
A Comprehensive Review on Chemical Synthesis and Chemotherapeutic Potential of 3-Heteroaryl Fluoroquinolone Hybrids


 , and
, and
Abstract
:1. Introduction
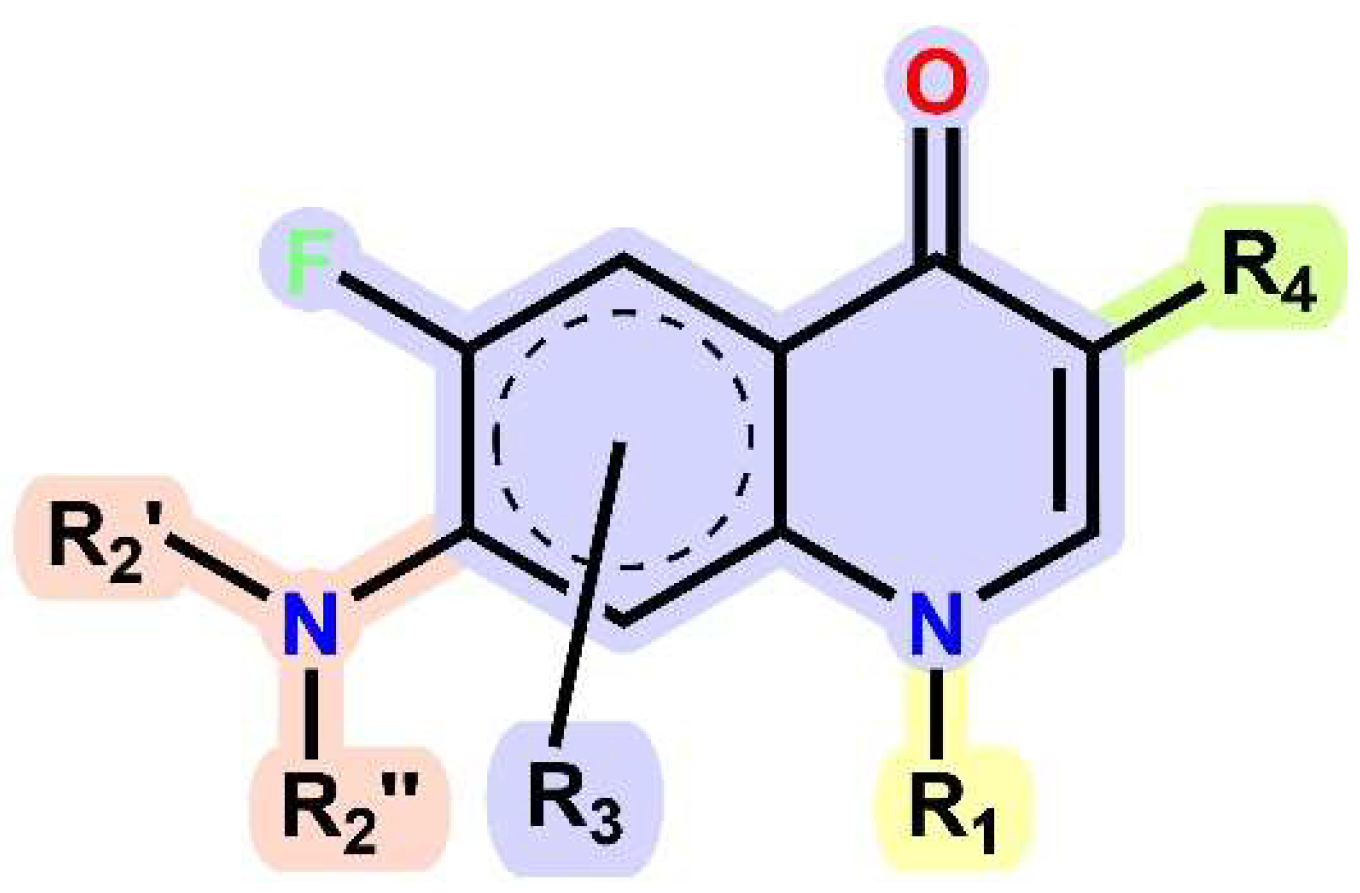
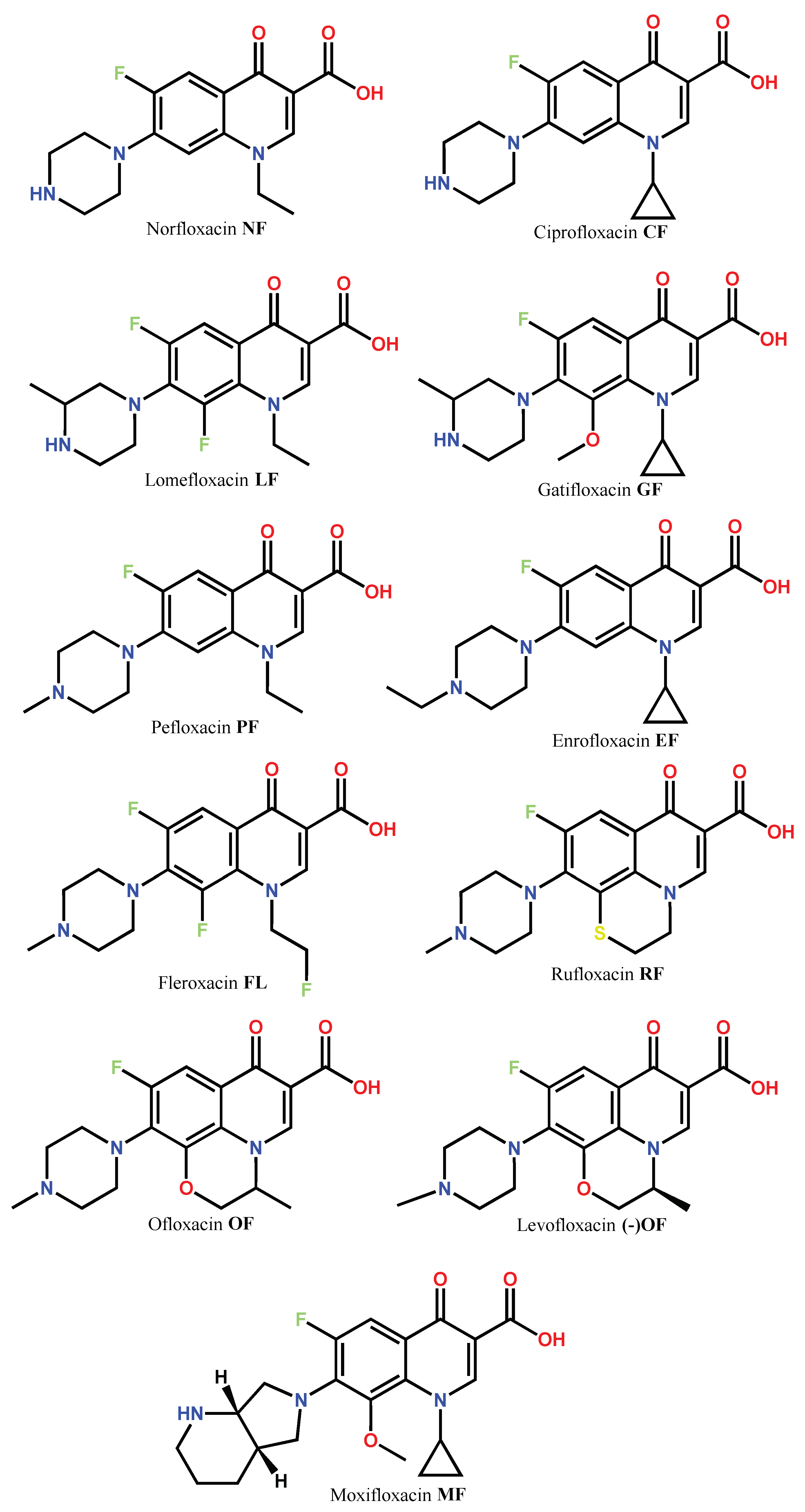
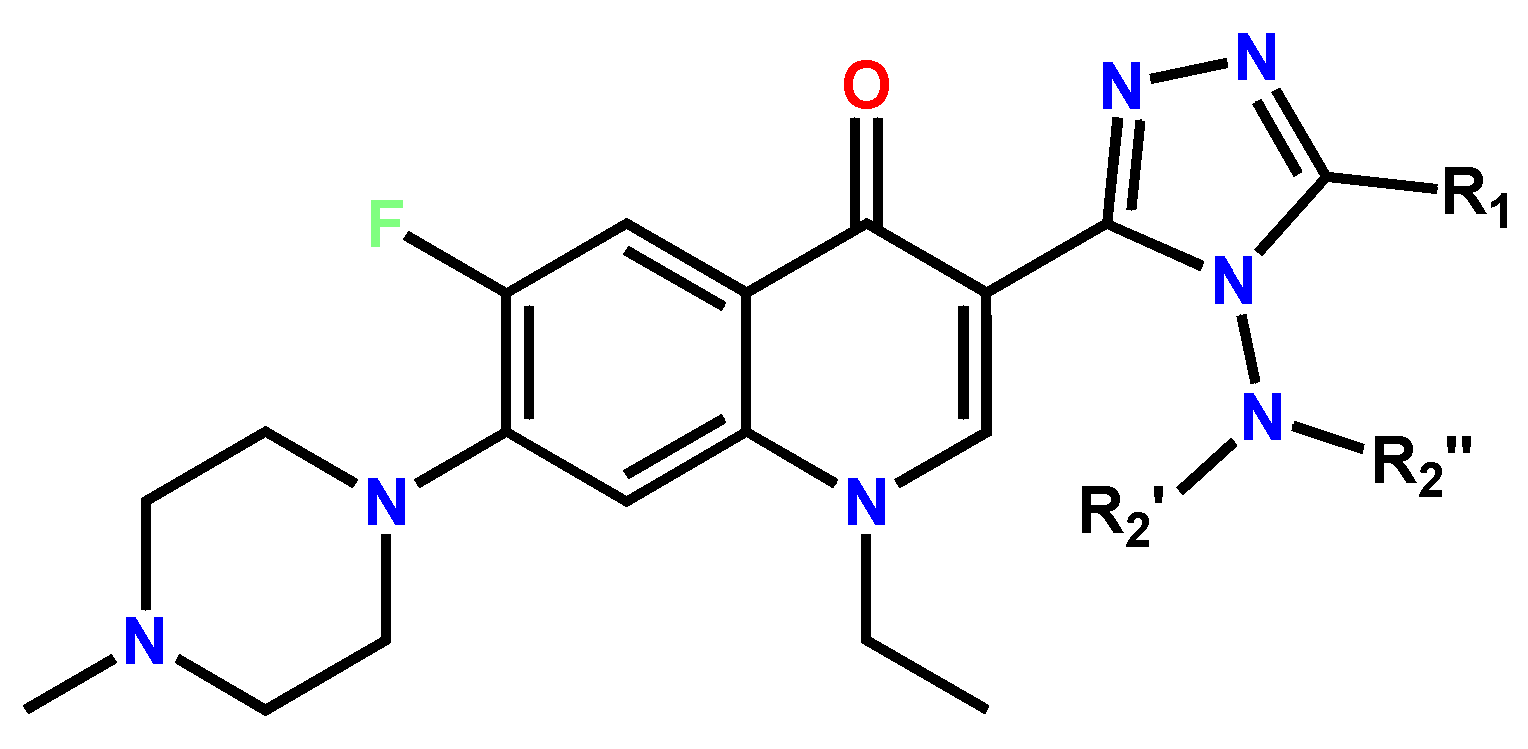
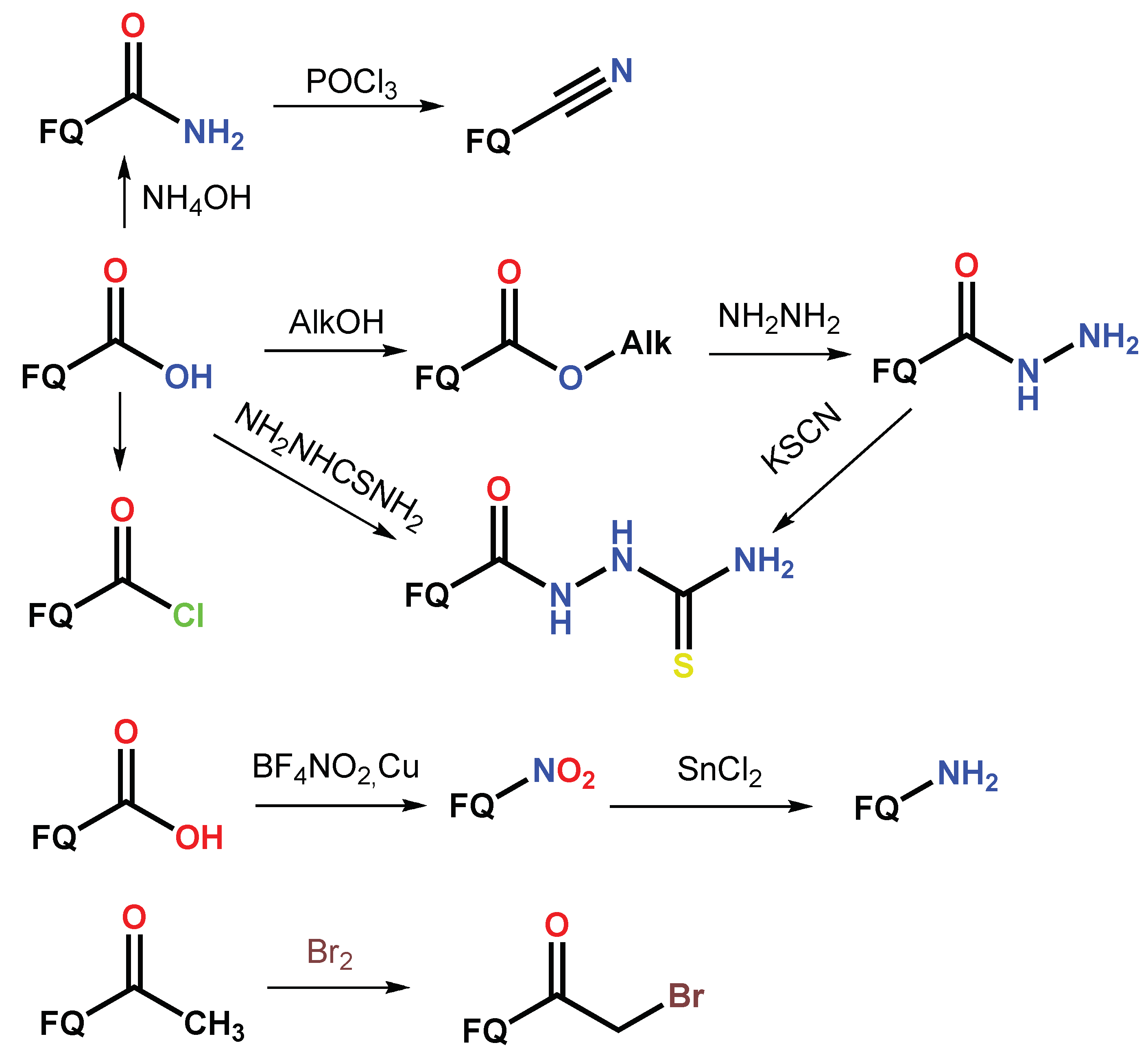
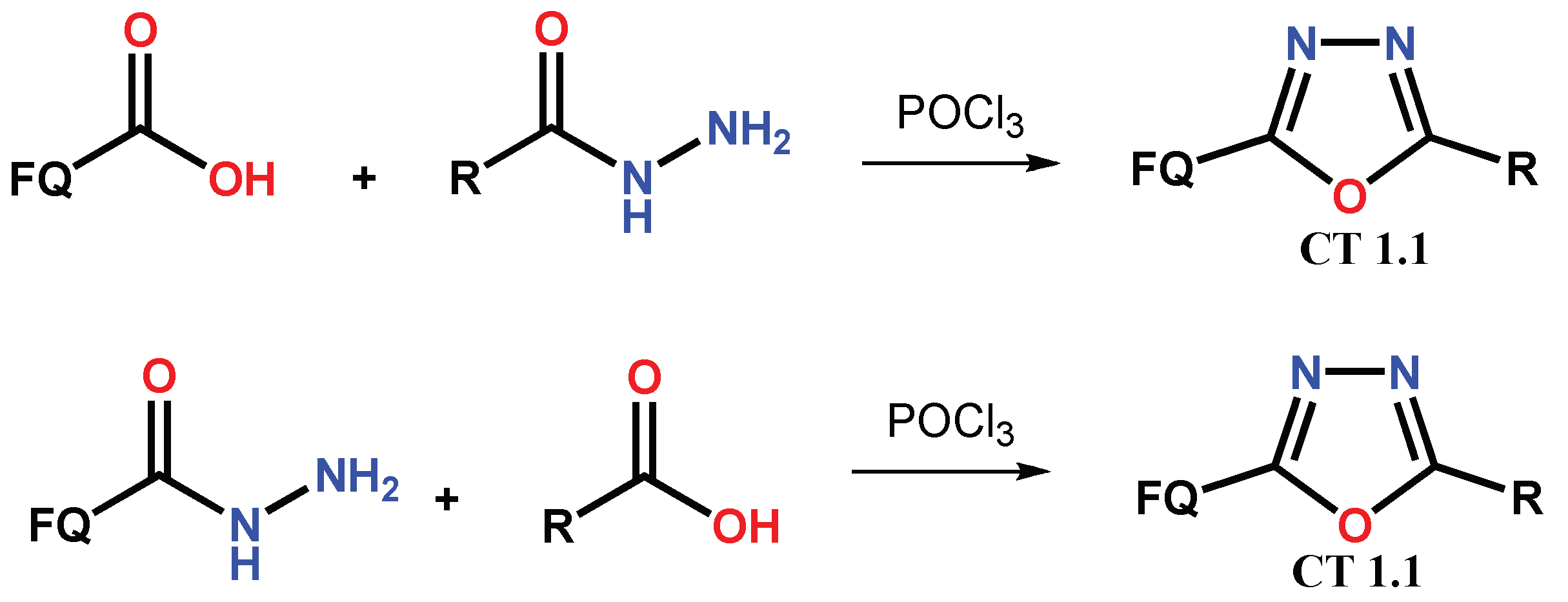
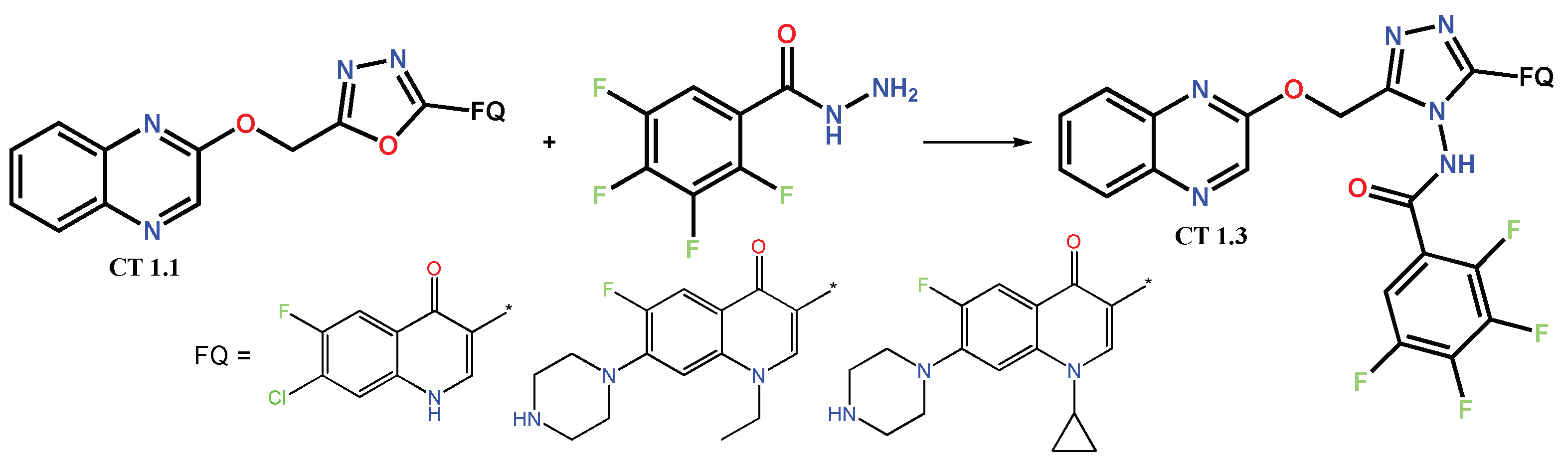
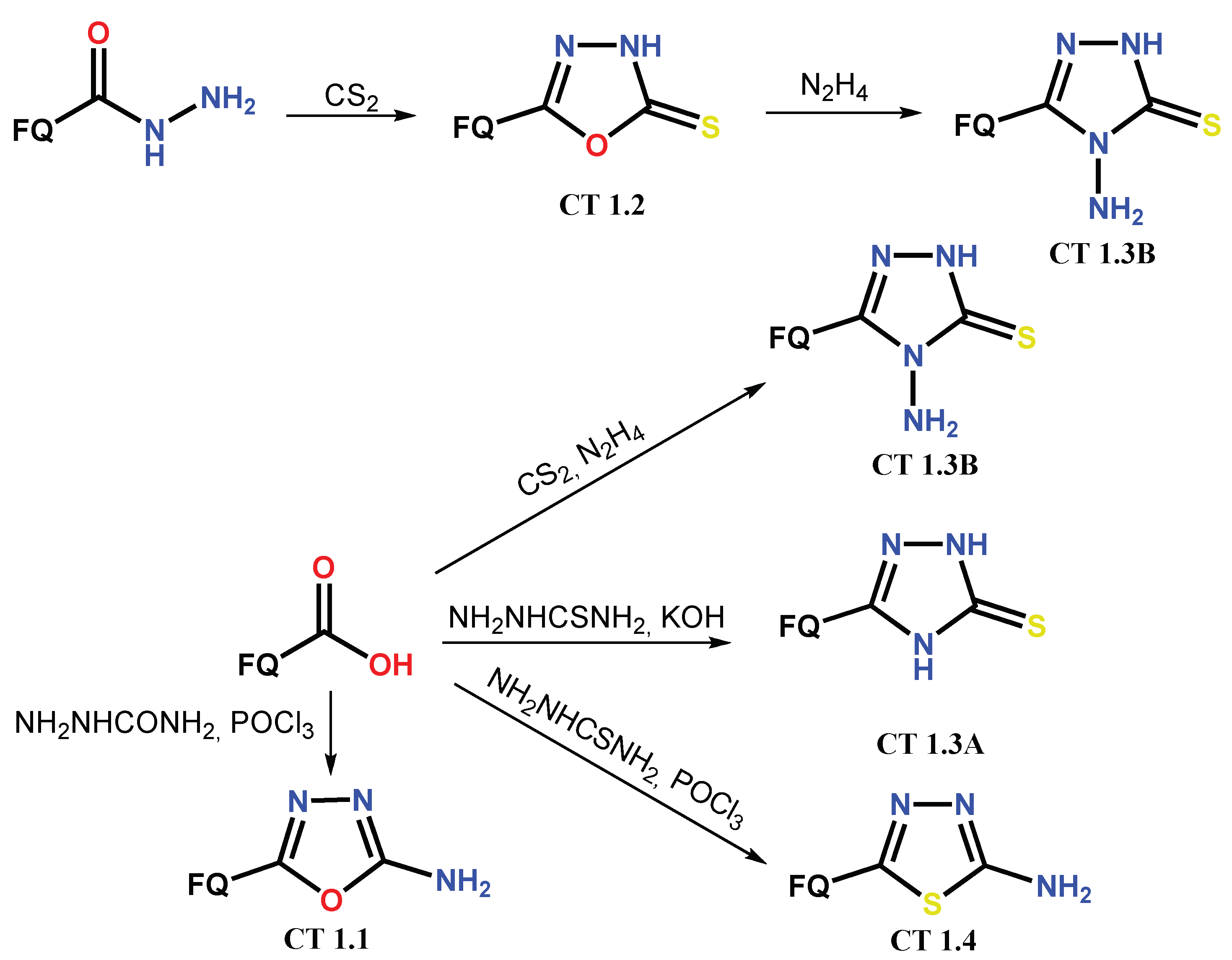
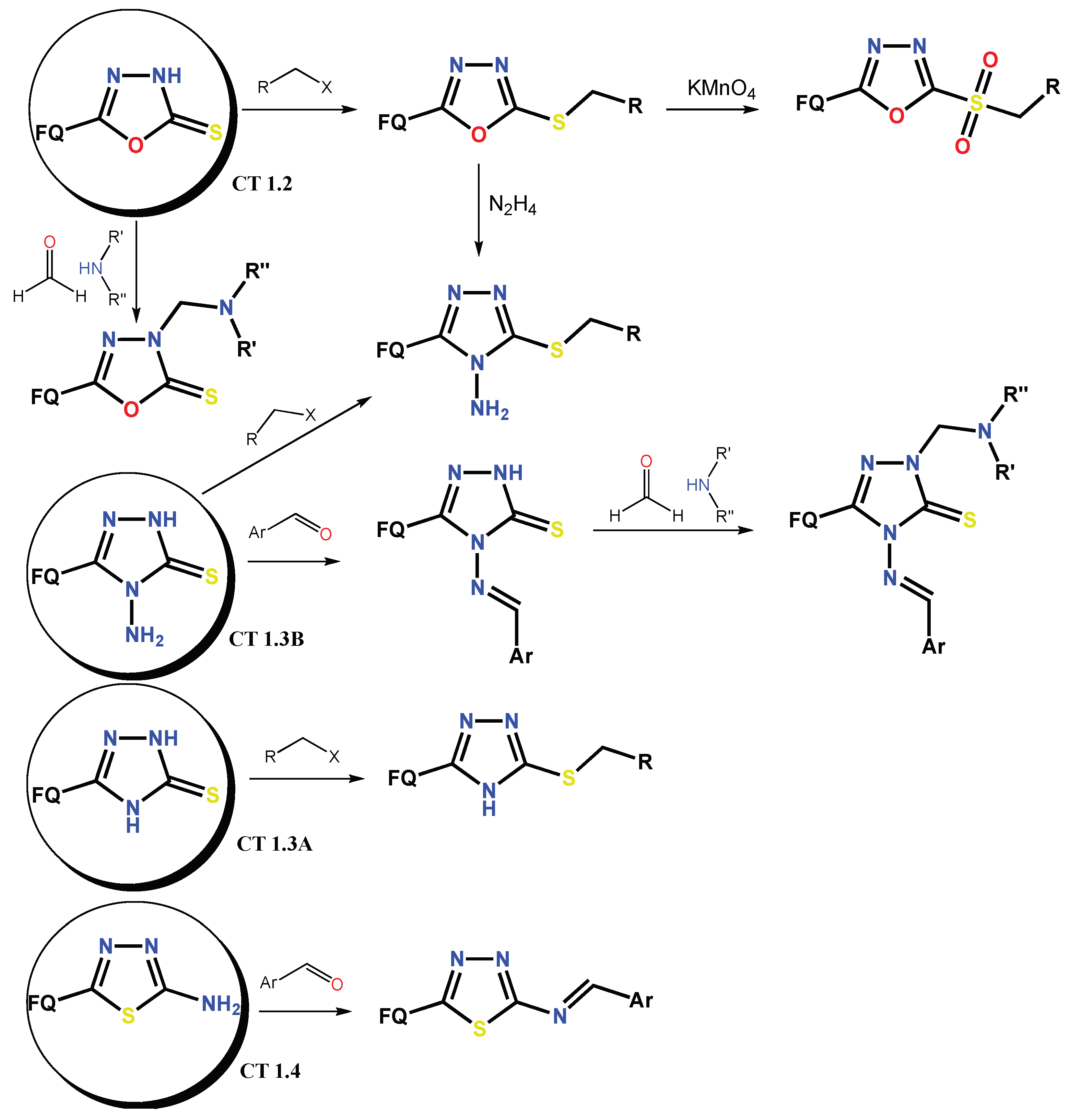
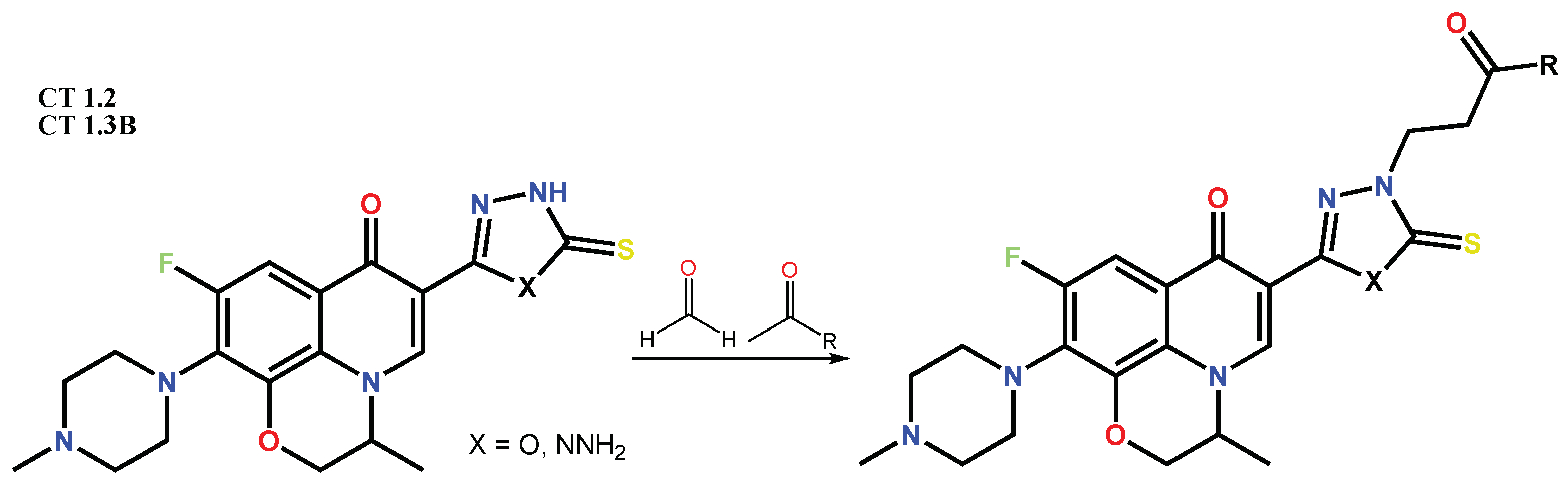
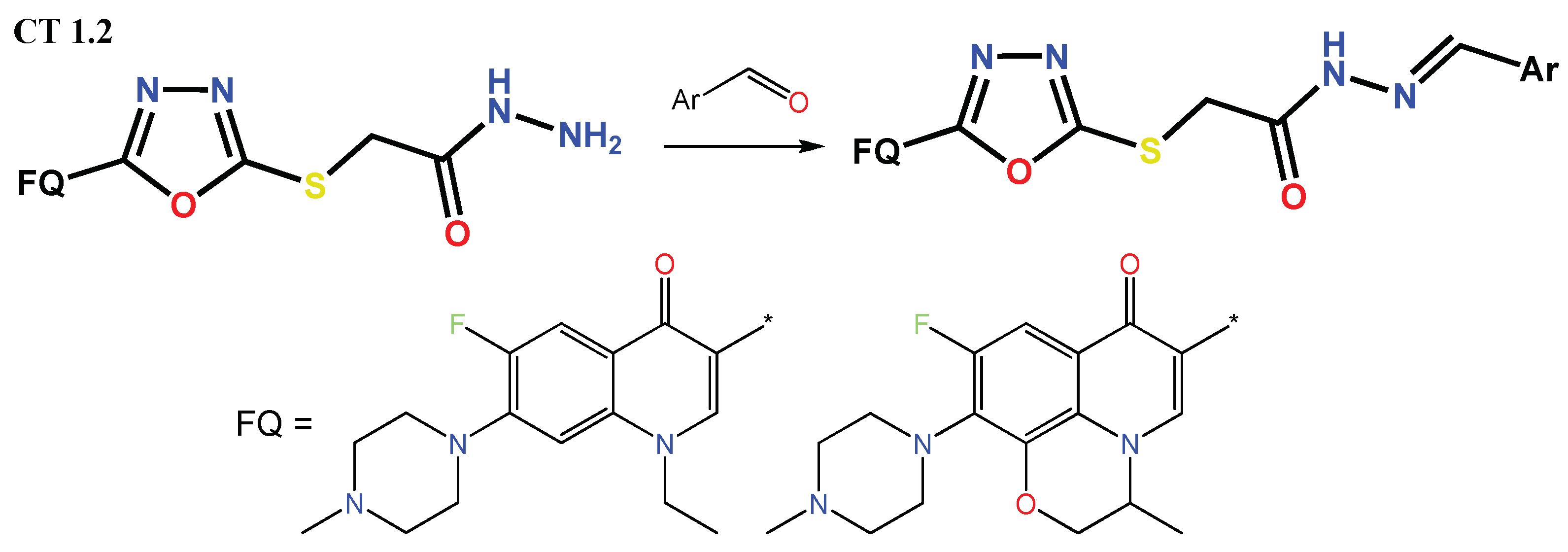
1.1. Modification of FQs with Five-Membered Heterocycles
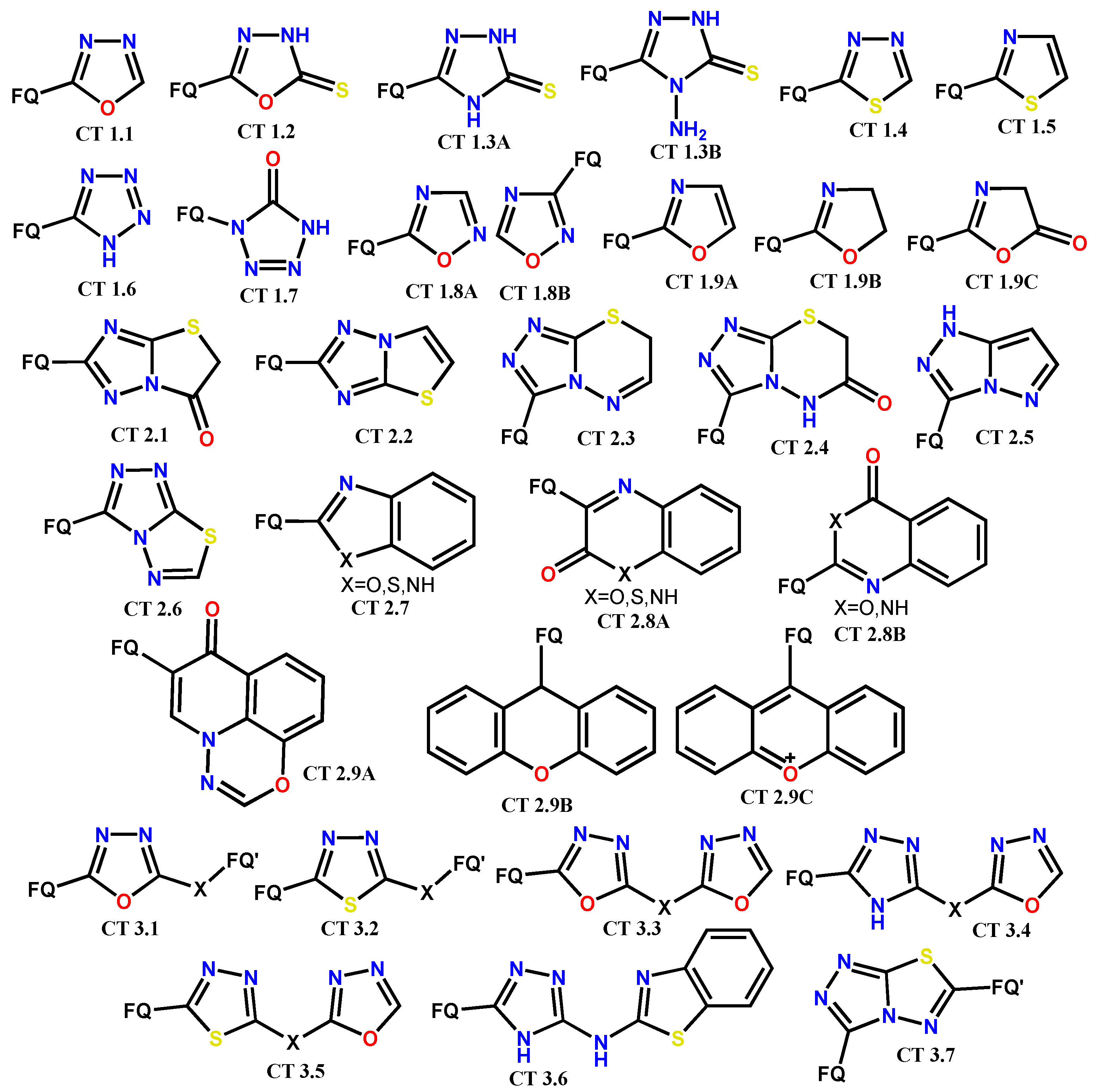
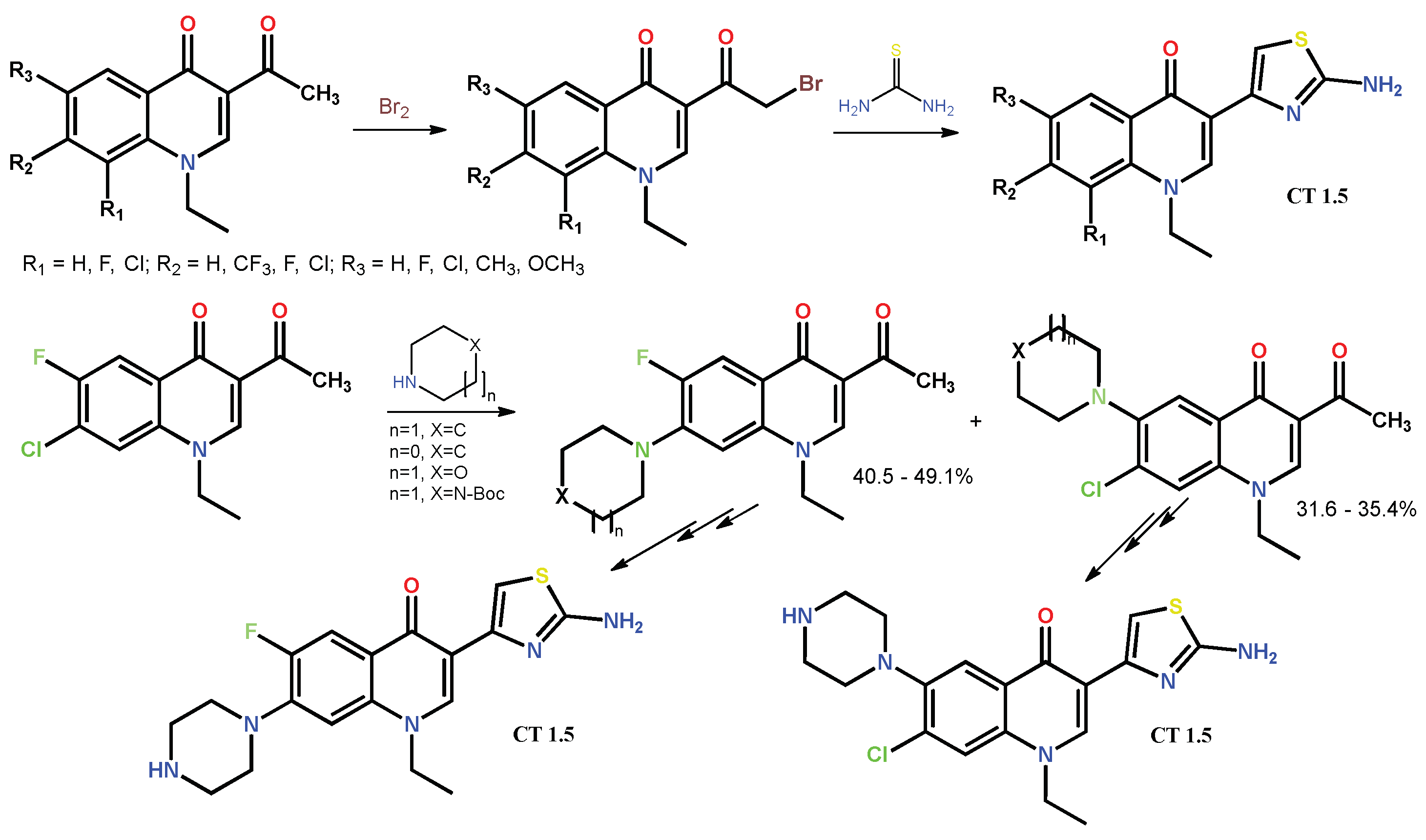
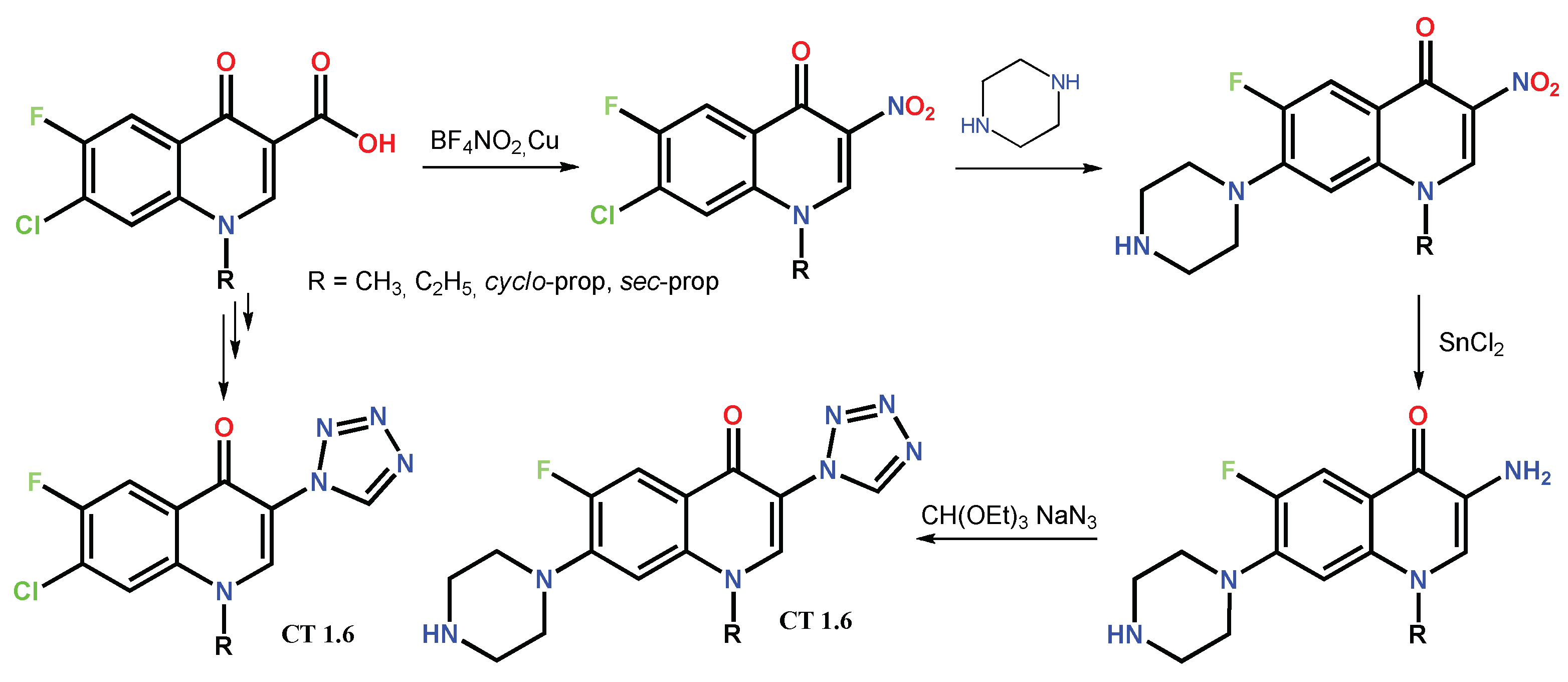
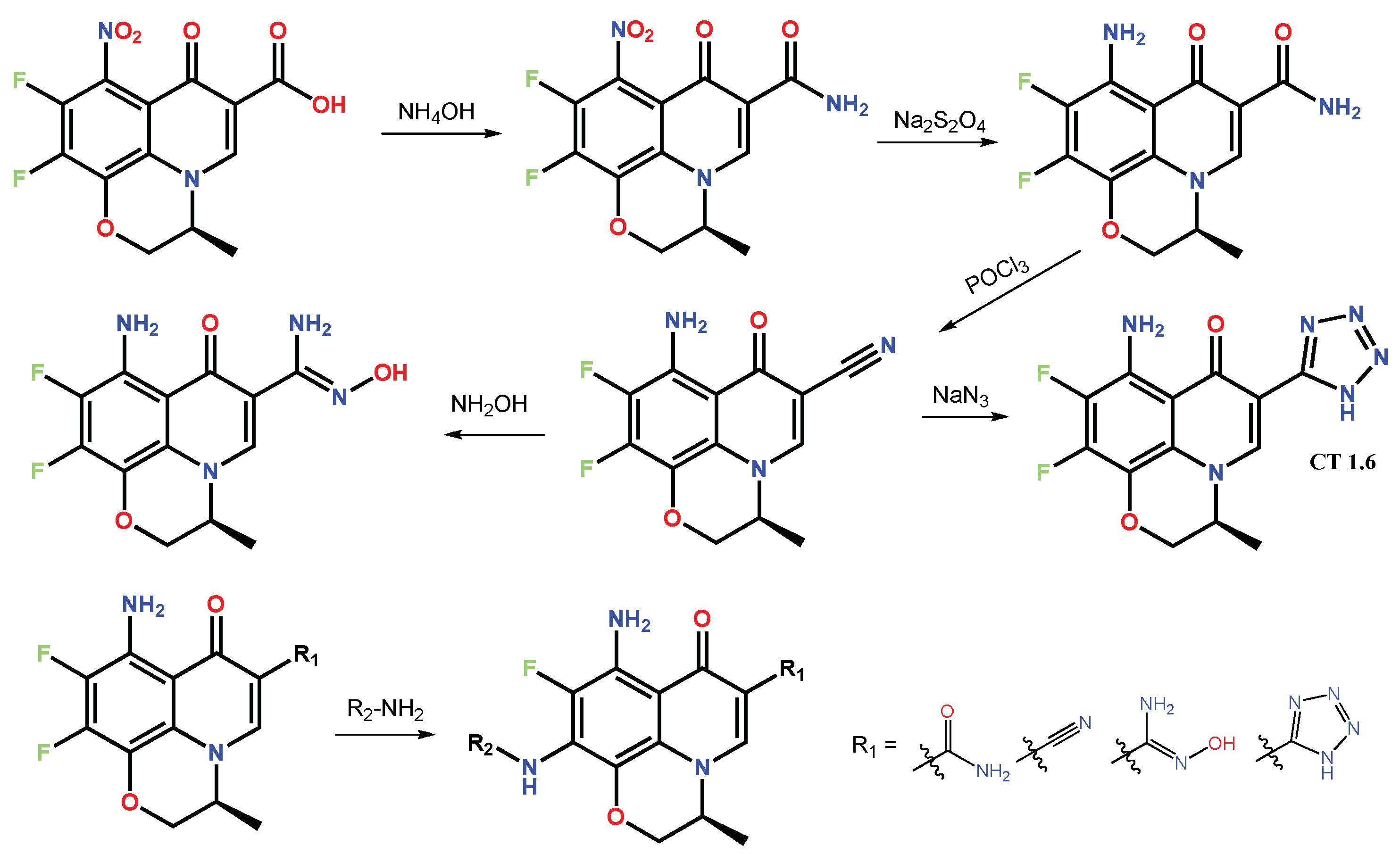
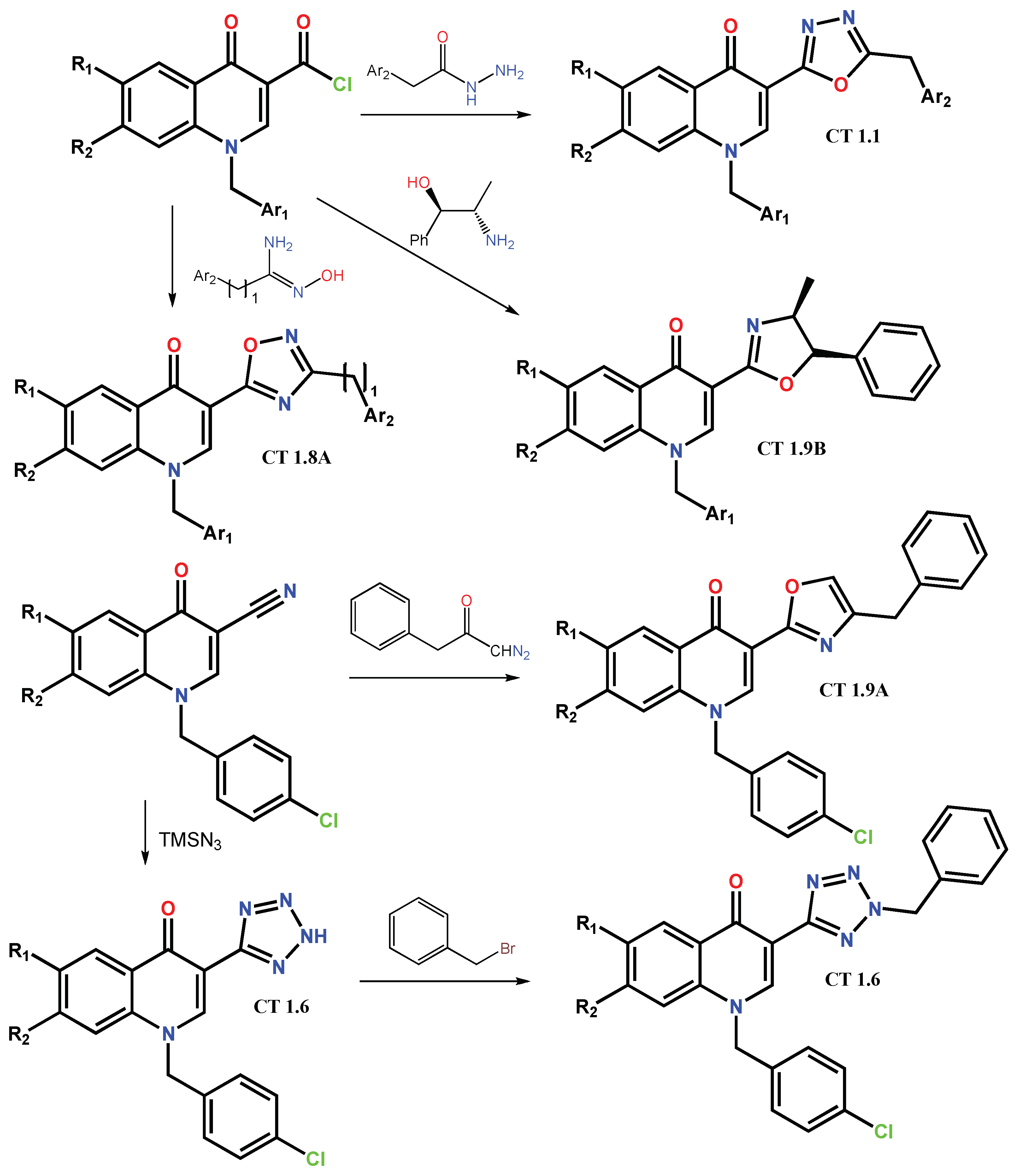
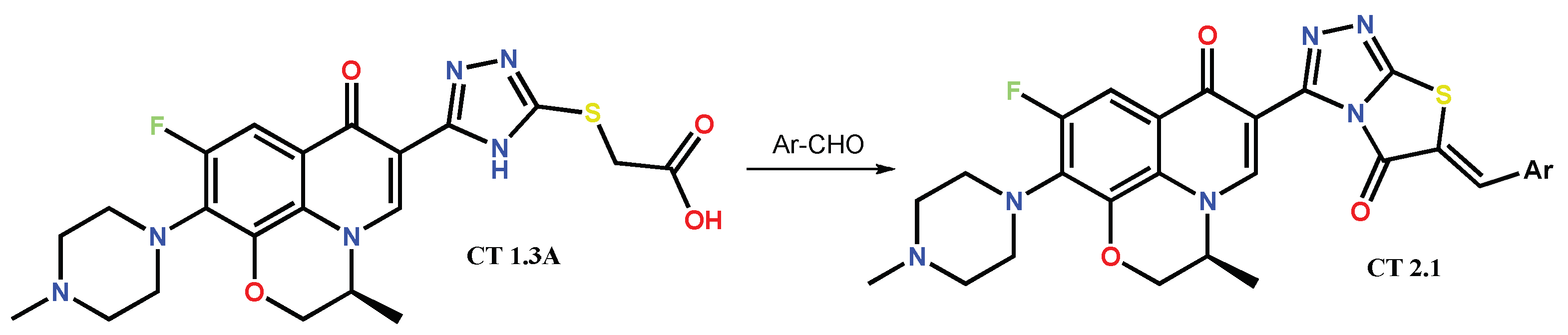
1.2. Modification of the C-3 Carboxylic Acid Group with Fused Heterocycles
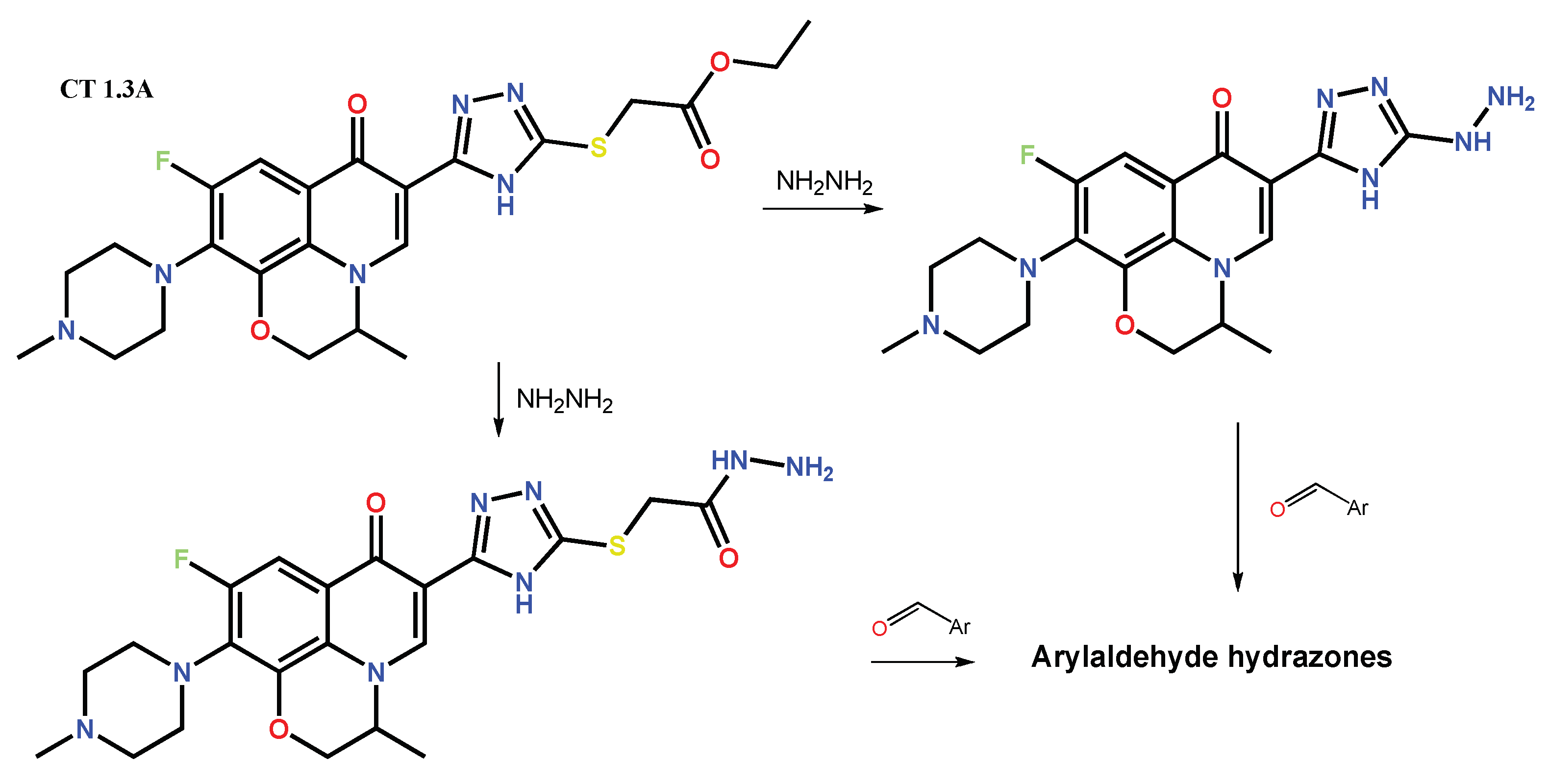
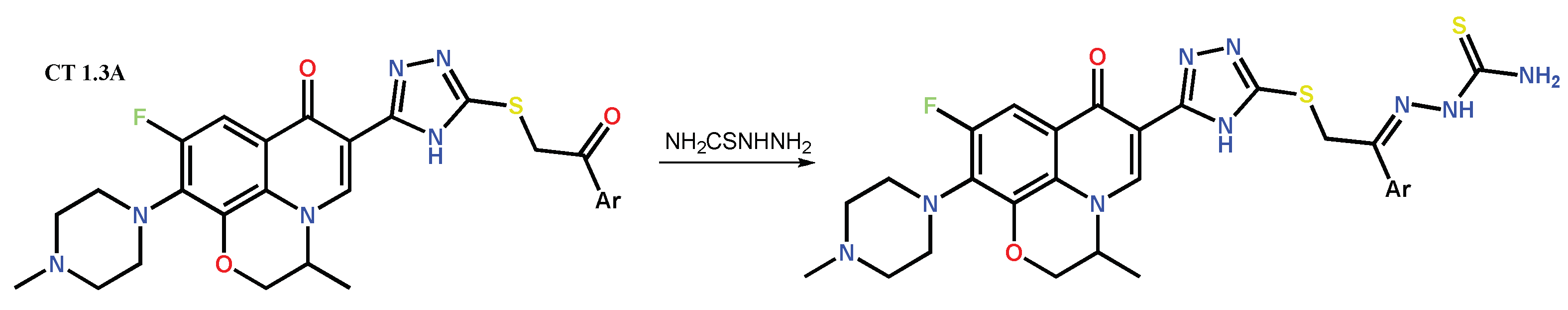
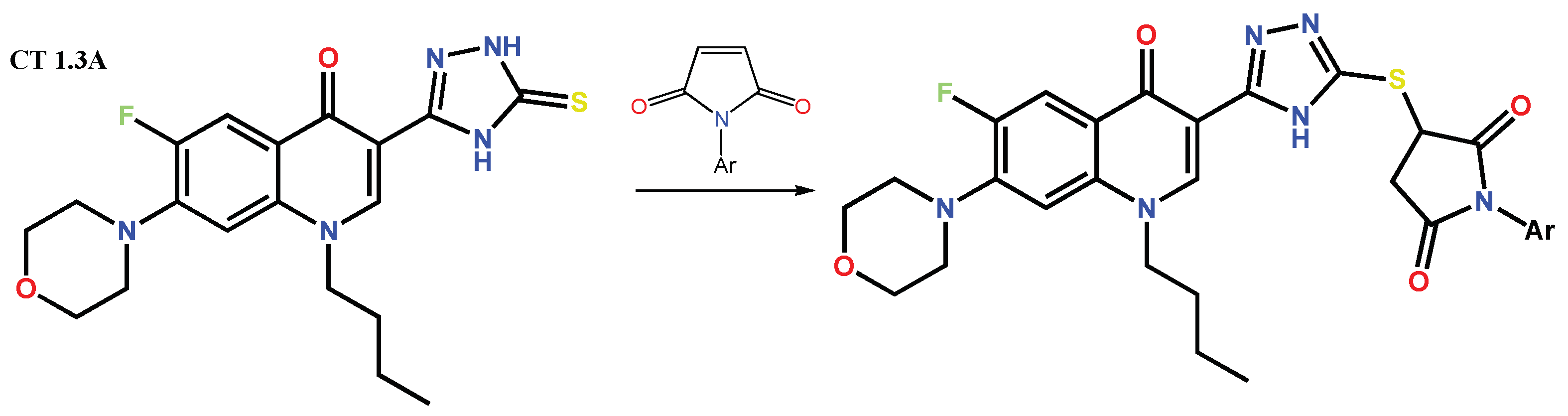
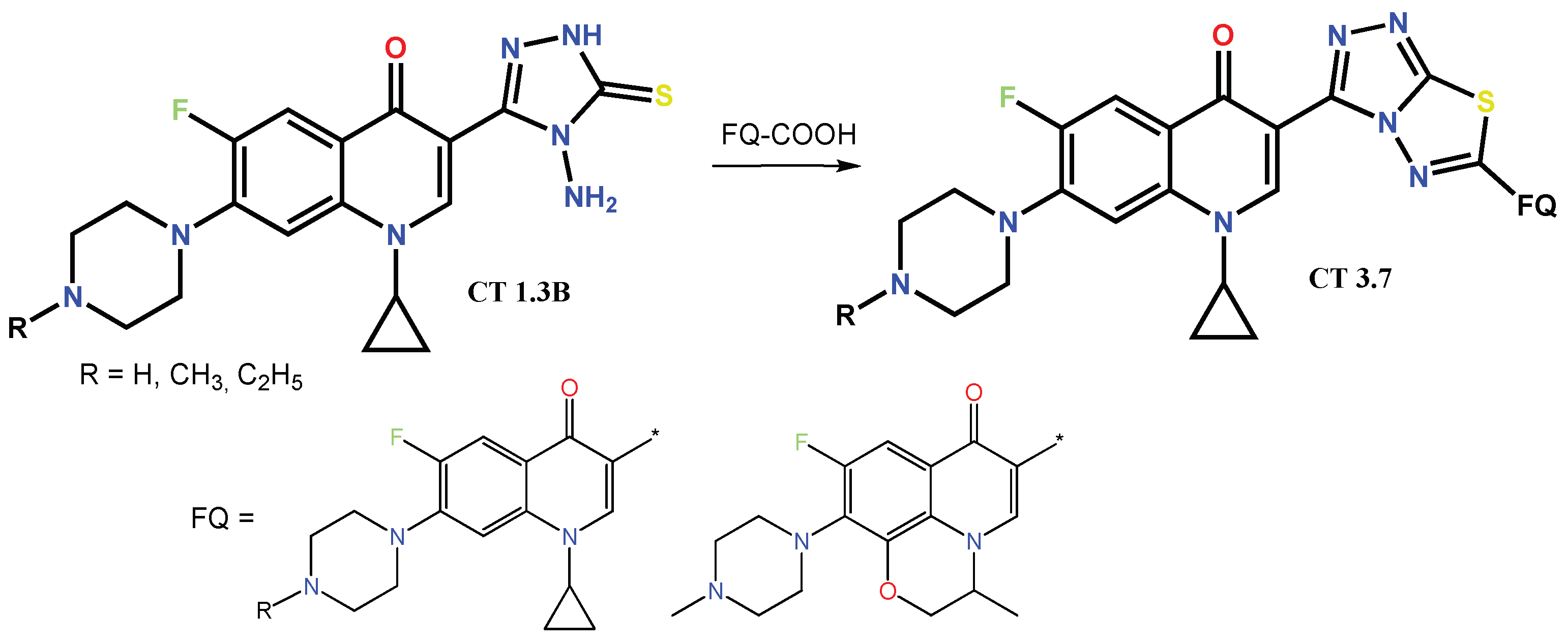
1.3. Bis-Fluoroquinolones and FQs with Two Heterocyclic Moieties as a Special Case in the Investigated Area
2. Biological Activity of the 3-Heteroaryl FQ Hybrids
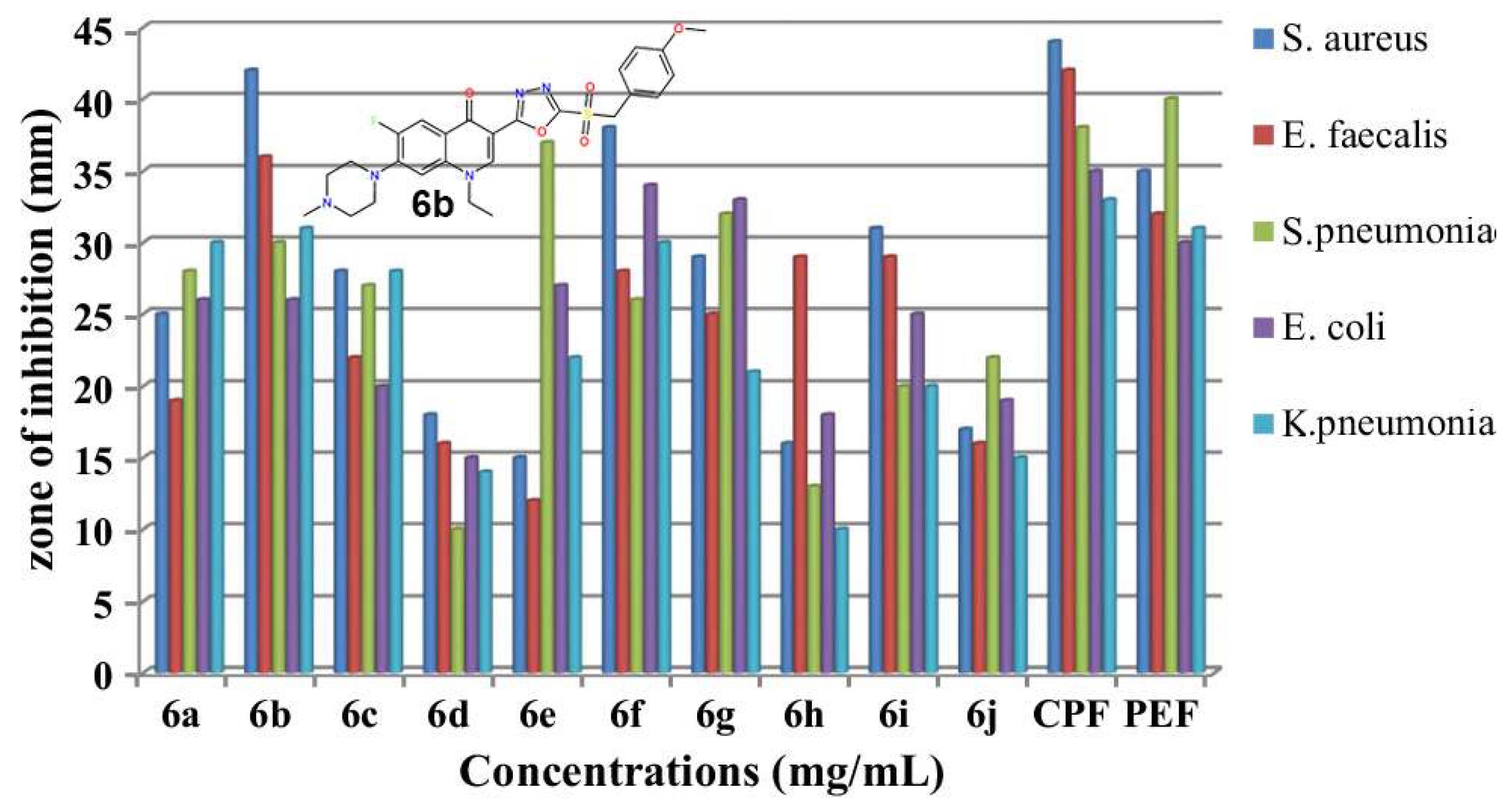
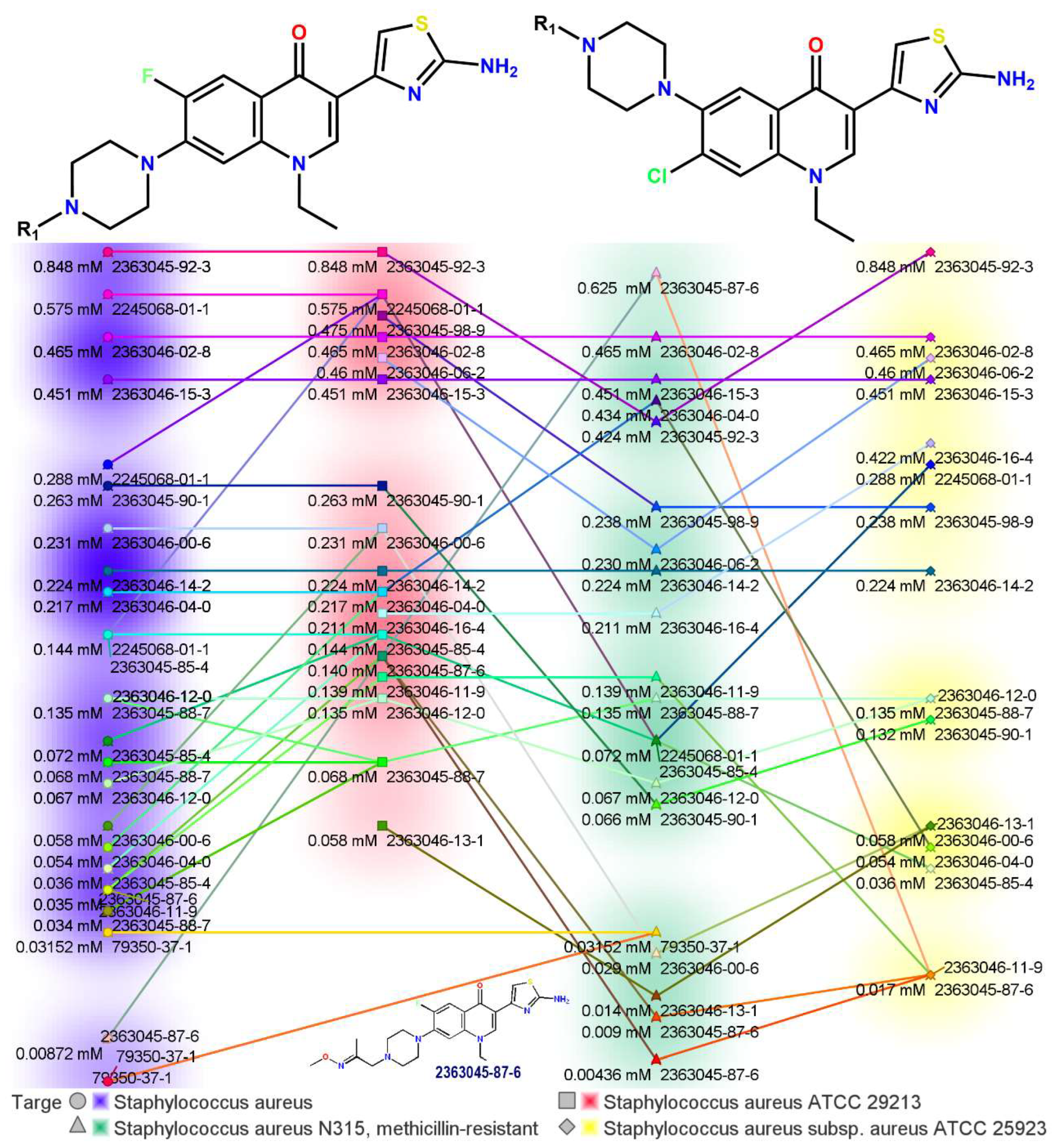
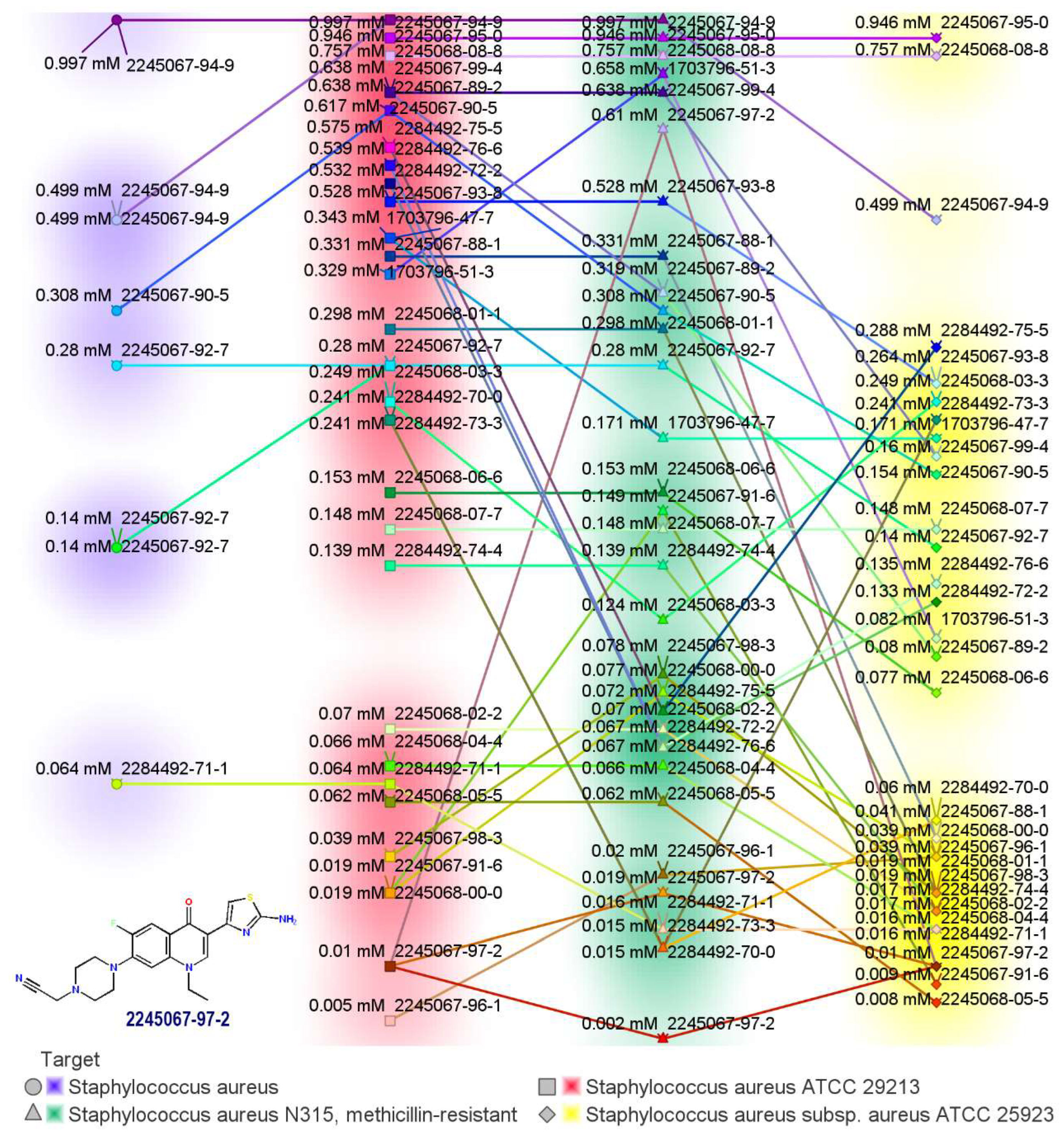
2.1. Novel FQ Hybrids as Antimicrobials and Antiviral Medicines
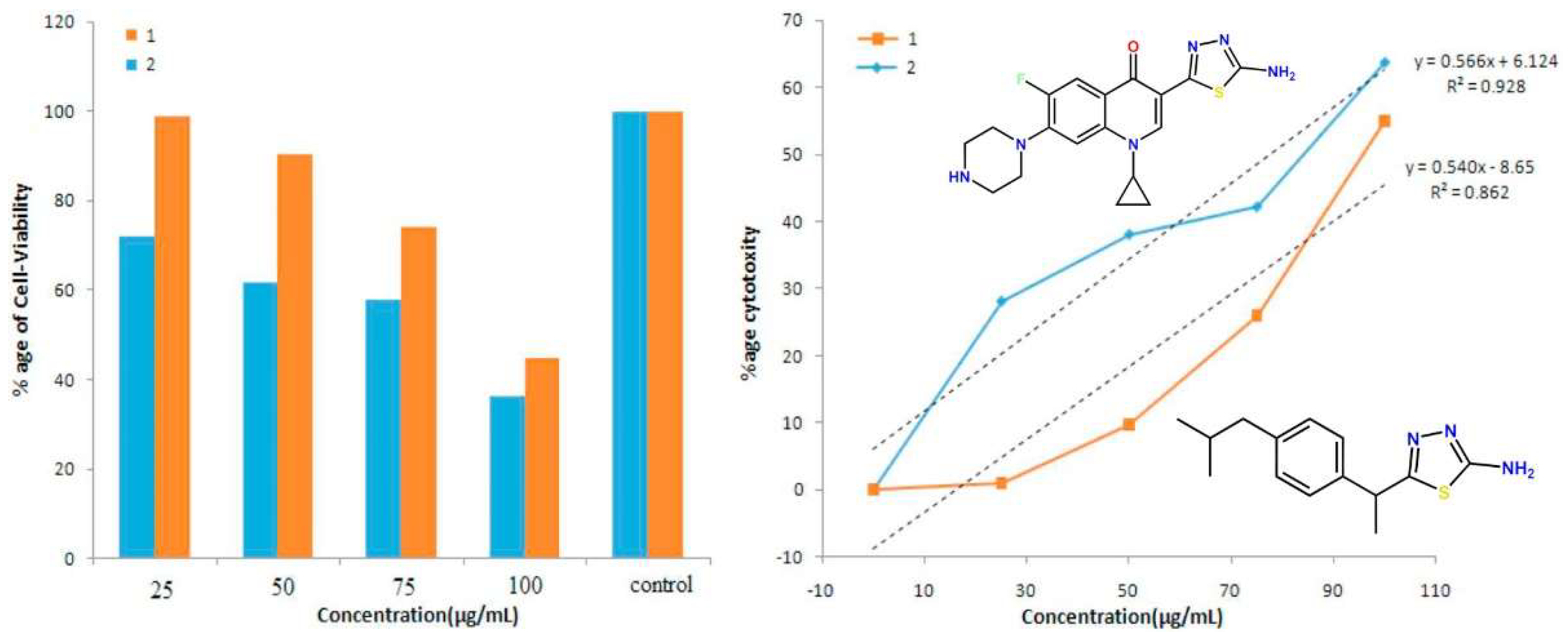
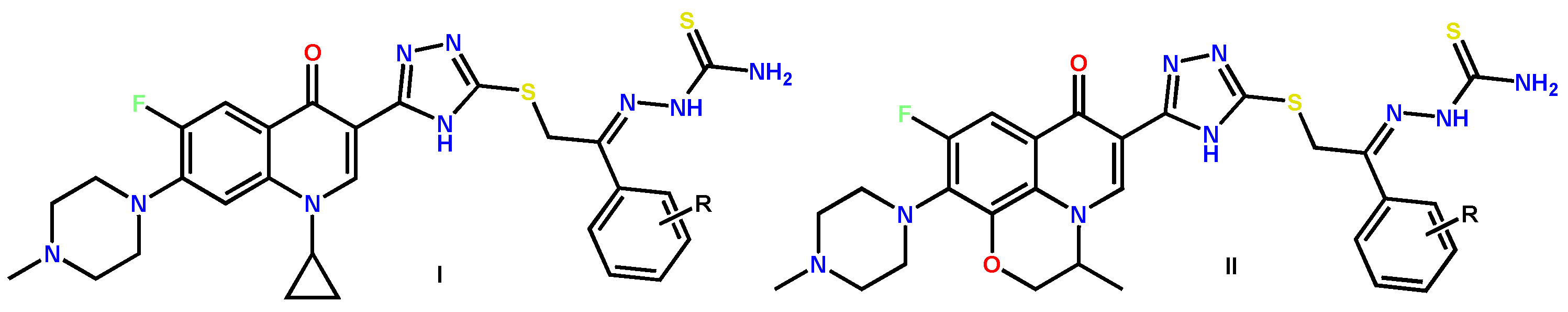
2.2. Novel FQ Hybrids as Promising Antitumor Agents
2.3. Other Types of Biological Activity

| Chemotype | Scheme | FQ * | Activities Studied ** |
|---|---|---|---|
 | Scheme 2 and Scheme 4 | (-)OF [25] amino | AM [25], AO [25] |
| CF [29,64,66,70] | AM [29], AF [29], AT/MTT [66] | ||
| NF [26,28,29,64,66] | AM [26,29], AF [26,29], AT/MTT [26,28,66] | ||
| EF [35] | AT/MTT [35] | ||
| OF [37,69] | AT/MTT [37] | ||
| FQ analogues [29] | AM [31], AF [29] | ||
| Scheme 15 | FQ analogues [57] | AV [57] | |
 | Scheme 4, Scheme 5, Scheme 6 and Scheme 7 | PF [31,36] | AM [31], ATB [31], AT [36] |
| NF citrate [30] | AC (HeLa) [30], AM [30], AF [26,29,30] | ||
| CF [32,37,49] | AM [32,37], ATB [32] | ||
| OF [27,46] | AT/MTT [27], AM [46] | ||
| EF [34,50] | AT/MTT [34] | ||
| (-)OF [38,67] | AT/MTT [38,67] | ||
| NF [39] | AT/MTT [39] | ||
 | Scheme 3, Scheme 4, Scheme 5 and Scheme 6 and Scheme 8, Scheme 9, Scheme 10 and Scheme 11 | (-)OF [25,40,48,67,74,88] | AM [25], AO [25], AT/MTT [67,74] |
| NF [26,27,82] NF citrate [30] | AM [26,27,30,82], AF [26,30,82], AT/MTT [26,30], AI [82], ATB [27] | ||
| CF [27,49,65] CF,N-Me [72] | AM [27,65], AT [72], ATB [27] | ||
| OF [41,44,45,46,60,73,92] | AT/MTT [41,45,58,60], AM [46] | ||
| PF [42,43] | - | ||
| EF [50] | - | ||
| GHQ168 [59] | ATr [59] | ||
 | Scheme 4 and Scheme 5 | (-)OF [25,67,81] (-)OF, desmethyl [81] | AM [25], AO [25,81], AT/MTT [67], AChE inhibitors [81] |
| NF [26] NF, N-acetyl [80] | AM [26], AF [26], AT/MTT [26,80] | ||
| CF [77,79,81] | AT [77,79]; AO [81], AChE inhibitors [81] | ||
| GHQ168 [59] | ATr [59] | ||
 | Scheme 12 | NF [61,62,83,84,85,86,87] | AM [61,62,83,84,85,86], AT [83], AF [84,86] |
| CF [84,85,86] | AM [84,85,86], AF [84,85] | ||
 | Scheme 13, Scheme 14 and Scheme 15 | FQ analogues [56,57] | AV [57], AM [56], GSK-3b inhibitor [56] |
| NF [63] | AM [63] | ||
| CF [63] | AM [63] | ||
| (-)OF [90] | - | ||
 | Scheme 15 | FQ analogues [55,57] | AV [55,57] |
 | Scheme 4 | (-)OF [25] | AM [25], AO [25] |
| Scheme 15 | FQ analogues [57] | AV [57] | |
 | Scheme 16 | (-)OF [48,88] | AT [48,88] |
| OF [92] | AT [92] | ||
 | Scheme 17 and Scheme 18 | CF [49,91] | AT/MTT [49,91] |
| OF [91] | AT/MTT [91] | ||
| EF [50,91] | AT/MTT [50,91] | ||
 | Scheme 17 | CF [49,91] | AT/MTT [49,91] |
| OF [91] | AT/MTT [91] | ||
| EF [91] | AT/MTT [91] | ||
 | Scheme 19 | CF [47] | AM [70], AT/MTT [47] |
 | Scheme 20 and Scheme 21 | CF [94] | ABCB1 inhibitors [93], AD [94], AF [94] |
| OF [94] | ABCB1 inhibitors [93], AD [94], AF [94] | ||
| NF [95,96] | ABCB1 inhibitors [96], AT [95,96] | ||
 | Scheme 22 and Scheme 23 | FQ analogues [89] | - |
| NF [58] | AM [58], ATB [58], AF [58] | ||
| CF [58] | AM [58], ATB [58], AF [58] | ||
 | Scheme 24 | NF analogues [51] | - |
| FQ analogues [119,120] | AM [119,120] | ||
 | Scheme 25 | NF [52,97,105] | AT/MTT [52,97,105], AM [97] |
| EF [52,97,105] | AT/MTT [52,97,105], AM [97] | ||
