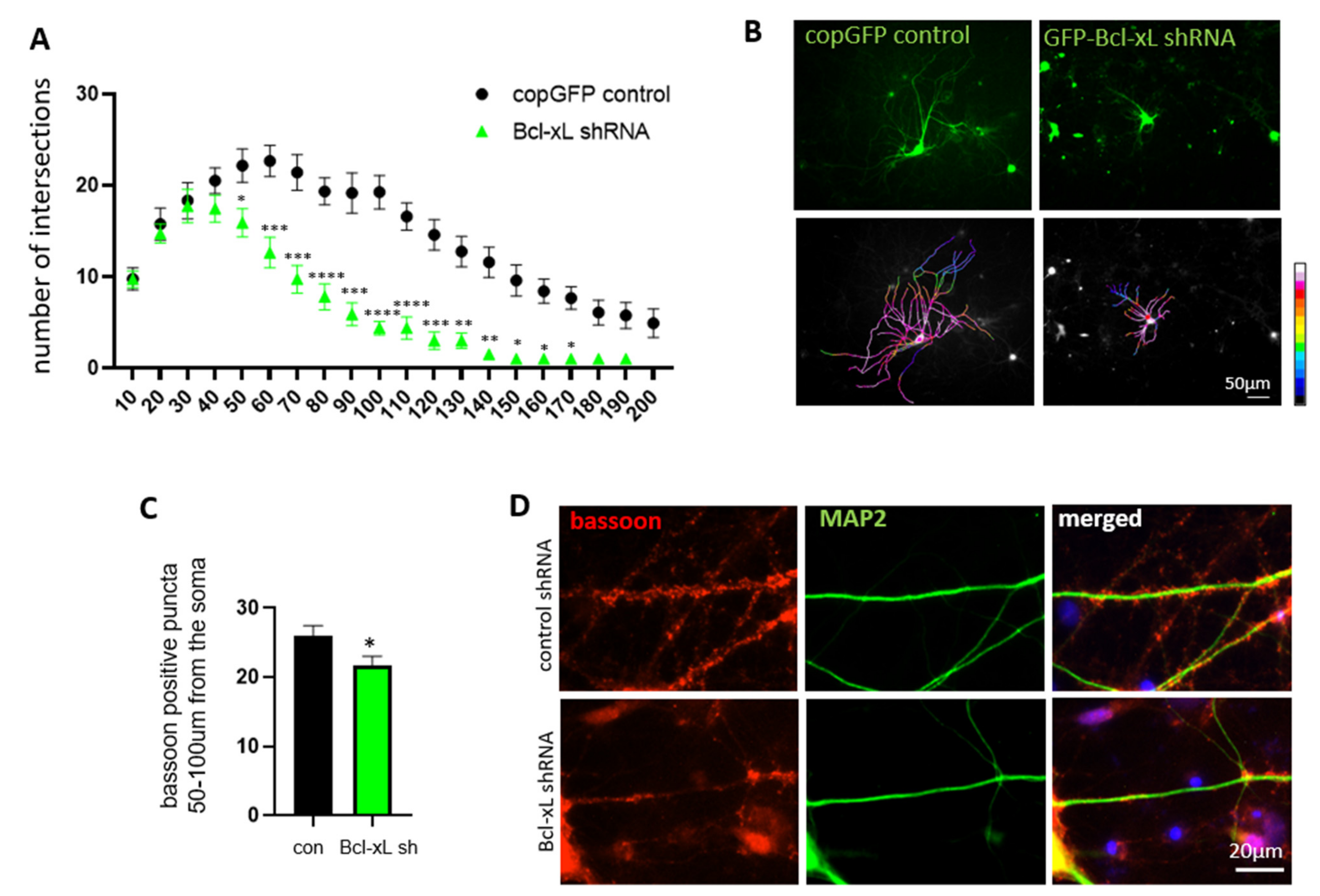
3.4. Bcl-xL Depletion Impairs Neurite Arborization and Synapse Formation in Primary Hippocampal Neurons
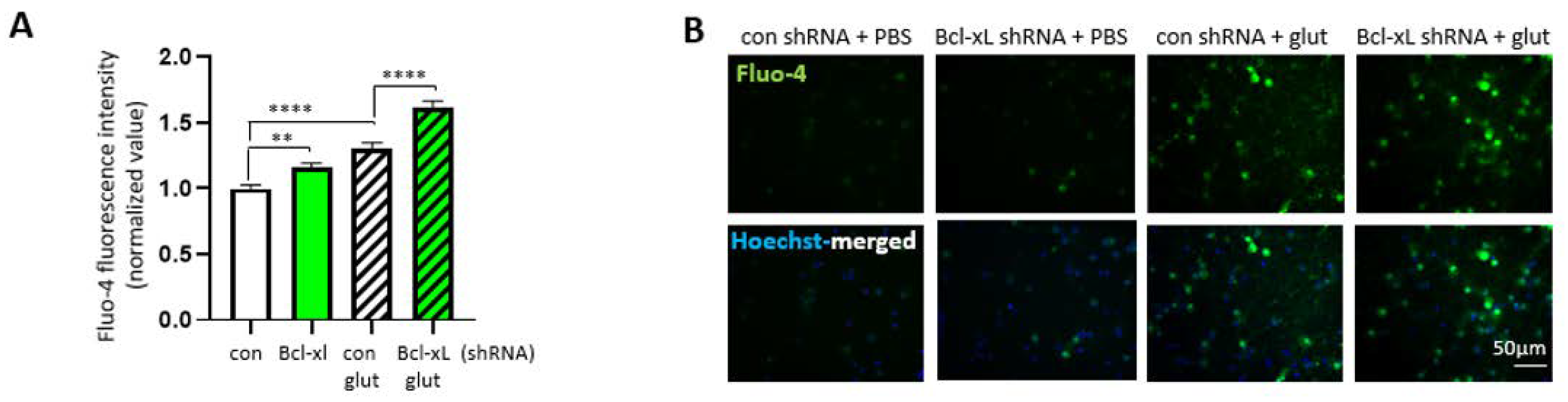
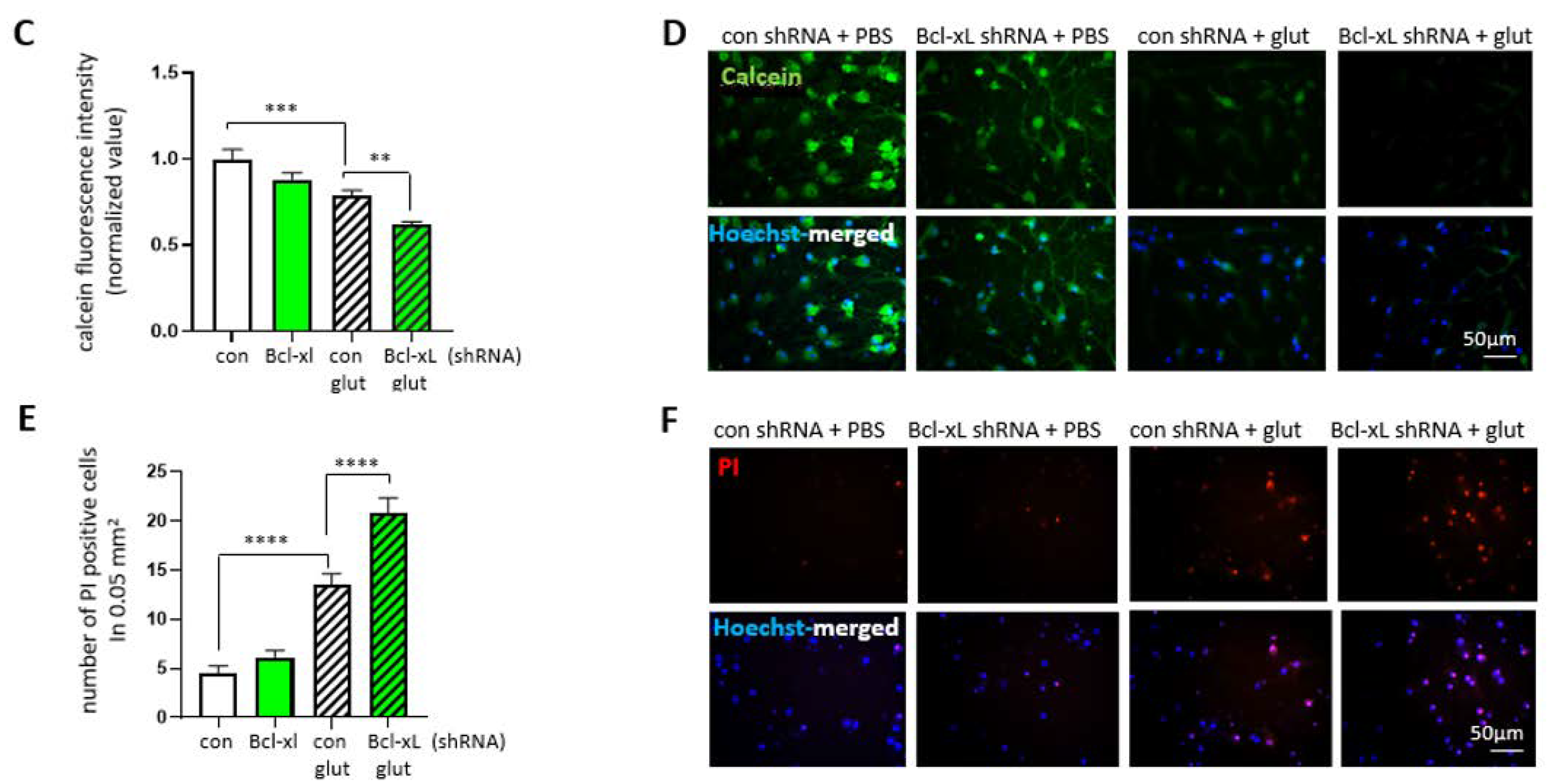
3.5. Bcl-xL Depletion Increases the Susceptibility of Primary Hippocampal Neuron to Excitotoxicity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boise, L.H.; Gonzalez-Garcia, M.; Postema, C.E.; Ding, L.; Lindsten, T.; Turka, L.A.; Mao, X.; Nunez, G.; Thompson, C.B. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993, 74, 597–608. [Google Scholar] [CrossRef] [Green Version]
- Soane, L.; Siegel, Z.T.; Schuh, R.A.; Fiskum, G. Postnatal developmental regulation of Bcl-2 family proteins in brain mitochondria. J. Neurosci. Res. 2008, 86, 1267–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattler, M.; Liang, H.; Nettesheim, D.; Meadows, R.P.; Harlan, J.E.; Eberstadt, M.; Yoon, H.S.; Shuker, S.B.; Chang, B.S.; Minn, A.J.; et al. Structure of Bcl-xL-Bak peptide complex: Recognition between regulators of apoptosis. Science 1997, 275, 983–986. [Google Scholar] [CrossRef] [PubMed]
- Cheng, E.H.; Levine, B.; Boise, L.H.; Thompson, C.B.; Hardwick, J.M. Bax-independent inhibition of apoptosis by Bcl-XL. Nature 1996, 379, 554–556. [Google Scholar] [CrossRef] [PubMed]
- Park, H.A.; Licznerski, P.; Mnatsakanyan, N.; Niu, Y.; Sacchetti, S.; Wu, J.; Polster, B.M.; Alavian, K.N.; Jonas, E.A. Inhibition of Bcl-xL prevents pro-death actions of DeltaN-Bcl-xL at the mitochondrial inner membrane during glutamate excitotoxicity. Cell Death Differ. 2017, 24, 1963–1974. [Google Scholar] [CrossRef] [Green Version]
- Arena, G.; Gelmetti, V.; Torosantucci, L.; Vignone, D.; Lamorte, G.; De Rosa, P.; Cilia, E.; Jonas, E.A.; Valente, E.M. PINK1 protects against cell death induced by mitochondrial depolarization, by phosphorylating Bcl-xL and impairing its pro-apoptotic cleavage. Cell Death Differ. 2013, 20, 920–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsadanian, A.S.; Cheng, Y.; Keller-Peck, C.R.; Holtzman, D.M.; Snider, W.D. Bcl-xL is an antiapoptotic regulator for postnatal CNS neurons. J. Neurosci. 1998, 18, 1009–1019. [Google Scholar] [CrossRef] [Green Version]
