


pH-Induced Orthogonal Photoresponse of trans-Chalcone Isomers and Related Compounds in Equilibria

1
Department of Chemistry, St. Joseph’s University, 245 Clinton Ave., Brooklyn, NY 11205, USA
2
Department of Chemistry, Hunter College-City University of New York, 695 Park Avenue, New York, NY 10065, USA
3
Department of Chemistry, York College-City University of New York, 94-20 Guy R. Brewer Blvd. Jamaica, New York, NY 11451, USA
*
Author to whom correspondence should be addressed.
Colorants 2023, 2(1), 58-72; https://doi.org/10.3390/colorants2010005
Submission received: 20 December 2022
/
Revised: 13 January 2023
/
Accepted: 31 January 2023
/
Published: 17 February 2023
(This article belongs to the Special Issue Recent Progress on Functional Dyes and Their Applications)
Abstract
:Photoresponsive molecular devices can be a valuable tool to promote chemical changes in response to multiple signals, such as photons and pH, to deliver drugs or to detect physiological conditions in vivo. For example, trans-chalcones (Ct) from 4′-hydroxyflavylium (F1) and 7-hydroxyflavylium (F2) can undergo cis-trans isomerization by photoreaction into many different structures. The isomerization takes place at a slow rate in response to pH change; however, it can be done in seconds by photoreaction. In the investigation, as confirming the previous reports, 3-(2-hydroxy-phenyl)-1-(4-hydroxy-phenyl)-propenone, the trans-chalcone (CtF1) from F1, produces flavylium ions in pH = 1–4.5. Then, we further discovered that the flavylium quickly releases protons to yield the corresponding quinoidal base (A) in a solution of pH = 5.2 during irradiation with 350 nm. Meanwhile, the photolysis of 3-(2,4-dihydroxy-phenyl)-1-phenyl-propenone, the trans-chalcone (CtF2) from F2 at pH = 5.6, induces photoacid behavior by losing a proton from the trans-chalcone to generate Ct2−. The different outcomes of these nearly colorless chalcones under similar pH conditions and with the same photochemical conditions can be useful when yielding colored AH+, A, or Ct2− in a mildly acidic pH environment with temporal and spatial control using photochemical means.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Photoresponsive molecular devices have great potential in the development of smart materials. They utilize various photoreactions, such as photofragmentation [1,2] and cis-trans isomerization [3]. The devices can have complex functions if multiple input-responsive mechanisms can be coupled. However, the main challenge is in achieving complete control of each output signal without the undesired interference of others. Therefore, it would be crucial to have orthogonal chemical reactions. Fortunately, orthogonal photoreactions can provide a temporally and spatially controlled response with specificity. Furthermore, multiple wavelengths in conjunction with other inputs, such as pH, can promote more diversified chemical responses. This endeavor has currently expanded to areas such as 3D printing [4], conductive hydrogels [5], photo-switchable nanosystems and materials [6], the hybridization of DNA/RNA [7], host–guest chemistry [8], self-assembly [9], uncaging biologically active compounds [10], and more [11].
Arguably, cis-trans isomerization is the most studied photoresponsive mechanism using stilbenes [12], diazostilbenes [3], chalcones [13], etc. The photochemical isomerization of chalcones has been studied as one of the mechanistic steps for pH-responsive flavylium ions (AH+). The distinct behavior patterns of the flavylium and its conjugate bases (CB: shown in Figure 1 as A, B2, Cc, Ct, and their anionic species) are well documented by Brouillard [14], McClelland/Gedge [15], Pina, and others [13]. The flavylium cation ring (AH+) in natural anthocyanins has different hydroxy and alkoxy groups commonly on 3, 5, 7, 3’, 4’, and 5’, demonstrating multiple functions in nature [16,17,18,19,20,21,22,23]. The pH-responding structural change gives a broad range of applications stemming from the different chromic and optical properties due to pH-adjusted structures. The optical property within the visible range of the colored flavylium ion ring for light-harvesting applications such as dye-sensitized solar cells (DSSCs) and photodynamic therapy, and its fluorescent property, make it suitable for applications to imaging, protein/DNA sensors, and optical diagnostic agents. Moreover, the flavylium derivatives show pharmaceutical potential in areas including antibacterial activity, antioxidant capability, and drug delivery systems [24,25,26,27,28,29,30,31,32,33,34,35,36,37,38].
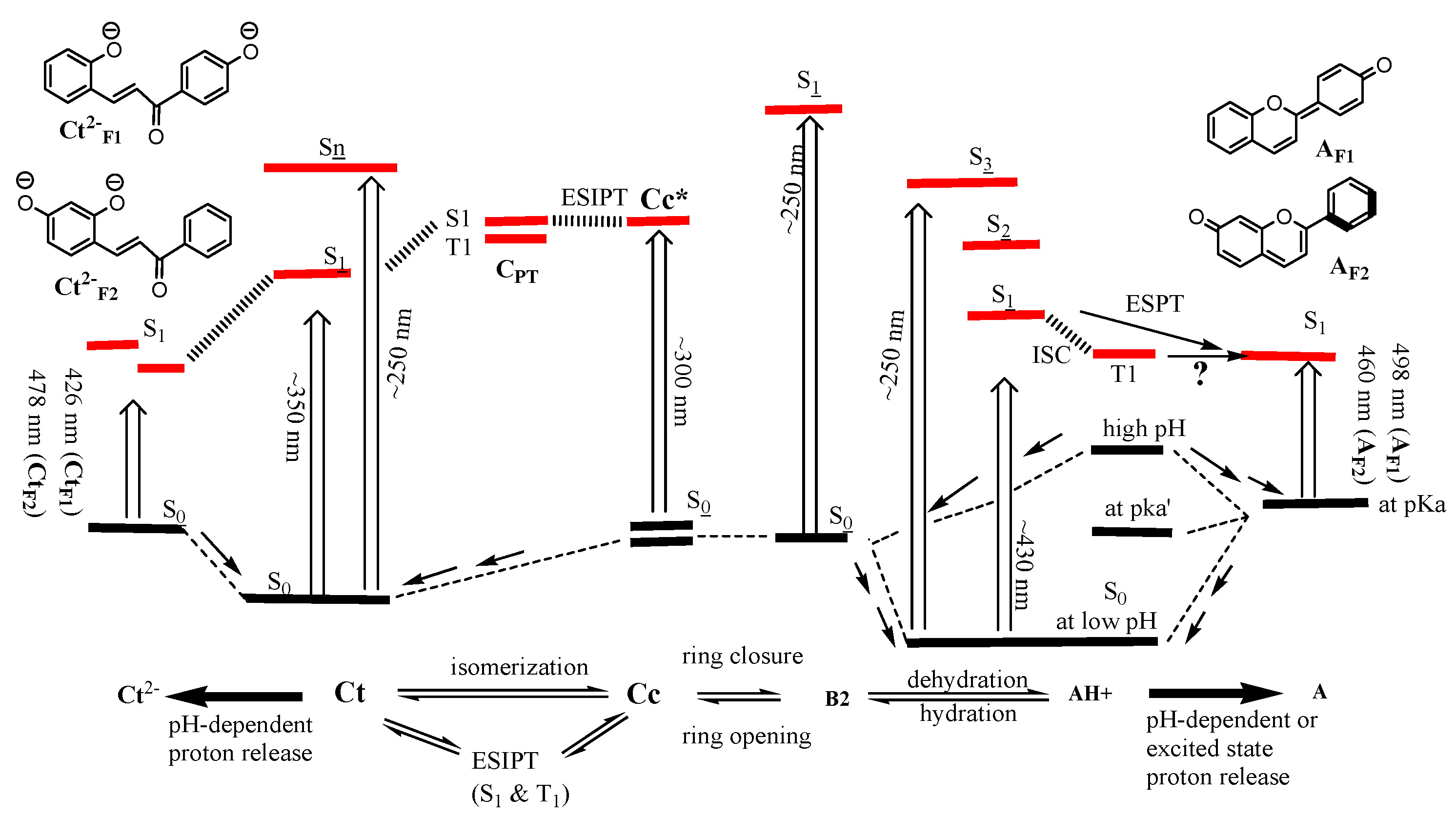
However, the flavyliums with specific OH substitutions are stable only in very acidic solutions, although it should be noted that an example such as deoxyanthocyanin (Dragon’s blood) with 7-OH and additional oxygenated substituents has AH+ stable over a wide range of pH [37]. The change in pH over 3 for most flavyliums can act as a stimulus for transforming flavylium ions to other relevant conjugate bases (CB) with de-coloration. Historically, a few mechanisms were proposed, such as the mechanism in Figure 1, by which A can form B2 via anionic B2− without going through AH+ in basic media (path b in Figure 1) using 6-hydroxyflavylium and 7-hydroxyflavylium [39,40,41], whereas the formation of AH+ from chalcone isomers via B2 when the pH drops to 1 (path a in Figure 1) is a widely accepted mechanism. On the other end of the mechanism, when the pH is raised above 3, AH+ goes through B2 by hydration (kh) and then tautomerization (kt), giving Cc, followed by isomerization (ki) to form Ct. This equilibrium above pH = 3 will provide a mixture of all the relevant compounds to our study. Eventually, at pH over 5 (near neutral conditions), trans-chalcone becomes the most stable species. Above pH = 8, anionic forms of Ct, Cc, and B2 begin forming depending on the basicity of the solution: typically Ct−, Cc−, and B2− at pH above 9 and Ct2−, Cc2− at pH above 12.
The rates for the steps in path b were measured by Brouillard et al. using Malvin in acidic media: ka ≈ 108 M−1s−1; ka−1 ≈ 104 s−1; kh ≈ 10 M−1s−1; kh−1 ≈ 10−2 M−1s−1; Kb = [B]/[A] ≈ 102 M−1s−1 [42]. The equilibrium constants and rates of the two flavylium ions synthesized for the investigation in this manuscript have already been measured by the Pina group: 4′-OH flavylium (F1): Ka = 3.16 × 10−6, Kh = 3.6 × 10−6, Kt = 1, Ki = 3500, kh = 8.9 × 10−2 M−1s−1, kh−1 = 2.5 × 104 M−1s−1, ki = 3.7 × 10−5 s−1, ki−1 < 10−7 s−1 [43]. 7-OH flavylium (F2): Ka = 2.8 × 10−4, Kh = 8 × 10−6, Ki = 500, kh = 0.48 M−1s−1, kh−1 = 3 × 104 M−1s−1, ki = 0.57 s−1, ki−1 = 8.3 × 10−4 s−1 [44]. The isomerization rate (ki) is generally the slowest step, with the hydration rate (kh) in a highly acidic condition as an exception.