| CF [52,97,105] CF, N-Me [107] CF, N-acetyl [108] | AT/MTT [52,97,105,107,108], AM [97] | ||
| PF [52,97,105,106] | AT/MTT [52,97,105,106], AM [97] | ||
| OF [52] | AT/MTT [52] | ||
| (-)OF [52,98] | AT/MTT [52,98] | ||
| RF [99] | AT/MTT [99] | ||
| MF, N-Me [100] | AT/MTT [100] | ||
| GF, N-Me [101] | AT/MTT [101] | ||
| LF [102,103] | AT/MTT [102,103] | ||
| FL [104] | AT [104] | ||
 | FL [109] | AT [109] | |
| RF [110] | AT [110] | ||
| CF, N-acetyl [111] | AT/MTT [111] | ||
| LF, N-Me [112] | AT/MTT [112] | ||
| MF, N-Me [113] | AT/MTT [113] | ||
| PF [114] | AT/MTT [114] | ||
| OF [115] | AT/MTT [115] | ||
| (-)OF [116] | AT/MTT [116] | ||
| GF, N-Me [117] | AT/MTT [117] | ||
 | Scheme 26 and Scheme 27 | (-)OF [38,67] | AT/MTT [38,67] |
| NF [39] | AT/MTT [39] | ||
| OF [69,75] | AT/MTT [69,75] | ||
| CF [70] | AT/MTT [70] | ||
 | Scheme 28 | (-)OF [67] | AT/MTT [67] |
| PF [42,43] | AT/MTT (SMMC-7721, L1210 and HL60) [42,43] | ||
| OF [44,75] | AT/MTT [44,75] | ||
 | (-)OF [67] | AT/MTT [67] | |
| OF [75] | AT/MTT [67] | ||
 | Scheme 29 | NF [82] | AI [82], AF [82], AM [82] |
 | Scheme 30 | CF [53,54,118] CF, N-Me [53] | AT/MTT [53,54,118] |
| EF [53,54,118] | AT/MTT [53,54,118] | ||
| NF [53,54,118] | AT/MTT [53,54,118] | ||
| OF [53,118] | AT/MTT [53,118] | ||
| (-)OF [53,118] | AT/MTT [53,118] | ||
| PF [54,118] | AT/MTT [54,118] |
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mohammed, H.H.; Abuo-Rahma, G.E.-D.A.; Abbas, S.; Abdelhafez, E.-S.M. Current Trends and Future Directions of Fluoroquinolones. Curr. Med. Chem. 2019, 26, 3132–3149. [Google Scholar] [CrossRef] [PubMed]
- Ezelarab, H.; Abbas, S.; Hassan, H.A.; Abuo-Rahma, G.E.-D.A. Recent updates of fluoroquinolones as antibacterial agents. Arch. Pharm. 2018, 351, e1800141. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-Q.; Zhang, S.; Xu, Z.; Lv, Z.-S.; Liu, M.-L.; Feng, L.-S. 4-Quinolone hybrids and their antibacterial activities. Eur. J. Med. Chem. 2017, 141, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Leyva, S.; Hernández, H. Fluoroquinolonas Perspectivas no antibacterianas. Rev. Esp. Quim. 2017, 30, 1–8. [Google Scholar]
- Jiang, D. 4-Quinolone Derivatives and Their Activities Against Gram-negative Pathogens. J. Heterocycl. Chem. 2018, 55, 2003–2018. [Google Scholar] [CrossRef]
- Zhang, G.-F.; Zhang, S.; Pan, B.; Liu, X.; Feng, L.-S. 4-Quinolone derivatives and their activities against Gram positive pathogens. Eur. J. Med. Chem. 2018, 143, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.-L.; Wu, J.-B.; Cheng, X.-W.; Zhang, F.-Z.; Feng, L.-S. Fluoroquinolone derivatives and their anti-tubercular activities. Eur. J. Med. Chem. 2018, 146, 554–563. [Google Scholar] [CrossRef]
- Pranger, A.D.; van der Werf, T.S.; Kosterink, J.G.W.; Alffenaar, J.W.C. The Role of Fluoroquinolones in the Treatment of Tuberculosis in 2019. Drugs 2019, 79, 161–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P. A review on quinoline derivatives as anti-methicillin resistant Staphylococcus aureus (MRSA) agents. BMC Chem. 2020, 14, 17. [Google Scholar] [CrossRef] [Green Version]
- Jassem, A.M.; Dhumad, A.M.; Almashal, F.A.; Alshawi, J. Microwave-assisted synthesis, molecular docking and anti-HIV activities of some drug-like quinolone derivatives. Med. Chem. Res. 2020, 29, 1067–1076. [Google Scholar] [CrossRef]
- Shahbazi, A.; Mostafavi, H.; Zarrini, G.; Mahdavi, M. Novel N-4-Piperazinyl Ciprofloxacin-Ester Hybrids: Synthesis, Biological Evaluation, and Molecular Docking Studies. Russ. J. Gen. Chem. 2020, 90, 1558–1565. [Google Scholar] [CrossRef]
- Abdel-Aal, M.A.A.; Abdel-Aziz, S.A.; Shaykoon, M.S.A.; Abuo-Rahma, G.E.-D.A. Towards anticancer fluoroquinolones: A review article. Arch. Pharm. 2019, 352, e1800376. [Google Scholar] [CrossRef] [PubMed]
- Idowu, T.; Schweizer, F. Ubiquitous Nature of Fluoroquinolones: The Oscillation between Antibacterial and Anticancer Activities. Antibiotics 2017, 6, 26. [Google Scholar] [CrossRef] [Green Version]
- Ajjah, N.T.; Naser, N.H.; Alard, A.A.A.; Diwan, M.F. Design, Synthesis, Docking Study and Preliminary Pharmacological Assessment of New Norfloxacin Analogues Having Thiazole Nucleus. J. Biochem. Technol. 2020, 11, 58–67. [Google Scholar]
- Majalekar, P.P.; Shirote, P.K.; Nalawade, V.; Shelake, P. Design, synthesis and comparative pharmacological assessment of novel fluoroquinolone derivatives. Int. J. Pharm. Sci. Res. 2019, 25, 3735–3740. [Google Scholar]