- Panickar, K.S.; Nonner, D.; Barrett, J.N. Overexpression of Bcl-xl protects septal neurons from prolonged hypoglycemia and from acute ischemia-like stress. Neuroscience 2005, 135, 73–80. [Google Scholar] [CrossRef]
- Jonas, E.A. Contributions of Bcl-xL to acute and long term changes in bioenergetics during neuronal plasticity. Biochim. Biophys. Acta 2013, 1842, 1168–1178. [Google Scholar] [CrossRef] [Green Version]
- Park, H.A.; Jonas, E.A. Mitochondrial membrane protein Bcl-xL, a regulator of adult neuronal growth and synaptic plasticity: Multiple functions beyond apoptosis. Neural Regen. Res. 2014, 9, 1706–1707. [Google Scholar] [CrossRef]
- Chen, Y.B.; Aon, M.A.; Hsu, Y.T.; Soane, L.; Teng, X.; McCaffery, J.M.; Cheng, W.C.; Qi, B.; Li, H.; Alavian, K.N.; et al. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J. Cell Biol. 2011, 195, 263–276. [Google Scholar] [CrossRef]
- Alavian, K.N.; Li, H.; Collis, L.; Bonanni, L.; Zeng, L.; Sacchetti, S.; Lazrove, E.; Nabili, P.; Flaherty, B.; Graham, M.; et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat. Cell Biol. 2011, 13, 1224–1233. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Park, H.A.; Mnatsakanyan, N.; Niu, Y.; Licznerski, P.; Wu, J.; Miranda, P.; Graham, M.; Tang, J.; Boon, A.J.W.; et al. Parkinson’s disease protein DJ-1 regulates ATP synthase protein components to increase neuronal process outgrowth. Cell Death Dis. 2019, 10, 469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Chen, Y.; Jones, A.F.; Sanger, R.H.; Collis, L.P.; Flannery, R.; McNay, E.C.; Yu, T.; Schwarzenbacher, R.; Bossy, B.; et al. Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 2169–2174. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Alavian, K.N.; Lazrove, E.; Mehta, N.; Jones, A.; Zhang, P.; Licznerski, P.; Graham, M.; Uo, T.; Guo, J.; et al. A Bcl-xL-Drp1 complex regulates synaptic vesicle membrane dynamics during endocytosis. Nat. Cell Biol. 2013, 15, 773–785. [Google Scholar] [CrossRef]