In short, a pH jump from pH = 1 to 6 transforms AH+ to A and B2, then Cc and Ct. The reversal pH jump exhibits the reverse equilibrium from Ct to AH+ at pH below 3, and the varying ratio mixtures of AH+, A, B2, and Cc are produced at pH = 4–7. However, irradiating Ct in the near-neutral pH (Figure 1) of 4′-hydroxyflavylium in pH = 5–7 established the pH-dependent mixtures of Cc, B2, AH+, and A. It was accounted for in the previous report that a quick photochemical reaction produces Cc, resulting in thermal equilibrium in response to pH conditions establishing mixtures with different ratios of CB and AH+ [41,43]. Similarly, 7-hydroxyflavylium formed the same type of mixture photochemically. Moreover, a small amount of A was detected when irradiating AH+ in acidic conditions: excited-state proton transfer (ESPT; Figure 1) was attributed to the loss of protons [44]. Meanwhile, phosphorescence was observed with bromoderivatives of flavylium due to the heavy atom effect. Also, derivatives without heavy atoms in acetonitrile with TFA due to suppressed ESPT showed phosphorescence at 500–700 nm from their triplet states [45].
This investigation was focused on the behavior of trans-chalcones in equilibrium with other CB and flavyliums as an ongoing effort to design a multifunctional phototrigger that can respond to pH changes and photosignals. Noticeably increasing photochemical elements have been employed in various applications due to the merit of noninvasive temporal and spatial control of the photoinitiated chemical reaction. Therefore, our goal was to examine the benefit of disturbing the thermal equilibrium at various pHs using photochemical means to form unusual species with the influence of the given pH condition.
2. Materials and Methods
Reagents and solvents were purchased from Fisher Scientific as reagent-grade and used without further purification. 1H- and 13C-NMR spectra were recorded on a Bruker DPX 400 MHz FT NMR with QNP probe (Billerica, MA, USA). HRMS and MS analyses were performed on an Agilent 6520 Q-TOF (Santa Clara, CA, USA). UV–vis spectra were acquired using a JASCO V-650 spectrophotometer (Easton, MD, USA).
2.1. Synthetic Procedure [46]
F1 (4′-Hydroxyflavylium chloride): 2-Hydroxy-benzaldehyde (0.635 g, 5.2 mmol) and 1-(4-hydroxy-phenyl)-ethanone (0.708 g, 5.2 mmol) were dissolved in 14 mL of dry ethyl acetate, and the mixture was cooled by placing the beaker in a salt ice bath (CaCl2:Ice = 1:2.5) until the temperature reached −10 °C. Sulfuric acid (100 mL) was dropped on NaCl(s) (120 g) to generate dry HCl gas, and the gas was bubbled in the mixture for 1 h at −10 °C. The reaction mixture was stirred at room temperature for 24 h. The product was obtained by filtration (orange solid, 1.010 g, 3.9 mmol, 75.0%).
1H-NMR (MeOD-d4/TFA-d, 95/5, 400 MHz): δ (ppm) 7.19 (d, J = 8.0 Hz, 2H), 7.94 (t, J = 8.0 Hz, 1H), 8.28 (overlap of three peaks: d, d, and t, 3H), 8.62 (d, J = 8.0 Hz, 2H), 8.67 (d, J = 8.0 Hz, 1H), 9.24 (d, J = 8.0 Hz, 1H). 13C-NMR (MeOD-d4/TFA-d, 95/5, 100 MHz): δ (ppm) 115.11, 117.95, 119.21, 119.94, 120.98, 125.00, 130.77, 131.55, 135.94, 139.39, 156.85, 170.28. HRMS (ESI) calcd. 223.0754 for C15H11O2+: found (M+) 223.0752 (mass error: −0.8965 ppm). The spectra are available in the Supplementary Materials.
F2 (7-Hydroxyflavylium chloride): 2,4-Dihydroxybenzaldehyde (0.718 g, 5.2 mmol) and acetophenone (0.625 g, 5.2 mmol) were dissolved in 14 mL of dry ethyl acetate, and the mixture was cooled by placing the beaker in a salt ice bath (CaCl2:Ice = 1:2.5) until the temperature reached −10 °C. Sulfuric acid (100 mL) was dropped on NaCl(s) (120 g) to generate dry HCl gas, and the gas was bubbled in the mixture for 1 h at −10 °C. The reaction mixture was stirred at room temperature for 24 h. The product was obtained by filtration (brown solid, 0.968 g, 3.7 mmol, 71.0%).
1H NMR (MeOD-d4/TFA-d, 95/5, 400 MHz): δ (ppm) 7.55 (dd, J = 9.0 & 2.0 Hz, 1H), 7.63 (d, J = 1.8 Hz, 1H), 7.79 (t, J = 7.5 Hz, 2H), 7.89 (t, J = 7.3 Hz, 1H), 8.29 (d, J = 9.0 Hz, 1H), 8.51 (dd, J = 8.1 & 1.4 Hz, 2H), 8.54 (d, J = 8.5 Hz, 1H), 9.33 (d, J = 8.4 Hz, 1H). 13C NMR (MeOD-d4/TFA-d, 95/5, 100 MHz): δ (ppm) 103.67, 114.19, 117.96, 121.81, 123.91, 130.33, 131.28, 134.56, 137.20, 156.51, 161.68, 172.06, 173.37. HRMS (ESI) calcd. 223.0754 for C15H11O2+: found (M+) 223.0763 (mass error: 4.0345 ppm). The spectra are available in the Supplementary Materials.
2.2. General Photoreaction Procedure
The photoreaction samples of flavyliums (10−4 M), F1 and F2 (Figure 2), were prepared and stored in the dark for over a week (or two, if necessary, to reach the equilibrium) to achieve equilibrium with trans-chalcones and other CB. Then, the pH of the solution was adjusted multiple times to the desired pH (Figure 3) by adding NaOH or HCl a few times over the week(s), while reaching equilibrium since the pH changes as the equilibrium shifts. Afterward, the sample in a quartz reactor was placed in a Rayonet photochemical reactor (Branford, CT, USA) with eight medium-pressure bulbs (RPR-3500A and RPR-2537A lamps) of each wavelength (350 and 254 nm). The samples were air-equilibrated and were not treated with inert gas. The light intensity was measured multiple times at different points of irradiation using a ferrioxalate actinometer. The intensity was in the range of 0.8–2 × 10−7 einstein/min for 350 nm and 254 nm. The photochemical reaction was initiated by photolysis with a wavelength of 254 nm or 350 nm. The photoreaction was done with continuous irradiation at intervals determined by the reaction progress. The reaction in the aqueous solution was monitored using UV–vis spectrometry and a pH meter. The quenching study [47] was carried out with 0.1 and 3 mM of sorbic acid (Mw = 112; pKa = 4.7), HDA (2,4-hexadienoic acid), with the time interval depending on the light intensity and the reaction rate. HDA was dissolved in the flavylium stock solution; then, the pH was readjusted, and samples were kept in the dark over the week(s) for equilibrium, as described above.
2.3. Gaussian Calculation
Modeling experiments using the Gaussian 95 Package were done at the level of DFT//B3LYP, employing the 6311G++dp basis set for the ground state. In order to gain the vertical transition energy level of S1 and T1, TD-DFT with unrestricted spin was used. Each calculation for the excited states with conformations at varying dihedral angles was done with the redundant coordinate by freezing coordinates for the double-bonded areas, and also optimizing the rest of the molecule. All calculations were done using the IEFPCM solvent model for water.
3. Results and Discussion
3.1. trans-Chalcone in Equilibrium at Different pHs
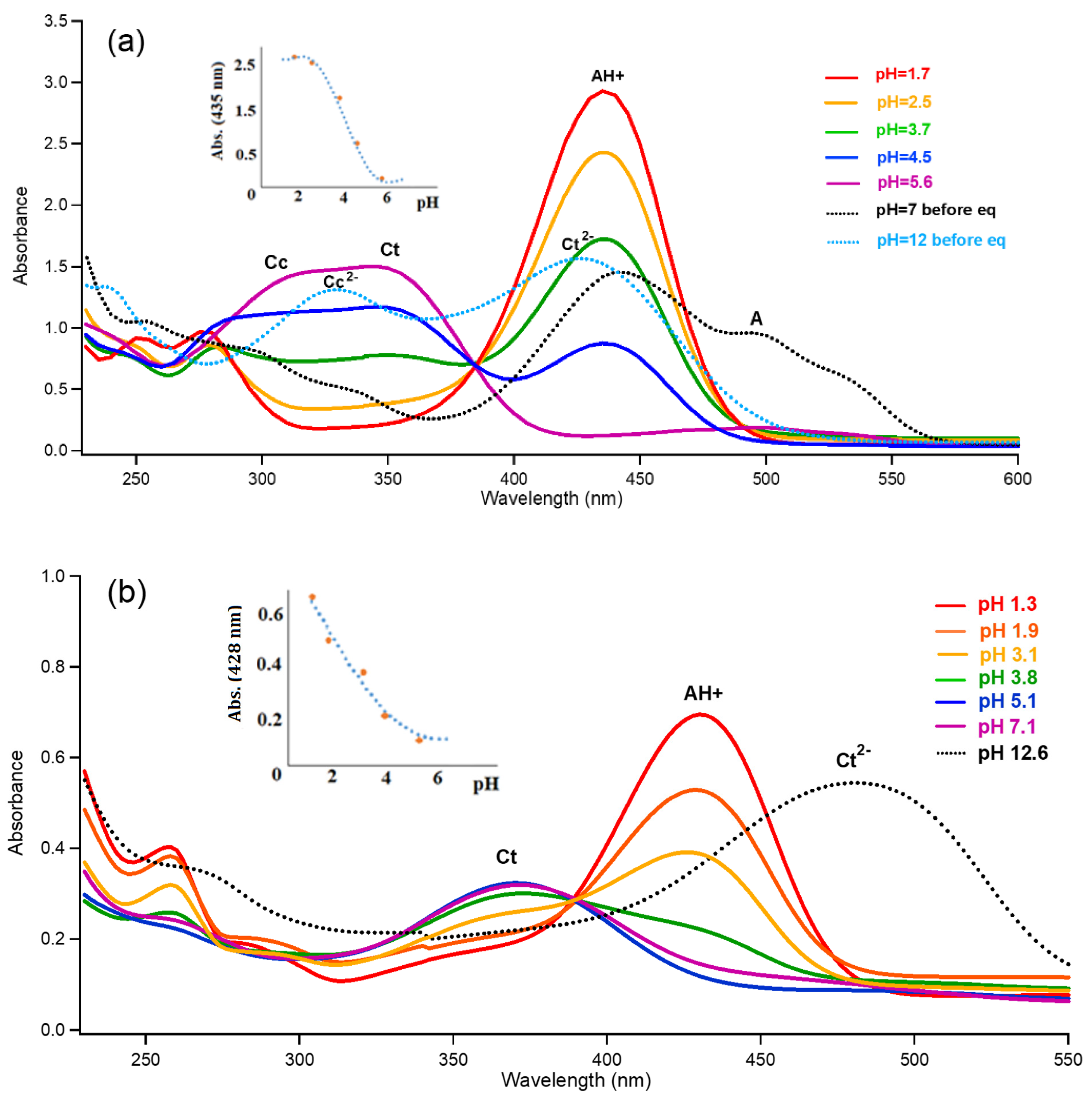
Both synthesized flavylium ions, F1 and F2, established equilibrium with trans-chalcones (CtF1 and CtF2) and other CB (mentioned above and shown in Figure 1 as A, B2, Cc, Ct, and their anionic forms) after several days. At a relatively high pH, as reported by Pina and other research groups [43,44,48], the cis-chalcone of F1 with a slow thermal isomerization rate (ki = 3.7 × 10−5 s−1) shows a mixture of cis and trans (at 345 nm) isomers. In contrast, F2 with a faster isomerization rate (ki = 0.57 s−1) shows mostly the trans isomer (368 nm) at a pH over 3 (Figure 3). Meanwhile, as shown in the inset of Figure 3, pKa′ (pH where [CB]/[AH+] = 1 using the concept of Henderson–Hasselbalch for acid–base reaction) was calculated by plotting the absorption of flavylium according to pH changes: 4.0 (±0.2) for F1 and 2.9 (±0.2) for F2. We were able to obtain chalcones, CtF1 at a pH from 4 to 7 for F1 and CtF2 at a pH from 3 to 7 for F2, at a manageable concentration level as mixtures for the photochemical reactions with thermal equilibria between relevant species.