- Aziz, H.A.; Moustafa, G.A.I.; Abuo-Rahma, G.E.A.; Rabea, S.M.; Hauk, G.; Krishna, V.S.; Sriram, D.; Berger, J.M.; Abbas, S.H. Synthesis and antimicrobial evaluation of new nitric oxide-donating fluoroquinolone/oxime hybrids. Arch. Pharm. 2020, 354, 2000180. [Google Scholar] [CrossRef]
- Bush, N.; Diez-Santos, I.; Abbott, L.; Maxwell, A. Quinolones: Mechanism, Lethality and Their Contributions to Antibiotic Resistance. Molecules 2020, 25, 5662. [Google Scholar] [CrossRef]
- Lucia, P. ChemInform Abstract: Quinolones: Synthesis and Antibacterial Activity. Cheminform 2013, 44. [Google Scholar] [CrossRef]
- Suaifan, G.A.R.Y.; Mohammed, A.A.M. Fluoroquinolones structural and medicinal developments (2013–2018): Where are we now? Bioorganic Med. Chem. 2019, 27, 3005–3060. [Google Scholar] [CrossRef]
- Zahoor, A.F.; Yousaf, M.; Siddique, R.; Ahmad, S.; Naqvi, S.A.R.; Rizvi, S.M.A. Synthetic strategies toward the synthesis of enoxacin-, levofloxacin-, and gatifloxacin-based compounds: A review. Synth. Commun. 2017, 47, 1021–1039. [Google Scholar] [CrossRef]
- Zhang, G.-F.; Liu, X.; Zhang, S.; Pan, B.; Liu, M.-L. Ciprofloxacin derivatives and their antibacterial activities. Eur. J. Med. Chem. 2018, 146, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Oniga, S.; Palage, M.; Araniciu, C.; Marc, G.; Oniga, O.; Vlase, L.; Prisăcari, V.; Valica, V.; Curlat, S.; Uncu, L. Design, synthesis, molecular docking, and antibacterial activity evaluation of some novel norfloxacin analogues. Farmacia 2018, 66, 1048–1058. [Google Scholar] [CrossRef] [Green Version]
- Hryhoriv, H.; Mariutsa, I.; Kovalenko, S.M.; Georgiyants, V.; Perekhoda, L.; Filimonova, N.; Geyderikh, O.; Sidorenko, L. The Search for New Antibacterial Agents among 1,2,3-Triazole Functionalized Ciprofloxacin and Norfloxacin Hybrids: Synthesis, Docking Studies, and Biological Activity Evaluation. Sci. Pharm. 2021, 90, 2. [Google Scholar] [CrossRef]
- Hryhoriv, H.; Mariutsa, I.; Kovalenko, S.M.; Sidorenko, L.; Perekhoda, L.; Filimonova, N.; Geyderikh, O.; Georgiyants, V. Structural modification of ciprofloxacin and norfloxacin for searching new antibiotics to combat drug-resistant bacteria. Sci. Pharm. Sci. 2021, 5, 4–11. [Google Scholar] [CrossRef]
- Lihumis, H.S.; Al Talebi, Z.A.; Khaleel, A.K. Synthesis and Identification of Some New Heterocyclic Compounds for Levofloxacin Drug Derivatives with Evaluating of Their Biological Efficiency and Antioxidant Activity. J. Med. Chem. Sci. 2022, 5, 596–606. [Google Scholar] [CrossRef]
- Arshad, M.; Khan, M.S.; Nami, S.A.A. Norfloxacin Analogues: Drug Likeness, Synthesis, Biological, and Molecular Docking Assessment. Russ. J. Bioorganic Chem. 2021, 47, 483–495. [Google Scholar] [CrossRef]
- Li, T.; Gao, L.-z.; Xie, Y.-s.; Hu, G.-q.; Huang, W.-l. Synthesis and antitumor activity fluoroquinolon-3-yloxadiazole sulfanylacetylhydrazone derivatives. Zhongguo Yaoxue Zazhi 2014, 49, 2206–2209. [Google Scholar]
- Chen, Y.; Wang, G.; Duan, N.; Cao, T.; Wen, X.; Yin, J.; Wang, W.; Xie, S.; Huang, W.; Hu, G. Synthesis and Antitumor Activity of Fluoroquinolone C3-Isostere Derivatives: Oxadiazole Mannich Base Derivatives. Acta Agron. Sin. 2012, 29, 1246. [Google Scholar] [CrossRef]
- Khan, N.M.; Kumar, P.; Hemanth, S.K.K.; Bharath, R.K.P. Synthesis, molecular bioinformatics modelling, and antimicrobial evaluation of some novel oxadiazole fluoroquinolone derivatives. Asian J. Pharm. Clin. Res. 2022, 15, 40–46. [Google Scholar] [CrossRef]
- Allaka, T.R.; Katari, N.K.; Jonnalagadda, S.B.; Malkhed, V.; Anireddy, J.S. Design, Synthesis and Biological Evaluation of Novel Heterocyclic Fluoroquinolone Citrate Conjugates as Potential Inhibitors of Topoisomerase IV: A Computational Molecular Modeling Study. Curr. Cancer Drug Targets 2021, 18, 11–30. [Google Scholar] [CrossRef]
- Allaka, T.R.; Kummari, B.; Polkam, N.; Kuntala, N.; Chepuri, K.; Anireddy, J.S. Novel heterocyclic 1,3,4-oxadiazole derivatives of fluoroquinolones as a potent antibacterial agent: Synthesis and computational molecular modeling. Mol. Divers. 2021, 26, 1581–1596. [Google Scholar] [CrossRef]
- Niveditha, N.; Begum, M.; Prathibha, D.; Sirisha, K.; Mahender, P.; Chitra, C.; Rao, V.R.; Reddy, V.M.; Achaiah, G. Design, Synthesis and Pharmacological Evaluation of Some C3 Heterocyclic-Substituted Ciprofloxacin Derivatives as Chimeric Antitubercular Agents. Chem. Pharm. Bull. 2020, 68, 1170–1177. [Google Scholar] [CrossRef]
- Singhai, A.; Gupta, M. Synthesis, characterization and biological evaluation of substituted 1,3,4-oxadiazole derivative: Derived from ciprofloxacin. Asian J. Pharm. Clin. Res. 2019, 12, 205–209. [Google Scholar] [CrossRef]