- Frederick, R.L.; Shaw, J.M. Moving mitochondria: Establishing distribution of an essential organelle. Traffic 2007, 8, 1668–1675. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Enriquez, S.; Kim, I.; Currin, R.T.; Lemasters, J.J. Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy 2006, 2, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chan, D.C. Mitochondrial dynamics--fusion, fission, movement, and mitophagy--in neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, T.; Xu, C.; Funahashi, Y.; Namba, T.; Kaibuchi, K. Neuronal polarization. Development 2015, 142, 2088–2093. [Google Scholar] [CrossRef] [Green Version]
- Safiulina, D.; Kaasik, A. Energetic and dynamic: How mitochondria meet neuronal energy demands. PLoS Biol. 2013, 11, e1001755. [Google Scholar] [CrossRef]
- Yu, S.B.; Pekkurnaz, G. Mechanisms Orchestrating Mitochondrial Dynamics for Energy Homeostasis. J. Mol. Biol. 2018, 430, 3922–3941. [Google Scholar] [CrossRef] [PubMed]
- Boldogh, I.R.; Pon, L.A. Mitochondria on the move. Trends Cell Biol. 2007, 17, 502–510. [Google Scholar] [CrossRef]
- Chen, Y.; Sheng, Z.H. Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. J. Cell Biol. 2013, 202, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Tian, J.H.; Pan, P.Y.; Zald, P.; Li, C.; Deng, C.; Sheng, Z.H. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 2008, 132, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, D.G.; Budd, S.L. Mitochondria and neuronal survival. Physiol. Rev. 2000, 80, 315–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, Z.H. The Interplay of Axonal Energy Homeostasis and Mitochondrial Trafficking and Anchoring. Trends Cell Biol. 2017, 27, 403–416. [Google Scholar] [CrossRef]
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef] [Green Version]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. 1), 216–231. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Swerdlow, R.H. Relationships Between Mitochondria and Neuroinflammation: Implications for Alzheimer’s Disease. Curr. Top. Med. Chem. 2016, 16, 849–857. [Google Scholar] [CrossRef]