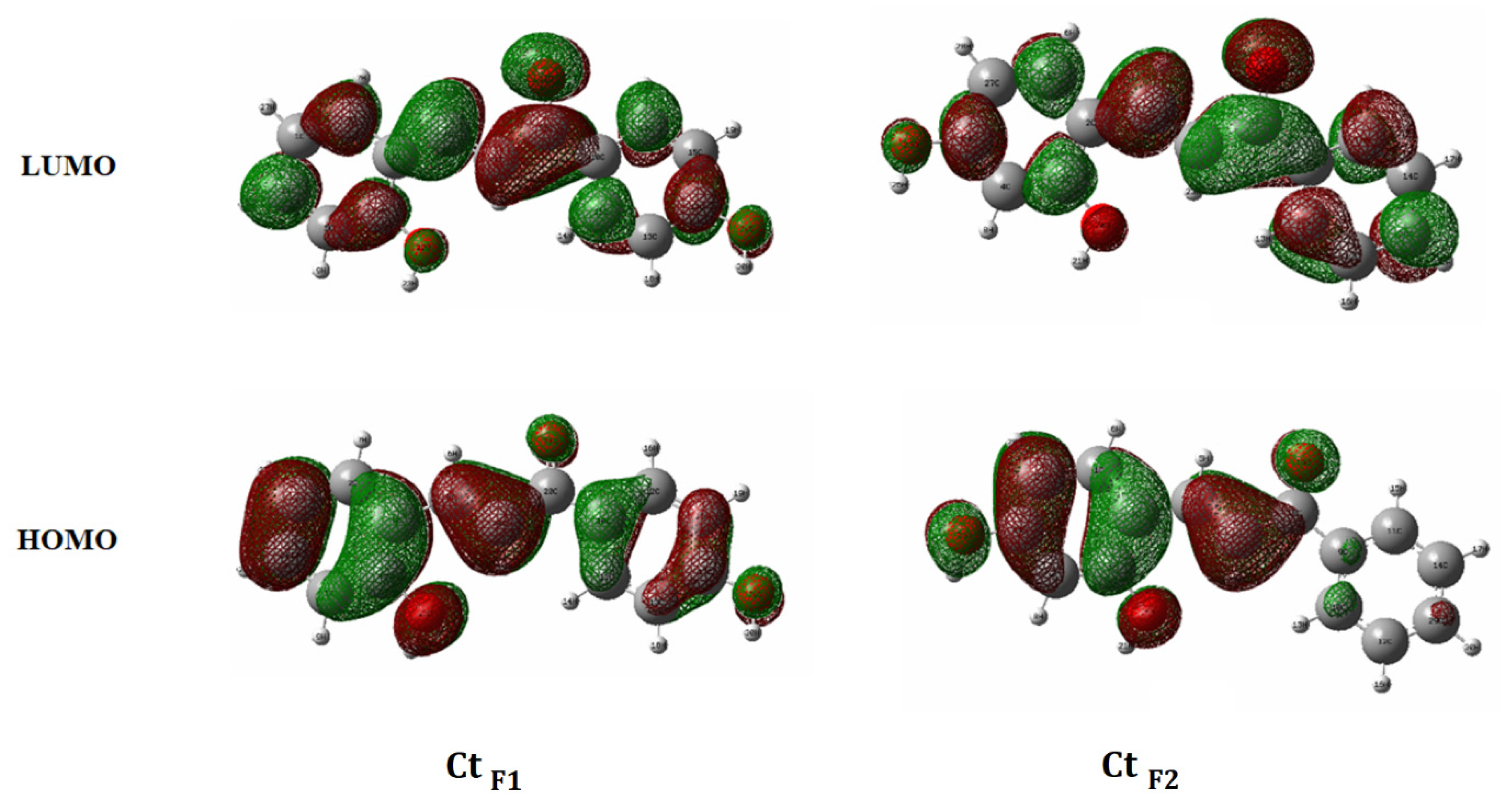
For F1 in Figure 3a, the peak from pH = 1.7 shows three main transitions from S0 to S1 at 435 nm, S2 at 275 nm, and S3 at 250 nm. AH+ and other species of CB show some degree of absorption below 300 nm, as also shown in Figure 4. The graph at pH = 5.6 shows Ct at 345 nm and Cc at 302 nm, and the immediately observed peak from the AH+ solid dissolution confirms Cc at 302 nm. The peak of the pH jump to 12 shows Ct2− (yellow solution) at 426 nm and Cc2− at 330 nm. Moreover, the absorption for the neutral A (orange solution) around 498 nm was observed immediately after dissolving the flavylium chloride solid (F1) in pH = 7–10 solutions, as previously reported [41]. It should be noted that pH = 7 is not basic enough to generate anions of Ct and Cc as major species. On the contrary, the corresponding peak of Ct2− for F2 around 478 nm in Figure 3b is only present at a pH over 10; thus, it is assigned to be Ct2− and not A, which was observed when the pH jumped to 12, as confirmed by NMR in the previous report [44]. Then, pH = 1.3 showed similar absorption to the F1 case—absorption (S0 →S1) of AH+ at 428 nm and higher excited states below 300 nm. UV–vis peaks for Cc2− and A were not identified for the F2 solution. It must be pointed out for later discussion that the quinoidal base (A) from F1 has more effective and extensive resonance, as shown in Figure 4, than the counterpart of F2; thus, it is understandable for AF1 to have absorption at 498 nm while AF2 with the twisted conformation around the single bond between rings B and C give absorption at 460 nm [44]. Meanwhile, Ct2− for F1 has negative oxygen atoms as powerful electron donors in the rings A and B to obtain an absorption peak at 426 nm. However, Ct2− for F2 has both electron donors on ring A; thus, its HOMO raised its energy to obtain absorption at 478 nm (Figure 4) according to the Woodward rule.
3.2. Photoisomerization of trans-Chalcone
Generally, photoreactions with 254 nm for trans-chalcones, 3-(2-hydroxy-phenyl)-1-(4-hydroxy-phenyl)-propenone (CtF1) from F1 and 3-(2,4-dihydroxy-phenyl)-1-phenyl-propenone (CtF2) from F2 (Figure 2), had prolonged reaction rates compared to the photolysis with 350 nm. However, as Figure 4 shows few potential excitations, irradiating the mixture with 254 nm should excite every type of CB and AH+, since almost every compound has an absorption in the region, as aforementioned in the discussion of Figure 3. Moreover, despite the greatest absorptivity of 350 nm of the trans-chalcone, AH+ and Cc formed in a mixture at a different pH, allowing them to absorb 350 nm light because their bands spread out into this region and lowered the absorbed light intensity for Ct. Thus, it must be considered that the reaction rate measure includes the error from this; in addition, the photoreaction causes a pH alteration by a unit of 0.2–0.3 due to the reaction with 10−4 M of the samples. At the NMR concentration (~10−2 M), the pH changes were greater by a unit of 0.3–0.6. The effect of the greater concentration on the photochemically induced pH change is disappointingly less than expected. It can be attributed to the self-quenching of their excited states at a higher concentration than 10−3 M. Additionally, as expected, the photoinduced pH jump is dependent on the solution pH. Thus, the jump is more significant near a neutral pH than at a more acidic pH solution.