- Wu, S.-m.; Hou, P.-p.; Wen, J.; Hu, G.-q. Synthesis and antitumor activity of oxadiazolone thione Mannich-base derivatives of enrofloxacin. Shenyang Yaoke Daxue Xuebao 2015, 32, 840–843, 858. [Google Scholar]
- Shang, H.-j.; Yan, Q.; Wu, S.-m.; Ni, L.-l.; Huang, W.-l.; Hu, G.-q. Synthesis and antitumor activity of novel oxadiazole derivatives of enrofloxacin. Shenyang Yaoke Daxue Xuebao 2015, 32, 604–608. [Google Scholar]
- Gao, L.-Z.; Xie, Y.-S.; Li, T.; Huang, W.-L.; Hu, G.-Q. [Synthesis, antitumor activity and SAR of C-3 oxadiazole sulfanylacetylhydrazone-substituted fluoroquinolone analogues]. Yao Xue Xue Bao 2014, 49, 1694–1698. [Google Scholar]
- Hou, L.-l.; Yin, J.; Wang, W.; Xie, S.-q.; Huang, W.-l.; Hu, G.-q. Synthesis and antitumor activity of fluoroquinolone C-3 heterocycles-oxadiazole derivatives (II). Zhongguo Yaoxue Zazhi 2013, 48, 1194–1196. [Google Scholar]
- Hu, G.-q.; Wang, G.-q.; Duan, N.-n.; Wen, X.-y.; Cao, T.-y.; Xie, S.-q.; Huang, W.-l. Design, Synthesis and Antitumor Activity of Fluoroquinolone C3 Heterocyclic Bis-oxadiazole Methylsulfide Derivatives Derived from Levofloxacin. Chem. Res. Chin. Univ. 2012, 28, 980–984. [Google Scholar]
- Wang, G.; Duan, N.; Cao, T.; Wen, X.; Yin, J.; Wang, W.; Xie, S.; Huang, W.; Hu, G. Antitumor fluoroquinolone C3-isostere derivatives(I)—Synthesis and activity of bis-oxadiazole methyl-sulfide derivatives. Yingyong Huaxue 2012, 29, 769–774. [Google Scholar] [CrossRef]
- Feng, H.; Feng, C.-J. CoMFA Model of Anti-tumor Activity for Fluoroquinolon-3-yl s-Triazole Sulfide-ketone Derivatives and Implications for Molecular Design. Chin. J. Struct. Chem. 2021, 40, 703–710. [Google Scholar]
- Xie, Y.; Gao, L.; Yan, Q.; Wu, S.; Ni, L.; Liu, Y.; Huang, W.; Hu, G. Synthesis and antitumor activity of fluoroquinolon-3-yl s-triazole sulfide ketone thiosemicarbazone derivatives of ofloxacin. Yingyong Huaxue 2016, 33, 25–31. [Google Scholar]
- Sun, Y.; Xu, Q.; Hou, L.; Yue, X.; Wu, Z.; Huang, W.; Xie, S.; Hu, G. Synthesis and antitumor activities of fluoroquinolone C-3 isosteres(IV):s-triazole-oxadiazole methylsulfide Mannich-base derivatives. Zhongguo Yaoke Daxue Xuebao 2014, 45, 39–42. [Google Scholar]
- Xu, Q.; Hou, L.; Wu, Z.; Yue, X.; Hu, G.; Huang, W. Synthesis and antitumor activity of fluoroquinolone C-3 isostere III: S-triazole oxadiazole methylsulfide derivatives from pefloxacin. Zhongguo Yaoke Daxue Xuebao 2013, 44, 511–514. [Google Scholar]
- Xu, Q.-j.; Hou, L.-l.; Wu, Z.-f.; Yue, X.-b.; Xie, S.-q.; Huang, W.-l.; Hu, G.-q. Synthesis and antitumor evaluation of fluoroquinolone C3 s-triazole oxadiazole methylsulfide derivatives of ofloxacin. Zhongguo Yaoxue Zazhi 2014, 49, 609–612. [Google Scholar]
- Hu, G.; Wang, G.; Duan, N.; Wen, X.; Cao, T.; Xie, S.; Huang, W. Design, synthesis and antitumor activities of fluoroquinolone C-3 heterocycles (IV): S-triazole Schiff–Mannich bases derived from ofloxacin. Acta Pharm. Sin. B 2012, 2, 312–317. [Google Scholar] [CrossRef] [Green Version]
- Jubie, S.; Prabitha, P.; Kumar, R.R.; Kalirajan, R.; Gayathri, R.; Sankar, S.; Elango, K. Design, synthesis, and docking studies of novel ofloxacin analogues as antimicrobial agents. Med. Chem. Res. 2011, 21, 1403–1410. [Google Scholar] [CrossRef]
- Hu, G.-Q.; Wu, X.-K.; Xie, S.-Q.; Du, G.-J.; Huang, W.-L.; Zhang, H.-B. Synthesis and bioactivity of water-soluble fused s-triazolothiadiazole systems (II): Fluoroquinolone piperazine derivatives. Huaxue Xuebao 2008, 66, 2157–2162. [Google Scholar]
- Li, Y.; Zhang, C.; Huang, W.; Chen, C.; Hu, G. Synthesis and Antitumor Activities of C-3 Thiazolotriazole Unsaturated Ketone Derivatives of Levofloxacoin. Chin. J. Appl. Chem. 2019, 36, 671–676. [Google Scholar]
- Xie, S.; Chen, Y.; Wang, G.; Duan, N.; Wen, X.; Cao, T.; Yin, J.; Wang, W.; Hu, G.; Huang, W. Synthesis and antitumor evaluation of s-triazolothiadiazines and pyrazolo s-triazoles derived from ciproxacin. Yaoxue Xuebao 2012, 47, 66–71. [Google Scholar]
- Hu, G.; Zhang, Z.; Wang, H.; Wu, X.; Wang, X.; Du, G.; Xie, S.; Huang, W.; Zhang, H. Synthesis and antitumor activity of fluoroquinolon-3-yl fused heterocyclic systems (III): S-triazolothiadiazinone derivatives derived enrofloxacin. Huaxue Xuebao 2009, 67, 2592–2596. [Google Scholar]
- Nosova, E.V.; Sidorova, L.P.; Lipunova, G.N.; Mochul’skaya, N.N.; Chasovskikh, O.M.; Charushin, V.N. Synthesis of new fluorinated derivatives of quinolinecarboxylic acids. Chem. Heterocycl. Compd. 2002, 38, 922–928. [Google Scholar] [CrossRef]