- Berman, S.B.; Chen, Y.B.; Qi, B.; McCaffery, J.M.; Rucker, E.B., III; Goebbels, S.; Nave, K.A.; Arnold, B.A.; Jonas, E.A.; Pineda, F.J.; et al. Bcl-x L increases mitochondrial fission, fusion, and biomass in neurons. J. Cell Biol. 2009, 184, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Park, H.A.; Licznerski, P.; Alavian, K.N.; Shanabrough, M.; Jonas, E.A. Bcl-xL Is Necessary for Neurite Outgrowth in Hippocampal Neurons. Antioxid. Redox Signal. 2015, 22, 93–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaudoin, G.M., III; Lee, S.H.; Singh, D.; Yuan, Y.; Ng, Y.G.; Reichardt, L.F.; Arikkath, J. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat. Protoc. 2012, 7, 1741–1754. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.; Banker, G. Culturing hippocampal neurons. Nat. Protoc. 2006, 1, 2406–2415. [Google Scholar] [CrossRef]
- Neumann, S.; Chassefeyre, R.; Campbell, G.E.; Encalada, S.E. KymoAnalyzer: A software tool for the quantitative analysis of intracellular transport in neurons. Traffic 2017, 18, 71–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longair, M.H.; Baker, D.A.; Armstrong, J.D. Simple Neurite Tracer: Open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics 2011, 27, 2453–2454. [Google Scholar] [CrossRef]
- Pool, M.; Thiemann, J.; Bar-Or, A.; Fournier, A.E. NeuriteTracer: A novel ImageJ plugin for automated quantification of neurite outgrowth. J. Neurosci. Methods 2008, 168, 134–139. [Google Scholar] [CrossRef]
- Tantama, M.; Martinez-Francois, J.R.; Mongeon, R.; Yellen, G. Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat. Commun. 2013, 4, 2550. [Google Scholar] [CrossRef] [Green Version]
- Berg, J.; Hung, Y.P.; Yellen, G. A genetically encoded fluorescent reporter of ATP:ADP ratio. Nat. Methods 2009, 6, 161–166. [Google Scholar] [CrossRef] [Green Version]
- Vaarmann, A.; Mandel, M.; Zeb, A.; Wareski, P.; Liiv, J.; Kuum, M.; Antsov, E.; Liiv, M.; Cagalinec, M.; Choubey, V.; et al. Mitochondrial biogenesis is required for axonal growth. Development 2016, 143, 1981–1992. [Google Scholar] [CrossRef] [Green Version]