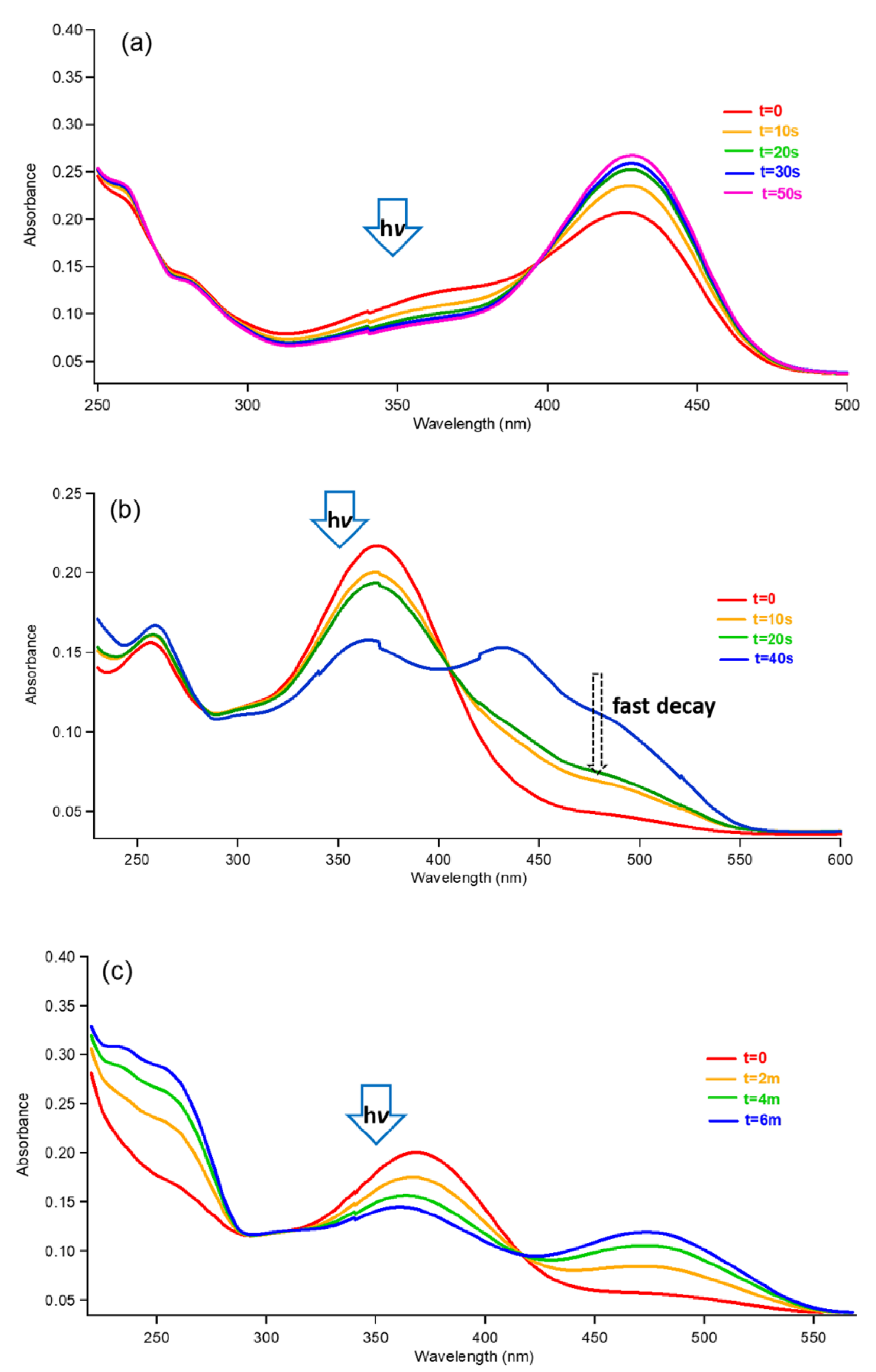
When the solution (initial pH = 4.5) of CtF1 (AH+, Ct, Cc, and others in equilibrium) was reacted only for 60 s under a photolytic condition with 350 nm light in the photoreactor, the majority of the trans-chalcone (345 nm) was converted to AH+ (435 nm) within a couple of minutes; as shown in Figure 5b, this leaves other CB forms in a steady-state concentration as a minor component. The ratio of AH+(435 nm)/A(498 nm) remained unaffected by the photolysis. This suggests that the trans isomer (Ct) was effectively converted to a cis isomer (Cc) photochemically, and then the increased concentration of Cc forced the equilibrium to elevate the concentration of B2 to the equilibrium constant. Afterward, the AH+/A mixture was generated from B2 with the influence of pH by the equilibrium favoring AH+ over A [13].
However, at initial pH = 5.2, CtF1 transformed (Figure 4) to A via Cc, B2, and AH+. The first hypothesis could be the ESPT mechanism. After conversion to AH+, as pointed out above, AH+ absorbs 350 nm to release protons via the ESPT mechanism from the excited state of AH+* (Figure 4) [44]. The excited phenolic hydroxy group on AH+ photochemically elevated its acidity at this pH to release H+, producing more stable excited A* at a fast rate before relaxation to its ground state (Figure 4) [49,50,51]. After the flavylium ion releases a proton, the negative charge on the oxygen for the conjugate base of AH+F1 is more efficiently delocalized compared to its counterpart of AH+F2; this could be the reason that the same behavior is not observed for AH+F2. Another hypothesis is the simple proton loss from the ground-state AH+ because of the instability of AH+ due to its instability affected by the pH condition after being photochemically formed from Ct. AH+ is less stable than B2 in the solution due to the higher pH than pKa′ (Figure 4), where it is much easier to release H+ to produce A. Therefore, AH+ quickly releases protons to reach equilibrium with A without forming a detectable amount of AH+ by the conventional UV–vis spectrometer.
However, interestingly, as shown by the black dotted line (3 mM HDA) and the solid black line (0.1 mM HDA) in Figure 5c, the photoreaction produced AH+ when a quencher, 2,4-hexadienoic acid (HDA; absorption λmax = 260 nm; ET = 50–60 kcal/mol), was used. The pH of the solution with HDA was readjusted over the weeks of equilibrium. With 1 eq. quencher, a relatively smaller amount of AH+, along with minor A, was generated. However, an excessive amount of quencher (30 times) gave almost exclusively AH+ with a trace amount of A. It could be possible that the intersystem crossing of the first singlet excited state of AH+ became competitive enough to form the T1 state under the limited ESPT condition in the buffer, as reported by Silva et al. [45]. The mixture consisted of carboxylic acid, and its conjugate base formed from the quencher during pH adjustment can act as a buffer. Therefore, unlike other tested solutions, these solutions with the quencher–buffer solution can limit deprotonation to accumulate AH+ without converting to A. Since both singlet and triplet states could release protons slowly in the buffer, it is a possible hypothesis that the energy level of T1 was quenched by HDA with a low triplet energy level (50–60 kcal/mol) before the proton release from AH+ as a photoacid to result in forming more AH+ with more HDA. It is also possible that the anionic quencher molecule with two π bonds can have a copigmentation interaction with a positively charged flavylium ion. It can stabilize AH+ to drive the equilibrium towards AH+. Nonetheless, the selective photoreaction result towards AH+ or A at this pH level can be beneficial to an application. Further study is underway with time-resolved spectroscopy to detect AH+ and the responsible spin multiplicity of the excited state at this pH level under photolysis conditions.
Due to the lower pKa′ value for F2, the trans-chalcone was detected even at pH = 1.3 (Figure 3b), and the cis-chalcone was very minor at all pH levels, which confirms the fast isomerization rate (ki = 0.57 s−1). Figure 6a shows the results at pH = 3.1; the photolysis with 350 nm follows a similar reaction path as CtF1 by growing the amount of AH+ at 428 nm. Afterward, the process of the photoproduct mixture returning to the thermal equilibrium state was much faster for CtF2.
When we examined the photochemical result from pH = 5.6 (Figure 6c), photolysis with 350 nm produced a Ct2− anion peak exclusively at 478 nm; then, the peak decayed within minutes (over 30 min) rather than seconds or hours. As shown in Figure 3b, this peak was formed when Ct was reacted with NaOH to reach pH = 12 or even higher. When AH+ was reacted with excessive NaOH, the observed result was the same as for Ct2−. A report from the Pina group confirmed the absorption of A to be 460 nm instead of 478 nm, as mentioned above [44]. In addition, the pH jump from both the photoproduct mixture (Figure 6c) and the equilibrated pH = 12 solution to pH = 5 generated only Ct at 368 nm, as Ct2− at 478 nm is consumed in the acid–base reaction.
The photolysis at pH = 4.2 in Figure 6b appears to show two bands growing between 400 and 500 nm and new species (represented by the blue peak shoulder) that show the same absorptivity of the remaining trans-chalcone after 40 s. These blue peaks that decay in two stages hint that the new species is a transient molecule, with noteworthy decay behaviors: (1) quickly decaying to the same intensity as the green peak, observed after 20 s photolysis, and then (2) slowly decaying to return to thermal equilibrium (red peak). It is possible that this shoulder peak around 432 nm can be AH+. However, the other broad peak between 400 and 500 nm could be Ct2− and/or A, as discussed with the photoreaction from pH = 5.6 solution (Figure 6c). Although the decay rate is certainly faster than the aforementioned rate at a less acidic pH = 5.6, it is reasonable to believe that the conjugate base of trans-chalcone Ct2− at pH = 5.6 lived longer than the case in a more acidic pH = 4.2.
While CtF1 photochemically formed the A species in equilibrium, this result from the trans-chalcone (CtF2) raises the interesting photoacidity aspect of a phenolic compound with a meta-stable conjugate base that lasts longer than nanoseconds in a pH over 4 [48,49,50]. In conjunction with pH, we can drive the reaction from colorless CtF2 toward colored species (AH+ or Ct2−) at much milder conditions by using a photochemical process. Without photochemical means, the pH jump must be close to 1 for CtF2 in order to form AH+ and over 10 to form Ct2−. If one wishes to alter their equilibrium noninvasively with spatial and temporal control, this can be a valuable tool with more development.
3.3. Insights from DFT Calculation
The detailed behavior such as photoacidity must be understood in conjunction with the role of pH; however, the modeling shows that, unlike CtF1, the HOMO of the C (middle) ring in CtF2 lacks electron density (Figure 7), while the A ring donates a significant portion of the density to the B ring during the excitation to LUMO. This can account for the easier loss of H+ from the phenolic CtF2 molecule on the excitation compared to CtF1, which has no difference in electron distribution for both rings.