- Hu, G.; Yang, Y.; Yi, L.; Wang, X.; Zhang, Z.; Xie, S.; Huang, W. Design, synthesis and antitumor action of C3/C3 bis-fluoroquinolones linked-cross 2,5-[1,3,4]oxadiazole. Yaoxue Xuebao 2010, 45, 1012–1016. [Google Scholar]
- Hu, G.-Q.; Yang, Y.; Yi, L.; Wang, G.-Q.; Duan, N.-N.; Wen, X.-Y.; Cao, T.-Y.; Xie, S.-Q.; Huang, W.-L. Design, synthesis and antitumor activity of C3/C3 bis-fluoroquinolones cross-linked with [1,2,4]triazolo[3,4-b][1,3,4]thiadiazole. Acta Pharm. Sin. B 2011, 1, 172–177. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.Q.; Zhang, Z.Q.; Xie, S.Q.; Huang, W. Long Synthesis and antitumor evaluation of C3/C3 fluoroquinolone dimers (I), tethered with a fused heterocyclic s-triazolo[2,1-b][1,3,4]thiadiazole. Chin. Chem. Lett. 2010, 21, 661–663. [Google Scholar] [CrossRef]
- Kumar, D.V.; Rai, R.; Young, W.B.; Hu, H.; Riggs, J.R.; Ton, T.L.; Green, M.J.; Hart, B.P.; Brameld, K.A.; Dener, J.M. Preparation of Quinolinones and Analogs as Antiviral Agents. WO2007130499A2, 15 November 2007. [Google Scholar]
- Li, B.; Cociorva, O.M.; Nomanbhoy, T.; Li, Q.; Nakamura, K.; Nomura, M.; Okada, K.; Yumoto, K.; Liyanage, M.; Zhang, M.C.; et al. 6-Position optimization of tricyclic 4-quinolone-based inhibitors of glycogen synthase kinase-3β: Discovery of nitrile derivatives with picomolar potency. Bioorganic Med. Chem. Lett. 2012, 22, 1005–1008. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.V.; Rai, R.; Brameld, K.A.; Riggs, J.; Somoza, J.R.; Rajagopalan, R.; Janc, J.W.; Xia, Y.M.; Ton, T.L.; Hu, H.; et al. 3-Heterocyclyl quinolone inhibitors of the HCV NS5B polymerase. Bioorganic Med. Chem. Lett. 2012, 22, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, A.R.; Patel, N.B.; Rajani, D. Antimycobacterial and antimicrobial studies of newly synthesized 3-(4-(6-methylbenzo[d]thiazol-2-yl) phenyl)quinazolin-4(3H)-ones. Indian J. Res. Pharm. Biotechnol. 2014, 2, 935–942. [Google Scholar]
- Pyrih, A.; Berninger, M.; Gzella, A.; Lesyk, R.; Holzgrabe, U. Synthesis and evaluation of antitrypanosomal activity of some thiosemicarbazide derivatives of 1-butyl-6-fluoro-7-morpholino-4-oxo-1,4-dihydroquinoline-3-carboxylic acid. Synth. Commun. 2018, 48, 1883–1891. [Google Scholar] [CrossRef]
- Gao, L.-z.; Li, T.; Xie, Y.-s.; Hu, G.-q.; Huang, W.-l. Synthesis and antitumor activity of fluoroquinolon-3-yl s-triazole sulfanylacetylhydrazones and s-triazole hydrazone derivatives (V). Zhongguo Yaoxue Zazhi 2015, 50, 545–549. [Google Scholar]
- Wang, L.-L.; Battini, N.; Bheemanaboina, R.R.Y.; Zhang, S.-L.; Zhou, C.-H. Design and synthesis of aminothiazolyl norfloxacin analogues as potential antimicrobial agents and their biological evaluation. Eur. J. Med. Chem. 2019, 167, 105–123. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.-F.; Addla, D.; Zhou, C.-H. Novel 3-aminothiazolylquinolones: Design, synthesis, bioactive evaluation, SARs, and preliminary antibacterial mechanism. J. Med. Chem. 2016, 59, 4488–4510. [Google Scholar] [CrossRef] [PubMed]
- Azad, C.S.; Narula, A.K. An operational transformation of 3-carboxy-4-quinolones into 3-nitro-4-quinolones via ipso-nitration using polysaccharide supported copper nanoparticles: Synthesis of 3-tetrazolyl bioisosteres of 3-carboxy-4-quinolones as antibacterial agents. RSC Adv. 2016, 6, 19052–19059. [Google Scholar] [CrossRef]
- Patel, N.B.; Shaikh, A.R.; Soni, H.; Parmar, R.B.; Patel, J.A. In vitro antimicrobial, antimycobacterial evaluation and synthesis of substituted 1,2,4-triazole motifs. Chem. Biol. Interface 2018, 8, 184–193. [Google Scholar]
- Jubie, S.; Kalirajan, R.; Yadav, P. Design, Synthesis and Docking Studies of a Novel Ciprofloxacin Analogue as an Antimicrobial AGENT. E-J. Chem. 2012, 9, 980–987. [Google Scholar] [CrossRef]
- Shaharyar, M.; Ali, M.A.; Abdullah, M.-M. Synthesis and antiproliferative activity of 1-[(sub)]-6-fluoro-3-[(sub)]-1,3,4-oxadiazol-2-yl-7-piperazino-1,4-dihydro-4-quinolinone derivatives. Med. Chem. Res. 2007, 16, 292–299. [Google Scholar] [CrossRef]
- Hu, G.; Ye, Q.; Yu, Z.; Chen, Y.; Liu, H.; Li, J.; Kang, J.; Huang, F. Levorotatory Fluoroquinolone-C3 Diazole Derivatives as Antitumor Agents and Their Preparation, Pharmaceutical Compositions and Use in the Treatment of Cancer. China. CN102391287A, 28 March 2012. [Google Scholar]
- Abdullah, M.; Guruprasad, L. Computational basis for the design of PLK-2 inhibitors. Struct. Chem. 2020, 31, 275–292. [Google Scholar] [CrossRef]