- Reid, A.B.; Kurten, R.C.; McCullough, S.S.; Brock, R.W.; Hinson, J.A. Mechanisms of acetaminophen-induced hepatotoxicity: Role of oxidative stress and mitochondrial permeability transition in freshly isolated mouse hepatocytes. J. Pharmacol. Exp. Ther. 2005, 312, 509–516. [Google Scholar] [CrossRef]
- Park, H.A.; Khanna, S.; Rink, C.; Gnyawali, S.; Roy, S.; Sen, C.K. Glutathione disulfide induces neural cell death via a 12-lipoxygenase pathway. Cell Death Differ. 2009, 16, 1167–1179. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Chen, S.; Luo, Y.; Chen, Z.; Liu, L.; Zhou, H.; Chen, W.; Shen, T.; Han, X.; Chen, L.; et al. Calcium signaling is involved in cadmium-induced neuronal apoptosis via induction of reactive oxygen species and activation of MAPK/mTOR network. PLoS ONE 2011, 6, e19052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gundelfinger, E.D.; Reissner, C.; Garner, C.C. Role of Bassoon and Piccolo in Assembly and Molecular Organization of the Active Zone. Front. Synaptic. Neurosci. 2015, 7, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.A.; Mnatsakanyan, N.; Broman, K.; Davis, A.U.; May, J.; Licznerski, P.; Crowe-White, K.M.; Lackey, K.H.; Jonas, E.A. Alpha-Tocotrienol Prevents Oxidative Stress-Mediated Post-Translational Cleavage of Bcl-xL in Primary Hippocampal Neurons. Int. J. Mol. Sci. 2019, 21, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mnatsakanyan, N.; Jonas, E.A. ATP synthase c-subunit ring as the channel of mitochondrial permeability transition: Regulator of metabolism in development and degeneration. J. Mol. Cell Cardiol. 2020, 144, 109–118. [Google Scholar] [CrossRef]
- Mnatsakanyan, N.; Llaguno, M.C.; Yang, Y.; Yan, Y.; Weber, J.; Sigworth, F.J.; Jonas, E.A. A mitochondrial megachannel resides in monomeric F1FO ATP synthase. Nat. Commun. 2019, 10, 5823. [Google Scholar] [CrossRef]