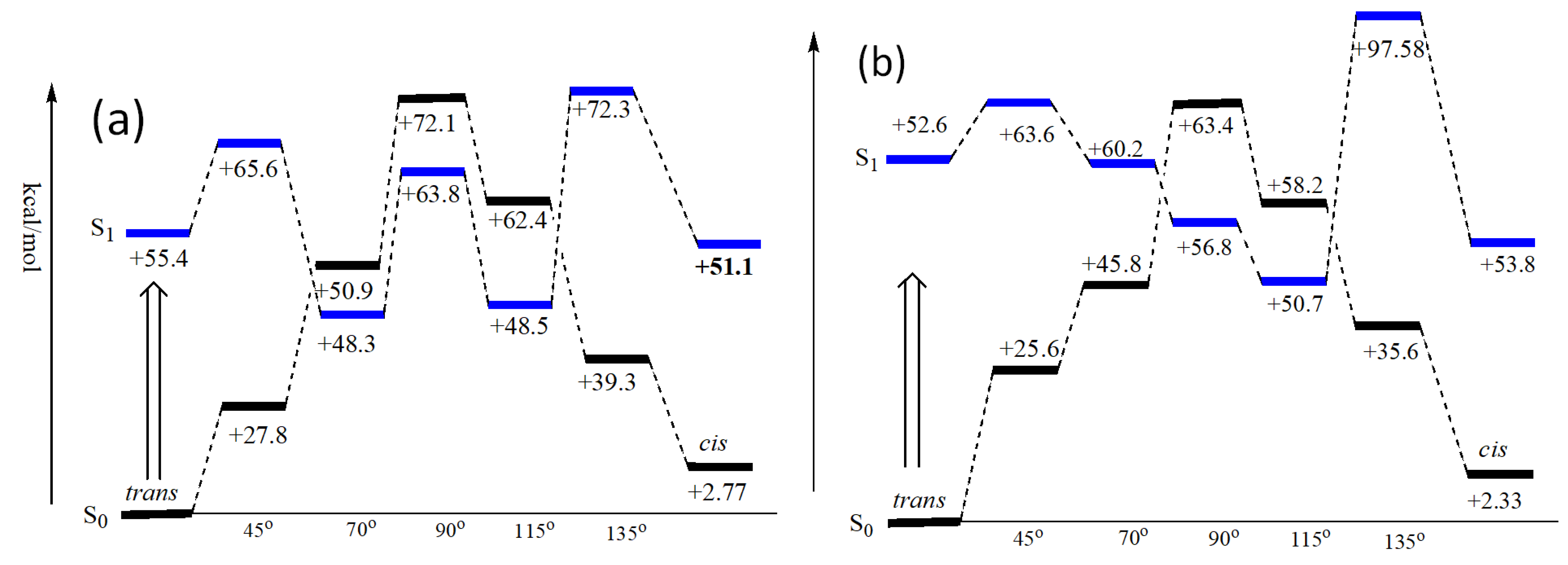
Moreover, modeling predicted the energy levels in Figure 8, demonstrating that the minima of the S1 surface do not stay above the TS of the ground-state surface for the isomerization. Therefore, the low-lying minima of the S1 surface can make it possible for the presence of conical intersections for the crossings of S0 and S1 energy surfaces during the photochemical reaction of the trans-chalcone to a cis-chalcone.
According to the schematic diagram in Figure 9, upon excitation to S1 by 350 nm, the trans-chalcone molecule changes the dihedral angle around the double bond over an activation barrier on the excited-state surface. Then, around 115–135° (Figure 8), the compound with a product-resembled structure moves over to the ground-state energy surface (S0) and faces downhill to the final structure of the cis-chalcone. Thus, the photochemical isomerization step occurs at a faster rate over only a small energy barrier. CtF2 shows an even easier path to the cis-chalcone isomer. The chalcone of F1 has a greater chance for relaxation back to trans at the conformations with the dihedral angles 45° and 70°, which is a reactant resembled structure, compared to the chalcone of F2 with the dihedral angle between 70° and 90°. Moreover, it is not likely to go back to the trans isomer because the S1 surface is still downhill toward the cis isomer. Finally, when the F2 chalcone reaches the dihedral angle 115°–135°, it moves on the S0 surface of the cis isomer.
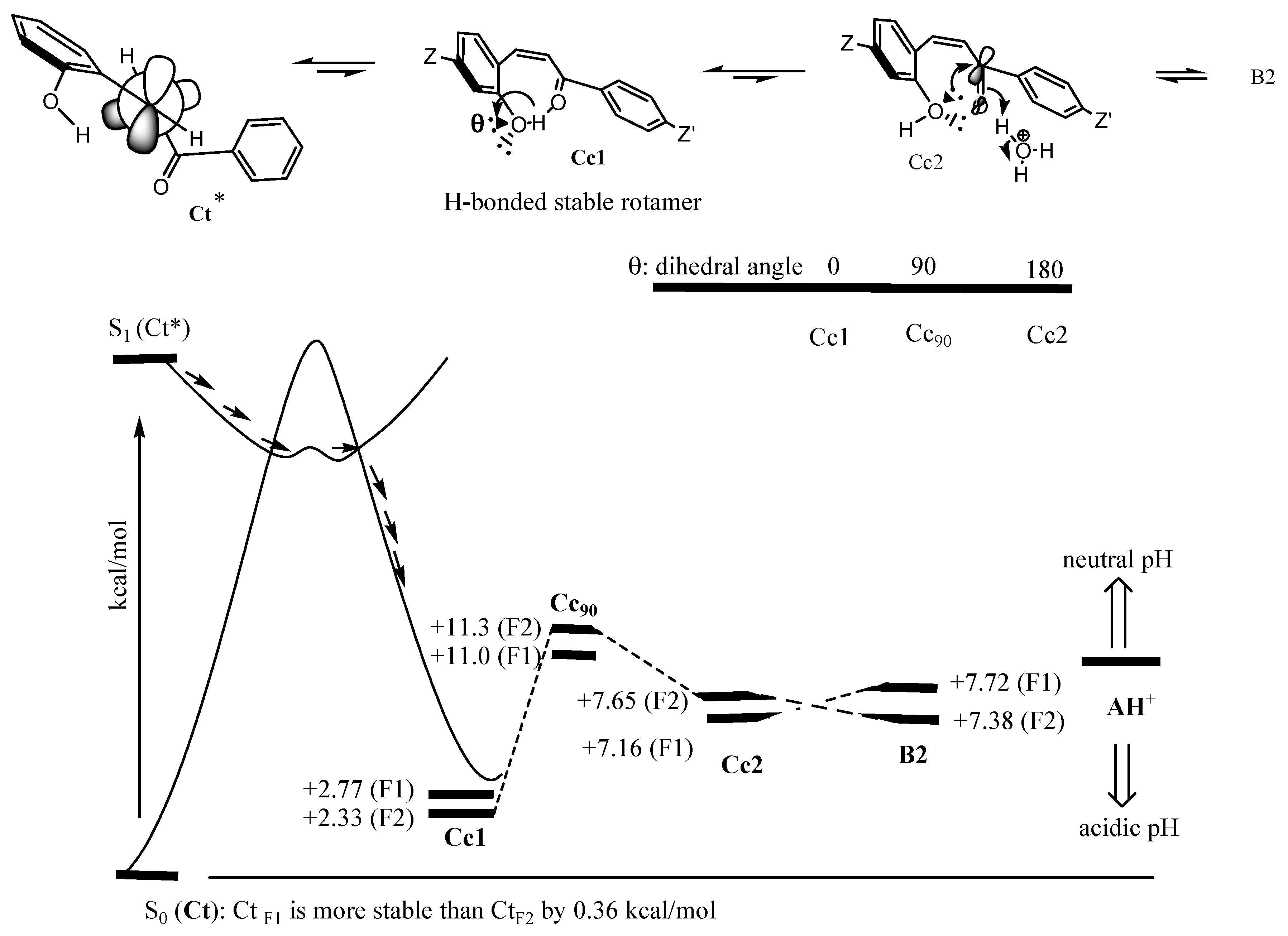
Moreover, it should be pointed out that the most stable conformer (Cc1) of Cc, photochemically forming from Ct, is not able to close the (middle) C ring of flavylium (Figure 9). According to modeling, energy around 9 kcal/mol is required to break the H-bond from the most stable conformer (Cc1) to produce Cc2, as shown in Figure 9. Afterward, the less stable Cc2 can use lone pairs to attack the carbonyl carbon for ring closure. To form B2 from Cc2, the calculation predicts a slight uphill for CtF1 and slight downhill for CtF2. Then, the reactivity and stability of AH+ are dependent on the pH level. It should be noted that the energy gaps are too small to be a major factor in the thermal equilibrium.
The modeling study revealed another interesting insight about photoisomerization, as shown in Figure 4 and Figure 10. The excited cis-chalcone could have a path in the excited state to give the structure with the enol form after excited-state intra-molecular proton transfer (ESIPT). The new structure can undergo relatively easy cis–trans isomerization due to the single bond rotations, as indicated in Figure 10. It was also found that, unlike the other CB of F1, the first singlet and triplet excited-state energies for Cc* and CPT* are very close, within 1 kcal/mol.