- Hu, G.; Chen, Y.; Wang, G.; Duan, N.; Wen, X.; Cao, T.; Yin, J.; Wang, W.; Xie, S.; Huang, W. Synthesis of fluoroquinolone C-3 heterocycles, bis-oxadiazole methylsulfides and methiodides and their antitumor activity. Zhongguo Yaoxue Zazhi 2012, 47, 72–76. [Google Scholar]
- Hu, G.-q.; Hou, L.-l.; Wang, G.-q.; Duan, N.-n.; Wen, X.-y.; Cao, T.-y.; Yin, J.; Wang, W.; Xie, S.-q.; Huang, W.-l. Design, synthesis and antitumor activity of fluoroquinolone C-3 heterocycles: Bis-oxadiazole methylsulfide derivatives derived from ciprofloxacin. Yaoxue Xuebao 2012, 47, 1017–1022. [Google Scholar]
- Al-Mathkuri, T.S.F.; Al-Jubori, H.M.S.; Saleh, A.T. Synthetic and Study the Chelating Activity of Some Polymers Containing Heterocyclic Rings Which Derivative from 1,2,4- Trizol Levofloxacin Acid. Orient. J. Chem. 2018, 34, 2031. [Google Scholar] [CrossRef]
- Jing, Y.; Wu, S.; Ni, L.; Yan, Q.; Gao, L.; Xie, Y.; Hu, G. Preparation of Cyclopropyl Fluoroquinolone C-3 S-Triazole Thioether Ketone Semicarbazone Compounds as Antitumor Agents. CN Patent CN104628702A, 20 May 2015. [Google Scholar]
- Jing, Y.; Zhao, H.; Wu, S.; Ni, L.; Yan, Q.; Hu, G. Preparation of OFLOXACIN Thiosemicarbazone Derivatives as Antitumor Agents. CN Patent CN104592252A, 6 May 2015. [Google Scholar]
- Hu, G.; Zhao, H.; Xie, Z.; Wu, S.; Ni, L.; Yan, Q. Chiral Fluoroquinolone C-3 1,2,4-triazole Thioether Thiosemicarbazone Useful in Treatment of Cancer and Its Preparation. CN Patent CN104557974A, 29 April 2015. [Google Scholar]
- Hu, G.; Pang, X.; Liu, H.; Xie, X.; Wang, G.; Duan, N.; Wen, X.; Cao, T.; Yin, J.; Wang, W. Preparation of Ofloxacin Derivatives as Antitumor Agents. CN Patent CN102443011A, 9 May 2012. [Google Scholar]
- Chao, W.; Feng, C.-J. QSAR Studies on the inhibitory activity of levofloxacin-thiadiazole HDACi Conjugates to histone deacetylases. Chin. J. Struct. Chem. 2018, 37, 1679–1688. [Google Scholar]
- Hu, G.; Wu, X.; Wang, X.; Zhang, Z.; Xie, S.; Huang, W.; Zhang, H. Synthesis and antitumor activity of C3 heterocyclic-substituted fluoroquinolone derivatives (I): Ciprofloxacin aminothiodiazole Schiff-bases. Yaoxue Xuebao 2008, 43, 1112–1115. [Google Scholar]
- Wan, Z.; Sun, R. Jing QSAR and molecular docking study on the biological activity of levofloxacin and thiodiazole histone deacetylase inhibitors. Chem. Rev. Lett. 2020, 3, 12–18. [Google Scholar]
- Farooqi, S.I.; Arshad, N.; Channar, P.A.; Perveen, F.; Saeed, A.; Larik, F.A.; Javeed, A. Synthesis, theoretical, spectroscopic and electrochemical DNA binding investigations of 1, 3, 4-thiadiazole derivatives of ibuprofen and ciprofloxacin: Cancer cell line studies. J. Photochem. Photobiol. B Biol. 2018, 189, 104–118. [Google Scholar] [CrossRef]
- Hu, G.; Zhang, C.; Sun, J.; Wang, N.; Shen, R. Preparation of Urea Compounds Containing Fluoroquinolone and N-acetylnorfloxacin Thiadiazole Useful as Anticancer Agents. CN Patent CN109678883A, 26 April 2019. [Google Scholar]
- Ujan, R.; Saeed, A.; Channar, P.A.; Larik, F.A.; Abbas, Q.; Alajmi, M.F.; El-Seedi, H.R.; Rind, M.A.; Hassan, M.; Raza, H.; et al. Drug-1,3,4-Thiadiazole Conjugates as Novel Mixed-Type Inhibitors of Acetylcholinesterase: Synthesis, Molecular Docking, Pharmacokinetics, and ADMET Evaluation. Molecules 2019, 24, 860. [Google Scholar] [CrossRef] [Green Version]
- Chandramouli; Shivanand, M.R.; Nayanbhai, T.B.; Bheemachari; Udupi, R.H. Synthesis and biological screening of certain new triazole Schiff bases and their derivatives bearing substituted benzothiazole moiety. J. Chem. Pharm. Res. 2012, 4, 1151–1159. [Google Scholar]
- Wang, L.-L.; Battini, N.; Bheemanaboina, R.R.Y.; Ansari, M.F.; Chen, J.-P.; Xie, Y.-P.; Cai, G.-X.; Zhang, S.-L.; Zhou, C.-H. A new exploration towards (aminothiazolyl)quinolone oximes as potentially multi-targeting antibacterial agents: Design, synthesis and evaluation acting on microbes, DNA, HSA and topoisomerase IV. Eur. J. Med. Chem. 2019, 179, 166–181. [Google Scholar] [CrossRef]
- Zhou, C.; Wang, L.; Battini, N.; Chen, J.; Xie, Y. Aminothiazole Quinolone Oxime Compound, Its Preparation Method and Application in Preparing Antibacterial Drug, Antifungal Drug and DNA Intercalator. CN Patent CN109651353A, 19 April 2019. [Google Scholar]
- Zhou, C.; Cui, S.; Dinesh, A. Quinolone Thiazole Compound and the Preparation Method and Application Thereof. CN Patent CN104530034A, 22 April 2015. [Google Scholar]
- Zhou, C.; Wang, L.; Battini, N. Preparation of the 3-(2-Aminothiazol-4-yl)-7-(4-substituent-piperazin-1-yl)quinolin-4(1H)-one Compound and Their Application as the Antifungal Drugs. CN Patent CN108440518A, 24 August 2018. [Google Scholar]