- Alavian, K.N.; Beutner, G.; Lazrove, E.; Sacchetti, S.; Park, H.A.; Licznerski, P.; Li, H.; Nabili, P.; Hockensmith, K.; Graham, M.; et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10580–10585. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, H.; Carter, A.P. Review: Structure and mechanism of the dynein motor ATPase. Biopolymers 2016, 105, 557–567. [Google Scholar] [CrossRef]
- Gilbert, S.P.; Webb, M.R.; Brune, M.; Johnson, K.A. Pathway of processive ATP hydrolysis by kinesin. Nature 1995, 373, 671–676. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Edamatsu, M.; Toyoshima, Y.Y. Multiple ATP-hydrolyzing sites that potentially function in cytoplasmic dynein. Proc. Natl. Acad. Sci. USA 2004, 101, 12865–12869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 family reunion. Mol. Cell 2010, 37, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Dai, S.; Zhu, Y.; Marrack, P.; Kappler, J.W. The structure of a Bcl-xL/Bim fragment complex: Implications for Bim function. Immunity 2003, 19, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Puthalakath, H.; Huang, D.C.; O’Reilly, L.A.; King, S.M.; Strasser, A. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol. Cell 1999, 3, 287–296. [Google Scholar] [CrossRef]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jansen, J.; Scott, M.; Amjad, E.; Stumpf, A.; Lackey, K.H.; Caldwell, K.A.; Park, H.-A. Bcl-xL Is Required by Primary Hippocampal Neurons during Development to Support Local Energy Metabolism at Neurites. Biology 2021, 10, 772. https://doi.org/10.3390/biology10080772
Jansen J, Scott M, Amjad E, Stumpf A, Lackey KH, Caldwell KA, Park H-A. Bcl-xL Is Required by Primary Hippocampal Neurons during Development to Support Local Energy Metabolism at Neurites. Biology. 2021; 10(8):772. https://doi.org/10.3390/biology10080772
Chicago/Turabian StyleJansen, Joseph, Madison Scott, Emma Amjad, Allison Stumpf, Kimberly H. Lackey, Kim A. Caldwell, and Han-A Park. 2021. "Bcl-xL Is Required by Primary Hippocampal Neurons during Development to Support Local Energy Metabolism at Neurites" Biology 10, no. 8: 772. https://doi.org/10.3390/biology10080772