3.4. Conclusions
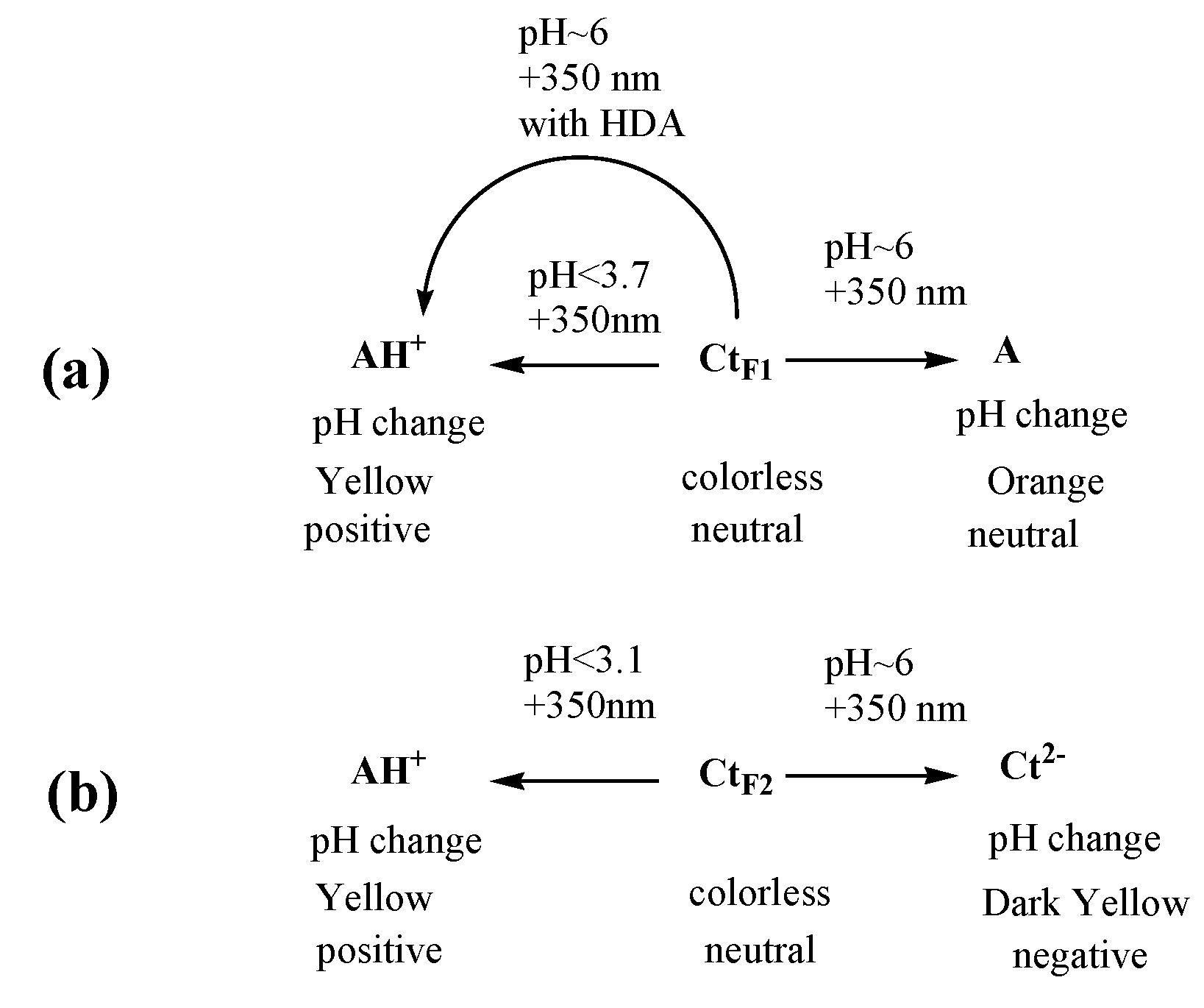
Two isomers of chalcones in mixtures with relevant CB are studied for their photobehavior in different pH solutions. The authors focused on the behavior of trans-chalcones in the ongoing effort to design a multifunctional phototrigger that can respond to pH changes and photosignals. Around the physiological pH, since the stability of flavylium is dependent on pH, the trans-chalcone becomes the major species, and its isomerization becomes essential for its application as a photoresponsive molecule. Utilizing photochemical selectivity, at pH 6, the equilibrium can be manipulated so that CtF1 can form either AH+ or A. Meanwhile, CtF2 shows a different pH-induced photochemical control to choose AH+ or Ct2− from the product in the opposite direction of the equilibrium. The capability to manipulate photochemical selectivity to give rise to either a positively charged AH+ or a completely opposing charged Ct2− gives rise to a multitude of windows of opportunity that are useful for applications; for example, a membrane with CtF2 units will be able to change from a cation to an anion to neutral. As summarized in Figure 11, CtF1 and CtF2 can be valuable tools to reversibly transform photochromism and other properties in response to pH and light. It also has great potential in the search for a smart phototrigger that can not only respond to photons for drug release but also shows the ability to change local acidity and color.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/colorants2010005/s1, Figure S1: Mass spectrum of F1. Figure S2: Mass spectrum of F2. Figure S3: 1H-NMR spectrum of F1 in MeOD-d4/TFA-d = 95/5. Figure S4: 13C-NMR spectrum of F1 in MeOD-d4/TFA-d = 95/5. Figure S5: 1H-NMR spectrum of F2 in MeOD-d4/TFA-d = 95/5. Figure S6: 13C-NMR spectrum of F2 in MeOD-d4/TFA-d = 95/5.
Author Contributions
J.K. synthesized the compounds and measured NMR, analyzed UV–vis data, and wrote the manuscript. K.B. carried out the synthesis of the compounds, and B.Y. performed mass spectrometric analysis. J.I.L. performed Gaussian calculations, UV–vis measurements, photoreactions, and manuscript writing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Faculty Development Grants and the Chemistry Departments at St. Joseph’s University and CUNY York College.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are contained within the article and are available in the Supplementary Materials.
Acknowledgments
The authors thank Gopal Subramaniam at Queens College-CUNY for assisting with the NMR measurements. The authors also thank Yarim Lee for proofreading this manuscript and the Department of Chemistry at York College for the facilities. We would also like to thank the Department of Chemistry and Biochemistry at Queens College-CUNY for allowing us to utilize the NMR facilities that made the research possible.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Givens, R.S.; Heger, D.; Hellrung, B.; Kamdzhilov, Y.; Mac, M.; Conrad, P.G., II; Cope, E.; Lee, J.I.; Mata-Segreda, J.F.; Showen, R.; et al. The photo-Favorskii reaction of p-hydroxyphenacyl compounds is initiated by water-Assisted, adiabatic extrusion of a triplet biradical. J. Am. Chem Soc. 2008, 130, 3307–3309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrella, A.; Rene, N.; Ali, K.; Kang, J.; Lee, J.I. Synthesis of first generation dendron, GABA-caged bis-1,2-(4-acetylphenylethynyl)-4,5-dimethoxybenzene for photorelease study. Lett. Org. Chem. 2019, 16, 718–722. [Google Scholar] [CrossRef]
- Cembran, A.; Bernardi, F.; Garavelli, M.; Gagliardi, L.; Orlandi, G. On the mechanism of the cis-trans isomerization in the lowest electronic states of azobenzene: S0, S1, and T1. J. Am. Chem. Soc. 2004, 126, 3234–3243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossegger, E.; Strasser, J.; Rita Höller, R.; Fleisch, M.; Berer, M.; Schlögl, S. Wavelength selective multi-material 3D printing of soft active devices using orthogonal photoreactions. Macromol. Rapid Commun. 2022, 44, 2200586. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Lei, M.; Zhang, P.; Leng, J.; Zheng, Z.; Yu, Y. Orthogonal photochemistry-assisted printing of 3D tough and stretchable conductive hydrogels. Nat. Commun. 2021, 12, 2082. [Google Scholar] [CrossRef]
- Yang, J.; Li, Z.; Gu, X.; Zhan, T.; Cui, J.; Zhang, K. Photogated photoswitchable [2]rotaxane based on orthogonal photoreactions. Tetrahedron 2021, 92, 132284. [Google Scholar] [CrossRef]
- Yamano, Y.; Murayama, K.; Asanuma, H. Dual crosslinking photo-switches for orthogonal photo-control of hybridization between serinol nucleic acid and RNA. Chem. A Eur. J. 2021, 27, 4475. [Google Scholar] [CrossRef]
- Wang, D.; Wagner, M.; Saydjari, A.; Mueller, J.; Winzen, S.; Butt, H.; Wu, S. A photoresponsive orthogonal supramolecular complex based on Host-Guest interactions. Chem. A Eur. J. 2017, 23, 2628–2634. [Google Scholar] [CrossRef]
- Manna, D.; Udayabhaskararao, T.; Zhao, H.; Klajn, R. Orthogonal light-induced self-assembly of nanoparticles using differently substituted azobenzenes. Angew. Chem. Int. Ed. 2015, 54, 12394–12397. [Google Scholar] [CrossRef]
- Olson, J.; Banghart, M.; Sabatini, B.; Ellis-Davies, G. Spectral evolution of a photochemical protecting group for orthogonal two-color uncaging with visible light. J. Am. Chem. Soc. 2013, 135, 15948–15954. [Google Scholar] [CrossRef] [Green Version]
- Corrigan, N.; Boyer, C. 100th anniversary of macromolecular science viewpoint: Photochemical reaction orthogonality in modern macromolecular science. ACS Macro Lett. 2019, 8, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Loffe, I.N.; Granovsky, A.A. Photoisomerization of Stilbene: The detailed XMCQDPT2 treatment. J. Chem. Theory Comput. 2013, 9, 4973–4990. [Google Scholar] [CrossRef]
- Pina, F.; Melo, M.J.; Laia, C.A.; Parola, J.; Lima, J.C. Chemistry and applications of flavylium compounds: A handful of colours. Chem. Soc. Rev. 2012, 41, 869–908. [Google Scholar] [CrossRef] [PubMed]
- Brouillard, R.; Iacobucci, G.A.; Sweeny, J.G. Chemistry of anthocyanin pigments. 9. UV-visible spectrophotometric determination of the acidity constants of apigeninidin and three related 3- deoxyflavylium salts. J. Am. Chem. Soc. 1982, 104, 7585–7590. [Google Scholar] [CrossRef]
- McClelland, R.A.; McGall, G.H. Hydration of the Flavylium Ion. 2. The 4′-Hydroxyflavylium Ion. J. Org. Chem. 1982, 47, 3730–3736. [Google Scholar] [CrossRef]
- Takeda, K.; Osakabe, A.; Saito, S.; Furuyama, D.; Tomita, A.; Kojima, Y.; Yamadera, M.; Sakuta, M. Components of protocyanin, a blue pigment from the blue flowers of Centaurea cyanus. Phytochemistry 2005, 66, 1607–1613. [Google Scholar] [CrossRef]
- Takeda, K. Blue metal complex pigments involved in blue flower color. Proc. Jpn. Acad. Ser. B Phys. Biol Sci. 2006, 82, 142–154. [Google Scholar] [CrossRef] [Green Version]
- Gould, K.S. Nature’s Swiss Army Knife: The Diverse Protective Roles of Anthocyanins in Leaves. J. Biomed. Biotechnol. 2004, 5, 314–320. [Google Scholar] [CrossRef] [Green Version]
- Lima, A.A.; Sussuchi, E.M.; Giovani, W.F. Electrochemical and Antioxidant Properties of Anthocyanins and Anthocyanidins. Croat. Chem. Acta 2007, 80, 29–34. [Google Scholar]
- Gould, K.S.; McKelvie, J.; Markham, K.R. Do anthocyanins function as antioxidants in leaves? Imaging of H2O2 in red and green leaves after mechanical injury. Plant Cell Environ. 2002, 25, 1261–1269. [Google Scholar] [CrossRef]