- Spyrydonova, N.; Silin, A.; Lebedynets, V.; Zavada, O.; Kalinenko, O. Development of specification for substance 7-azepan-1-il-1-ethil-6-ftor-3-(4-phenyl-1, 3-tiazol-2-il)-Quinoline-4(1h)-one. Pharma Innov. 2019, 8, 54–56. [Google Scholar]
- Hu, G.; Li, T.; Gao, L.; Xie, Y.; Feng, Y.; Yan, Q.; Wu, S.; Ni, L. Preparation of Fluoroquinolone Derivatives as Antitumor Agents. CN Patent CN104497014A, 8 April 2015. [Google Scholar]
- Fokin, A.; Burgart, Y.; Saloutin, V.; Chupakhin, O. Synthesis of 2-(1-alkyl(aryl)-4-oxo-5,6,7,8-tetrafluoro-1,4-dihydroquinolin-3-yl)glyoxylic acid derivatives. J. Fluor. Chem. 2001, 108, 187–194. [Google Scholar] [CrossRef]
- Duncton, M.; Singh, R. Preparation of Tetrazolones as Carboxylic acid Bioisosteres for the Treatment of Diseases. US Patent US20160213648A1, 28 July 2016. [Google Scholar]
- Hu, G.Q.; Hou, L.L.; Yang, Y.; Yi, L.; Xie, S.Q.; Wang, G.Q.; Duan, N.N.; Chao, T.Y.; Wen, X.Y.; Huang, W. Long Synthesis and antitumor evaluation of fluoroquinolone C3 fused heterocycles (II): From triazolothiadiazines to pyrazolotriazoles. Chin. Chem. Lett. 2011, 22, 804–806. [Google Scholar] [CrossRef]
- Wu, S.-m.; Yan, Q.; Ni, L.-l.; Xie, Y.-s.; Gao, L.-z.; Liu, Y.-j.; Huang, W.-l.; Hu, G.-q. Synthesis and antitumor activity of fluoroquinolone C-3 fused heterocyclic thiazolo[3,2-b][1,2,4]triazole derivatives (VI). Zhongguo Yaoxue Zazhi 2016, 51, 353–357. [Google Scholar]
- Zhang, S.-l.; Wei, Y.-x.; Li, Q.; Sun, H.-p.; Peng, H.; You, Q.-d. Pharmacophore-based drug design and biological evaluation of novel ABCB1 inhibitors. Chem. Biol. Drug Des. 2013, 81, 349–358. [Google Scholar] [CrossRef]
- Muluk, R.; Kothawade, P.; Kulkarni, G.; Ingale, P. Synthesis and evaluation of some novel benzimidazole-quinolinone derivatives for their antifungal and antidiabetic activity. World J. Pharm. Pharm. Sci. 2018, 7, 1263–1278. [Google Scholar]
- You, Q.-D.; Li, Z.-Y.; Huang, C.-H.; Yang, Q.; Wang, X.-J.; Guo, Q.-L.; Chen, X.-G.; He, X.-G.; Li, T.-K.; Chern, J.-W. Discovery of a Novel Series of Quinolone and Naphthyridine Derivatives as Potential Topoisomerase I Inhibitors by Scaffold Modification. J. Med. Chem. 2009, 52, 5649–5661. [Google Scholar] [PubMed]
- You, Q.; He, X.; Li, Z.; Chen, X.; Li, Y.; Li, H.; Zhang, W. Preparation of Quinolone Derivatives as Antitumor Agents. CN Patent CN1473827A, 11 February 2004. [Google Scholar]
- Hu, G.; Xie, S.; Hou, L.; Sun, M.; Zhang, J.; Yang, Y.; Yi, L. Preparation of 1,3,4-Oxadiazole Linked Fluoroquinolone Dimers for Treating Neoplasm and Microbial Infection. CN Patent CN101643471A, 10 February 2010. [Google Scholar]
- Li, K.; Cai, Y.; Zang, F.; Hu, G. Levofloxacin-Containing Bis-Fluoroquinolone Oxadiazole Carbamide Derivative Useful in Treatment of Cancer and Its Preparation. CN Patent CN109369674A, 22 February 2019. [Google Scholar]
- Cen, S.; Yang, L.; Li, X.; Hu, G. Preparation Method of Rufloxacin-Containing Bis-Fluoroquinolone Oxadiazole Urea Derivative Applied to Antitumor Drug. CN Patent CN109438482A, 8 March 2019. [Google Scholar]
- Cen, S.; Geng, S.; Yang, L.; Hu, G. N-Methyl Moxifloxacin-Containing Bis-Fluoroquinolone Oxadiazole Urea Derivative Useful in Treatment of Cancer and Its Preparation. CN Patent CN109438481A, 8 March 2019. [Google Scholar]
- Hu, G.; Feng, Y.; Liu, J.; Zhao, Y. Preparation Method of N-methyl Gatifloxacin-Containing Bis-Fluoroquinolone Oxadiazole Urea Derivative Applied to Antitumor Drug. CN Patent CN, 109438472A, 8 March 2019. [Google Scholar]
- Zhang, H.; Liu, J.; Li, D.; Zhao, Y. Preparation of Lomefloxacin Derivatives for Treatment of Cancer. CN Patent CN109400631A, 1 March 2019. [Google Scholar]
- Lu, L.; Zhang, H.; Wang, H.; Hu, G. Preparation of Lomefloxacin Derivatives for Treatment of Cancer. CN Patent CN109400627A, 1 March 2019. [Google Scholar]
- Wang, S.; Mao, Y.; Lu, L.; Hu, G. Preparation of Fluoroquinolone Oxadiazole Urea Fleroxacin Derivatives Useful for the Treatment of Cancer. CN Patent CN109400626A, 1 March 2019. [Google Scholar]
- Jiang, Y.; Liu, Q.; Shao, X.; Hu, G. Preparation of Bis-Fluoroquinolone Oxadiazole Urea Derivative as Antitumor Drug. CN Patent CN109369676A, 22 February 2019. [Google Scholar]
- Liu, Q.; Shao, X.; Mao, Y.; Hu, G. Bifluoroquinolone Oxadiazole Urea Pefloxacin Derivative Useful in Treatment of Cancer and Its Preparation. CN Patent CN109369675A, 22 February 2019. [Google Scholar]
- Li, Y.; Liang, J.; Zhou, J.; Hu, G. Bis-fluoroquinolone oxadiazole carbazide N-methyl-Ciprofloxacin derivative, its preparation method and application as antitumor agents. CN Patent CN109369677A, 22 February 2019. [Google Scholar]
- Feng, Y.; Jiang, Y.; Geng, S.; Hu, G. Bis-Fluoroquinolone Oxadiazole Urea N-Acetyl-Ciprofloxacin Derivative, Its Preparation Method and Application. CN Patent CN109336902A, 15 February 2019. [Google Scholar]
- Hou, L.; Zhang, C.; Hu, G.; Sun, J.; Wang, N.; Shen, R. Bis-Fluoroquinolone Thiadiazole Urea-Series Fleroxacin Derivative Preparation Method and Application as Antitumor Agents. CN Patent CN109761998A, 17 May 2019. [Google Scholar]
- Hou, L.; Du, L.; Li, Y.; Hu, G.; Sun, J.; Zhang, C.; Shen, R.; Wang, N. Preparation of Fluoroquinolone 1,3,4-Thiadiazole Urea Rufloxacin Derivatives Useful for the Treatment of Cancer. CN Patent CN109762005A, 17 May 2019. [Google Scholar]
- Hu, G.; Zhang, C.; Sun, J.; Wang, N.; Shen, R. Preparation Method and Application of Bis-Fluoroquinolone Thiadiazole Urea-series N-Acetyl-Ciprofloxacin Derivative. CN Patent CN109761999A, 17 May 2019. [Google Scholar]
- Hou, L.; Hu, G.; Zhang, C.; Sun, J.; Wang, N.; Shen, R. Process for Preparation and Application of Bis-Fluoroquinolone Thiadiazole Urea-Series N-Methyl Lomefloxacin Derivatives. CN Patent CN109761997A, 17 May 2019. [Google Scholar]
- Hou, L.; Du, L.; Li, Y.; Hu, G.; Sun, J.; Zhang, C.; Shen, R.; Wang, N. Process for Preparation and Application of Bis-Fluoroquinolone Thiadiazole Urea-Based N-Methyl Moxifloxacin Derivative. CN Patent CN109678890A, 26 April 2019. [Google Scholar]
- Hu, G.; Sun, J.; Zhang, C.; Wang, N.; Shen, R. Preparation of Bis-Fluoroquinolone Thiadiazole Urea-Based Pefloxacin Derivatives as Antitumor Agents. CN Patent CN109678882A, 26 April 2019. [Google Scholar]
- Hu, G.; Sun, J.; Zhang, C. Preparation of Bis-Fluoroquinolone Thiadiazole Urea-Based Ofloxacin Derivatives as Antitumor Agents. CN Patent CN109678881A, 26 April 2019. [Google Scholar]
- Hu, G.; Sun, J.; Zhang, C. Bis-Fluoroquinolone Thiadiazole Urea-Based Levofloxacin Derivative Useful in Treatment of Cancer and Its Preparation. CN Patent CN109678889A, 26 April 2019. [Google Scholar]
- Hou, L.; Li, Y.; Hu, G.; Sun, J.; Zhang, C.; Shen, R.; Wang, N. Bis-Fluoroquinolone Thiadiazole Urea-Based N-Methyl Gatifloxacin Derivative Useful in Treatment of Cancer and Its Preparation. CN Patent CN109678885A, 26 April 2019. [Google Scholar]
- Hu, G.; Xie, S.; Hou, L.; Sun, M.; Zhang, J.; Yang, Y.; Yi, L. Preparation of 1,2,4-Triazolo[3,4-b][1,3,4]thiadiazole Linked Fluoroquinolone Dimers for Treating Tumor and Antimicrobial Infection. CN Patent CN101648962A, 17 February 2010. [Google Scholar]
- Yang, Y.; Chen, D.; Qian, X.; Luo, X.; Shao, L.; Dong, X.; Qian, L. Preparation of Novel Rhodamine Dyes Useful as Antibacterial Agents. WO Patent WO2020019289A1, 30 January 2020. [Google Scholar]
- Yang, Y.; Chen, D.; Qian, X.; Luo, X.; Shao, L.; Dong, X.; Qian, L. Preparation of Novel Rhodamine Dyes Useful as Antibacterial Agents. CN Patent CN108570032A, 25 September 2018. [Google Scholar]
- Wang, Y.; Zhou, Q.; Wang, L.; Ma, J.-J. QSAR Analysis of 6-Fluoro-3-(4H-1,2,4-triazol-3-yl)quinolin-4(1H)-ones as Antileukemic Agents using Physicochemical and Alignment Independent Topological Based Descriptors. Lett. Org. Chem. 2018, 15, 551–558. [Google Scholar] [CrossRef]
- Zhao, C.; Huang, D.; Li, R.; Xu, Y.; Su, S.; Gu, Q.; Xu, J. Identifying Novel Anti-Osteoporosis Leads with a Chemotype-Assembly Approach. J. Med. Chem. 2019, 62, 5885–5900. [Google Scholar] [CrossRef]
































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hryhoriv, H.; Kovalenko, S.M.; Georgiyants, M.; Sidorenko, L.; Georgiyants, V. A Comprehensive Review on Chemical Synthesis and Chemotherapeutic Potential of 3-Heteroaryl Fluoroquinolone Hybrids. Antibiotics 2023, 12, 625. https://doi.org/10.3390/antibiotics12030625
Hryhoriv H, Kovalenko SM, Georgiyants M, Sidorenko L, Georgiyants V. A Comprehensive Review on Chemical Synthesis and Chemotherapeutic Potential of 3-Heteroaryl Fluoroquinolone Hybrids. Antibiotics. 2023; 12(3):625. https://doi.org/10.3390/antibiotics12030625
Chicago/Turabian StyleHryhoriv, Halyna, Sergiy M. Kovalenko, Marine Georgiyants, Lyudmila Sidorenko, and Victoriya Georgiyants. 2023. "A Comprehensive Review on Chemical Synthesis and Chemotherapeutic Potential of 3-Heteroaryl Fluoroquinolone Hybrids" Antibiotics 12, no. 3: 625. https://doi.org/10.3390/antibiotics12030625