- Hoch, W.A.; Singsaas, E.L.; McCown, B.H. Resorption protection. Anthocyanins facilitate nutrient recovery in autumn by shielding leaves from potentially damaging light levels. Plant Physiol. 2003, 133, 1296–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keskitalo, J.; Bergquist, G.; Gardeström, P.; Jansson, S.A. Cellular Timetable of Autumn Senescence. Plant Physiol. 2005, 139, 1635–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Close, D.C.; Beadle, C.L. The ecophysiology of foliar anthocyanin. Bot. Rev. 2003, 69, 149–161. [Google Scholar] [CrossRef]
- Rechner, A.R.; Kroner, C. Anthocyanins and colonic metabolites of dietary polyphenols inhibit platelet function. Thromb. Res. 2005, 116, 327–334. [Google Scholar] [CrossRef]
- Bell, D.R.; Gochenaur, K. Direct vasoactive and vasoprotective properties of anthocyanin-rich extracts. J. Appl. Physiol. 2006, 100, 1164–1170. [Google Scholar] [CrossRef] [Green Version]
- Toufektsian, M.C.; De Lorgeril, M.; Nagy, N.; Salen, P.; Donati, M.B.; Giordano, L.; Mock, H.-P.; Peterek, S.; Matros, A.; Petroni, K. Chronic dietary intake of plant-derived anthocyanins protects the rat heart against ischemia-reperfusion injury. J. Nutr. 2008, 138, 747–752. [Google Scholar] [CrossRef] [Green Version]
- Bontempo, P.; de Masi, L.; Carafa, V.; Rigano, D.; Scisciola, L.; Iside, C.; Grassi, R.; Molinari, A.M.; Aversano, R.; Nebbioso, A. Anticancer activities of anthocyanin extract from genotyped Solanum tuberosum L. “Vitelotte”. J. Funct. Foods 2015, 19, 584–593. [Google Scholar] [CrossRef] [Green Version]
- Côté, J.; Caillet, S.; Doyon, G.; Dussault, D.; Sylvain, J.-F.; Lacroix, M. Antimicrobial effect of cranberry juice and extracts. Food Control 2011, 22, 1413–1418. [Google Scholar] [CrossRef]
- Bors, W.; Heller, W.; Michel, C.; Saran, M. Flavonoids as antioxidants: Determination of radical-scavenging efficiencies. Methods Enzymol. 1990, 186, 343–355. [Google Scholar] [CrossRef]
- Hardin, B.E.; Hoke, E.T.; Amstrong, P.B.; Yum, J.H.; Comte, P.; Torres, T.; Frechet, J.M.J.; Nazeeruddin, M.K.; Gratzel, M.; McGehee, M.D. Increased light harvesting in dye-sensitized solar cells with energy relay dyes. Nat. Photonics 2009, 3, 406–411. [Google Scholar] [CrossRef]
- Moncada, M.C.; Fernandez, D.; Lima, J.C.; Parola, A.J.; Lodeiro, C.; Folgosa, F.; Melo, M.J.; Pina, F. Multistate properties of 7-(N,N-diethylamino)-40 -hydroxyflavylium. An example of an unidirectional reaction cycle driven by pH. Org. Biomol. Chem. 2004, 2, 2802–2808. [Google Scholar] [CrossRef] [PubMed]
- Giestas, L.; Folgosa, F.; Lima, J.C.; Parola, A.J.; Pina, F. Bio-Inspired Multistate Networks Responsive to Light, pH and Thermal Inputs–An Example of a Multistate System Operating Through Different Algorithms. Eur. J. Org. Chem. 2005, 19, 4187–4200. [Google Scholar] [CrossRef]
- Furtado, P.; Figueiredo, P.; Chaves das Neves, H.; Pina, F. Photochemical and thermal degradation of anthocyanidins. Photochemical and thermal degradation of anthocyanidins. J. Photochem. Photobiol. A 1993, 75, 113–118. [Google Scholar] [CrossRef]
- Alejo-Armijo, A.; Parola, A.J.; Pina, F. pH-Dependent Multistate System Generated by a Synthetic Furanoflavylium Compound: An Ancestor of the Anthocyanin Multistate of Chemical Species. ACS Omega 2019, 4, 4091–4100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, M.J.; Moura, S.; Roque, A.; Maestri, M.; Pina, F. Photochemistry of luteolinidin: “Write-lock-read-unlock-erase” with a natural compound. J. Photochem. Photobiol. A 2000, 135, 33–39. [Google Scholar] [CrossRef]
- Gomes, R.; Diniz, A.M.; Jesus, A.; Parola, A.J.; Pina, F. The synthesis and reaction network of 2-styryl-1-benzopyrylium salts: An unexploited class of potential colorants. Dye. Pigment. 2009, 81, 69–79. [Google Scholar] [CrossRef]
- Sweeny, J.G.; Iacobucci, G.A. Effect of Substitution on the stability of 3-Deoxyanthocyanidins in Aqueous Solutions. J. Agric. Food Chem. 1983, 31, 531–533. [Google Scholar] [CrossRef]
- Petrov, V.; Diniz, A.M.; Cunha-Silva, L.; Parola, A.J.; Pina, F. Kinetic and thermodynamic study of 2′-hydroxy-8-methoxyflavylium. Reaction network interconverting flavylium cation and flavanone. RSC Adv. 2013, 3, 10786–10794. [Google Scholar] [CrossRef]
- Jimenez, A.; Pinheiro, C.; Parola, A.J.; Maestri, M.; Pina, F. The chemistry of 6-hydroxyflavylium: Zwitterionic base and p-quinoidal chalcones. A multiswitchable system operated by proton, electron and photon inputs. Photochem. Photobiol. Sci. 2007, 6, 372–380. [Google Scholar] [CrossRef]
- Gago, S.; Petrov, V.; Parola, A.J.; Pina, F. Synthesis and characterization of 3′-butoxy-flavylium derivatives. J. Photochem. Photobiol. A Chem. 2012, 244, 54–64. [Google Scholar] [CrossRef]
- Fernandez, D.; Parola, A.J.; Branco, L.C.; Afonso, C.A.M.; Pina, F. Thermal and photochemical properties of 4′-hydroxyflavylium in water-ionic liquid biphasic systems. J. Photochem. Photobiol. A Chem. 2004, 168, 185–189. [Google Scholar] [CrossRef]
- Brouillard, R.; Dubois, J. Mechanism of the Structural Transformations of Anthocyanins in Acidic Media. J. Am. Chem. Soc. 1976, 99, 1359–1364. [Google Scholar] [CrossRef]
- Pina, F.; Roque, A.; Melo, M.J.; Maestri, M.; Belladelli, L.; Balzani, V. Multistate/Multifunctional Molecular-Level Systems: Light and pH Switching between the Various Forms of a Synthetic Flavylium Salt. Chem. Eur. J. 1998, 4, 1184. [Google Scholar] [CrossRef]
- Pina, F.; Melo, M.J.; Parola, A.J.; Maestri, M.; Balzani, V. pH-Controlled Photochromism of Hydroxyflavylium Ions. Chem. Eur. J. 1998, 4, 2001. [Google Scholar] [CrossRef]
- Silva, G.T.M.; Silva, C.; Silva, K.M.; Pioli, R.M.; Costa, T.S.; Marto, V.V.; Freitas, A.A.; Rozendo, J.; Martins, L.M.O.S.; Cavalcante, V.F.; et al. Fluorescence and Phosphorescence of Flavylium Cation Analogues of Anthocyanins. Photochem 2022, 2, 423–434. [Google Scholar] [CrossRef]
- Mora-Soumille, N.; Al Bittar, S.; Rosa, M.; Olivier Dangles, O. Analogs of anthocyanins with a 3′,4′-dihydroxy substitution: Synthesis and investigation of their acid–base, hydration, metal binding and hydrogen-donating properties in aqueous solution. Dye. Pigment. 2013, 96, 7–15. [Google Scholar] [CrossRef]
- Moor, K.J.; Schmitt, M.; Erickson, P.R.; McNeill, K. Sorbic acid as a triplet probe: Triplet energy and reactivity with triplet-state dissolved organic matter via 1O2. Environ. Sci. Technol. 2019, 53, 8078–8086. [Google Scholar] [CrossRef] [Green Version]
- Pina, F.; Melo, M.J.; Maestri, M.; Passaniti, P.; Camaioni, N.; Balzani, V. Photo- and pH-induced transformations of flavylium cation: “Write-Lock-Read-Unlock-Erase” cycles. Eur. J. Org. Chem. 1999, 1999, 3199–3207. [Google Scholar] [CrossRef]
- Johns, V.K.; Wang, Z.; Li, X.; Liao, Y. Physicochemical study of a metastable-state photoacid. J. Phys. Chem. A 2013, 117, 13101–13104. [Google Scholar] [CrossRef]
- Shi, Z.; Peng, P.; Strohecker, D.; Liao, Y. Long-lived photoacid based upon a photochromic reaction. J. Am. Chem. Soc. 2011, 133, 14699–14703. [Google Scholar] [CrossRef]
- Halbritter, T.; Kaiser, C.; Wachtveitl, J.; Heckel, A. Pyridine-spiropyran derivative as a persistent, reversible photoacid in water. J. Org. Chem. 2017, 82, 8040–8047. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Thermal mechanism of flavylium ion with CB using F1 and photochemical mechanism of AH+ and Ct as an example. Path a represents the reaction mechanism in an acidic solution, while path b represents the basic mechanism between AH+ and B2.
Figure 1.
Thermal mechanism of flavylium ion with CB using F1 and photochemical mechanism of AH+ and Ct as an example. Path a represents the reaction mechanism in an acidic solution, while path b represents the basic mechanism between AH+ and B2.

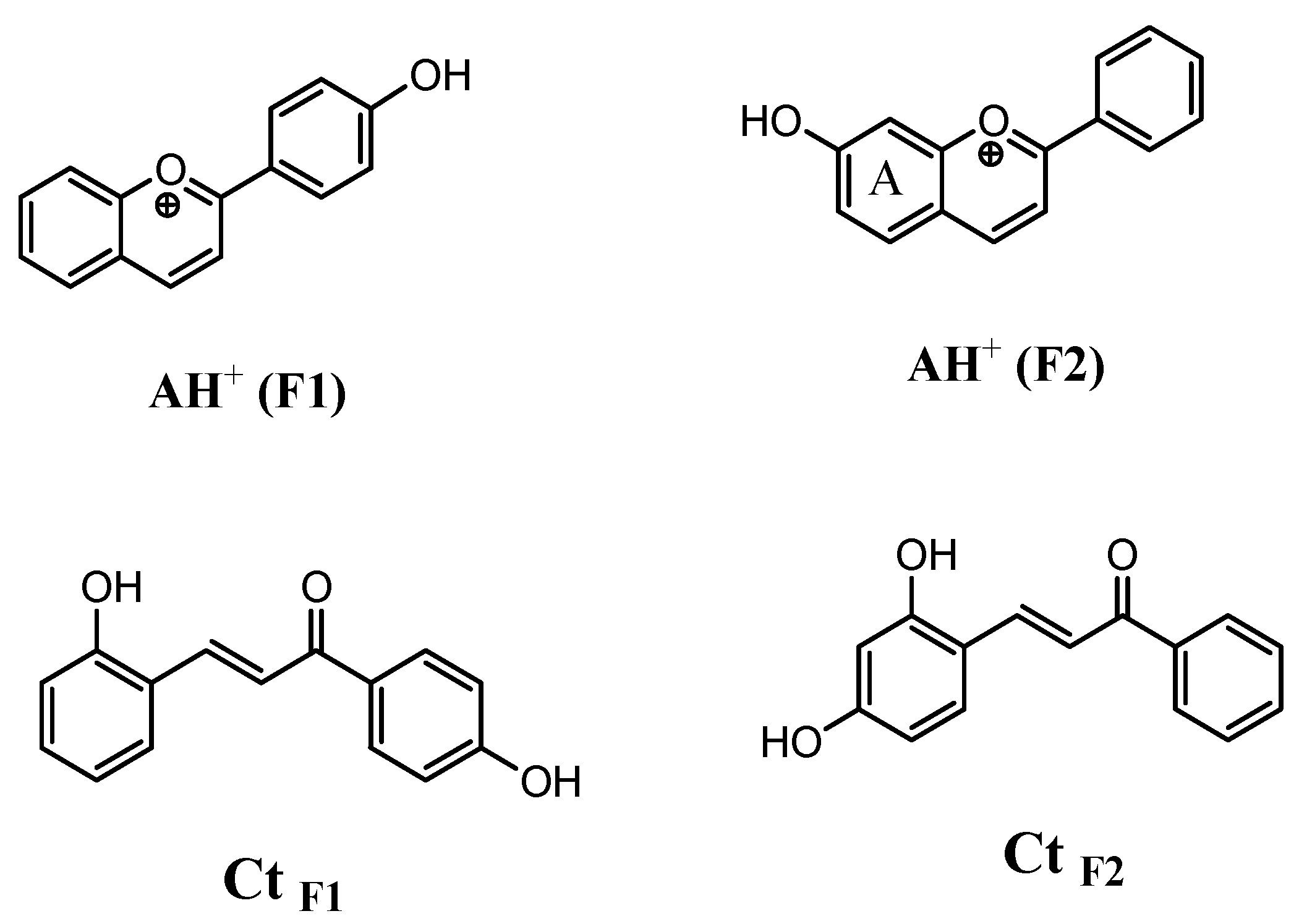
Figure 2.
Structure of flavylium (AH+) isomers 4′-hydroxyflavylium (F1) and 7-hydroxyflavylium (F2) and their corresponding trans-chalcone isomers CtF1 (3-(2-hydroxy-phenyl)-1-(4-hydroxy-phenyl)-propenone) and CtF2 (3-(2,4-Dihydroxy-phenyl)-1-phenyl-propenone) in the equilibrated mixture at different pH levels.
Figure 2.
Structure of flavylium (AH+) isomers 4′-hydroxyflavylium (F1) and 7-hydroxyflavylium (F2) and their corresponding trans-chalcone isomers CtF1 (3-(2-hydroxy-phenyl)-1-(4-hydroxy-phenyl)-propenone) and CtF2 (3-(2,4-Dihydroxy-phenyl)-1-phenyl-propenone) in the equilibrated mixture at different pH levels.

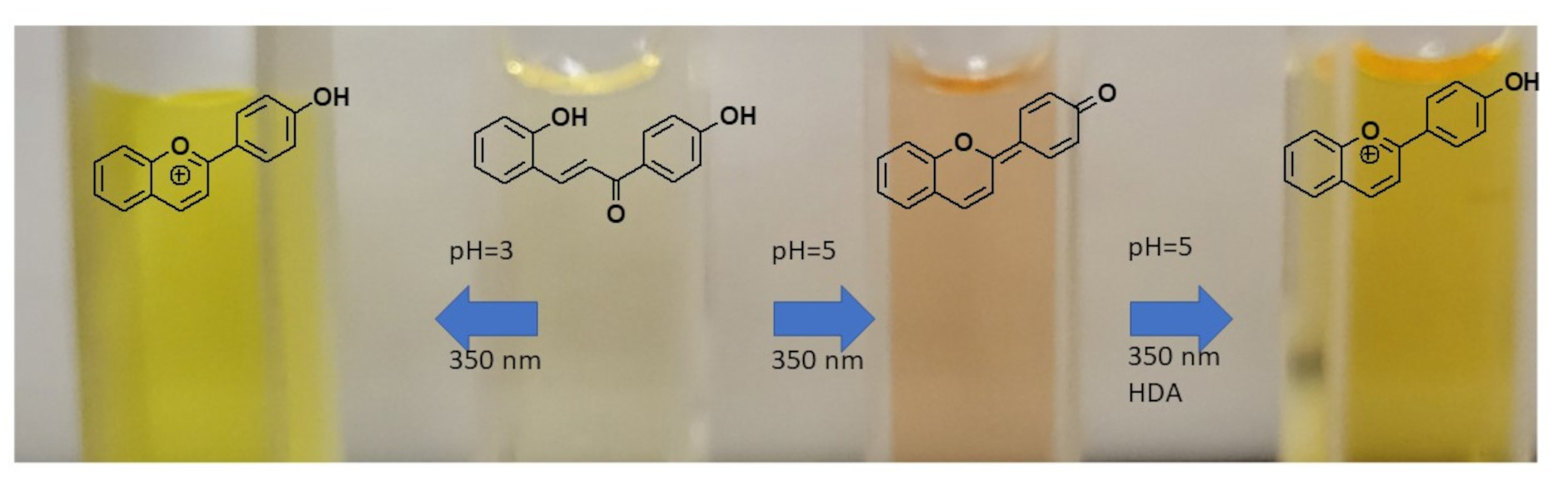
Figure 3.
UV–vis spectra of flavyliums F1 and F2 and their CB mixtures in various pH levels. (a) On the F1 spectrum, 435 nm is assigned for AH+, 302 nm for Cc, 345 nm for Ct, 426 nm for Ct2− (dotted blue curve at pH = 12), and 498 nm for A (dotted black curve obtained immediately after dissolving flavylium ion in pH = 7); (b) on the F2 spectrum, 428 nm is assigned for AH+, 368 nm for Ct, and 478 nm for Ct2−. pKa′ was found to be around 4.0 for F1 and 2.9 for F2.
Figure 3.
UV–vis spectra of flavyliums F1 and F2 and their CB mixtures in various pH levels. (a) On the F1 spectrum, 435 nm is assigned for AH+, 302 nm for Cc, 345 nm for Ct, 426 nm for Ct2− (dotted blue curve at pH = 12), and 498 nm for A (dotted black curve obtained immediately after dissolving flavylium ion in pH = 7); (b) on the F2 spectrum, 428 nm is assigned for AH+, 368 nm for Ct, and 478 nm for Ct2−. pKa′ was found to be around 4.0 for F1 and 2.9 for F2.

Figure 4.
Schematic representation for relative energy levels and approximate absorptions of AH+ and CB in pH range between 1 and 7. Ct is generally most stable at a pH greater than pKa′, whereas AH+ is most stable in a more acidic pH solution than pKa′.
Figure 4.
Schematic representation for relative energy levels and approximate absorptions of AH+ and CB in pH range between 1 and 7. Ct is generally most stable at a pH greater than pKa′, whereas AH+ is most stable in a more acidic pH solution than pKa′.

Figure 5.
Irradiation of F1 solutions (a) 350 nm at initial pH = 3.7; (b) 350 nm at initial pH = 4.5; (c) 350 nm at initial pH = 5.2 (AH+ peak in dotted black formed by quenching with excessive 3 mM HDA at pH = 5.2 and AH+/A peaks in black formed by quenching with 0.1 mM HDA at pH = 5.2).
Figure 5.
Irradiation of F1 solutions (a) 350 nm at initial pH = 3.7; (b) 350 nm at initial pH = 4.5; (c) 350 nm at initial pH = 5.2 (AH+ peak in dotted black formed by quenching with excessive 3 mM HDA at pH = 5.2 and AH+/A peaks in black formed by quenching with 0.1 mM HDA at pH = 5.2).

Figure 6.
Irradiation of F2 solutions (a) 350 nm at pH = 3.1; (b) 350 nm at pH = 4.2; (c) 350 nm at pH = 5.6.
Figure 6.
Irradiation of F2 solutions (a) 350 nm at pH = 3.1; (b) 350 nm at pH = 4.2; (c) 350 nm at pH = 5.6.

Figure 7.
DFT modeling for HOMO and LUMO orbitals of trans-chalcones for F1 and F2.

Figure 8.
Calculation of S0 and S1 energy levels for chalcones of F1 (a) and F2 (b) at different dihedral angles, showing snapshots of energy surfaces and intersections.
Figure 8.
Calculation of S0 and S1 energy levels for chalcones of F1 (a) and F2 (b) at different dihedral angles, showing snapshots of energy surfaces and intersections.

Figure 9.
Schematic drawing based on the calculation for the whole process from Ct to AH+. The numbers on the schematic illustration indicate how much more energy they have compared to the energy of corresponding trans-chalcones CtF1 and CtF2.
Figure 9.
Schematic drawing based on the calculation for the whole process from Ct to AH+. The numbers on the schematic illustration indicate how much more energy they have compared to the energy of corresponding trans-chalcones CtF1 and CtF2.

Figure 10.
Intra-molecular proton transfer reaction in the excited states (ESIPT) can lead to easier isomerization to trans-chalcone.
Figure 10.
Intra-molecular proton transfer reaction in the excited states (ESIPT) can lead to easier isomerization to trans-chalcone.

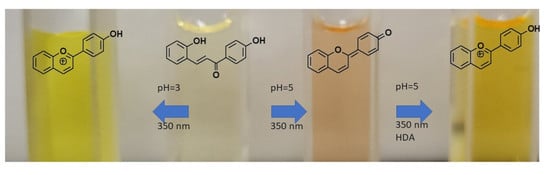
Figure 11.
Orthogonal response of Ct for F1 (a) and F2 (b) with two signals, pH and light.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kang, J.; Basilashvili, K.; Yoo, B.; Lee, J.I. pH-Induced Orthogonal Photoresponse of trans-Chalcone Isomers and Related Compounds in Equilibria. Colorants 2023, 2, 58-72. https://doi.org/10.3390/colorants2010005
AMA Style
Kang J, Basilashvili K, Yoo B, Lee JI. pH-Induced Orthogonal Photoresponse of trans-Chalcone Isomers and Related Compounds in Equilibria. Colorants. 2023; 2(1):58-72. https://doi.org/10.3390/colorants2010005
Chicago/Turabian StyleKang, Jeonghee, Ketevan Basilashvili, Barney Yoo, and Jong I. Lee. 2023. "pH-Induced Orthogonal Photoresponse of trans-Chalcone Isomers and Related Compounds in Equilibria" Colorants 2, no. 1: 58-72. https://doi.org/10.3390/colorants2010005