
Antimicrobial Activity of an Fmoc-Plantaricin 149 Derivative Peptide against Multidrug-Resistant Bacteria
 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Quantitative Peptide Comparison and Antimicrobial Susceptibility
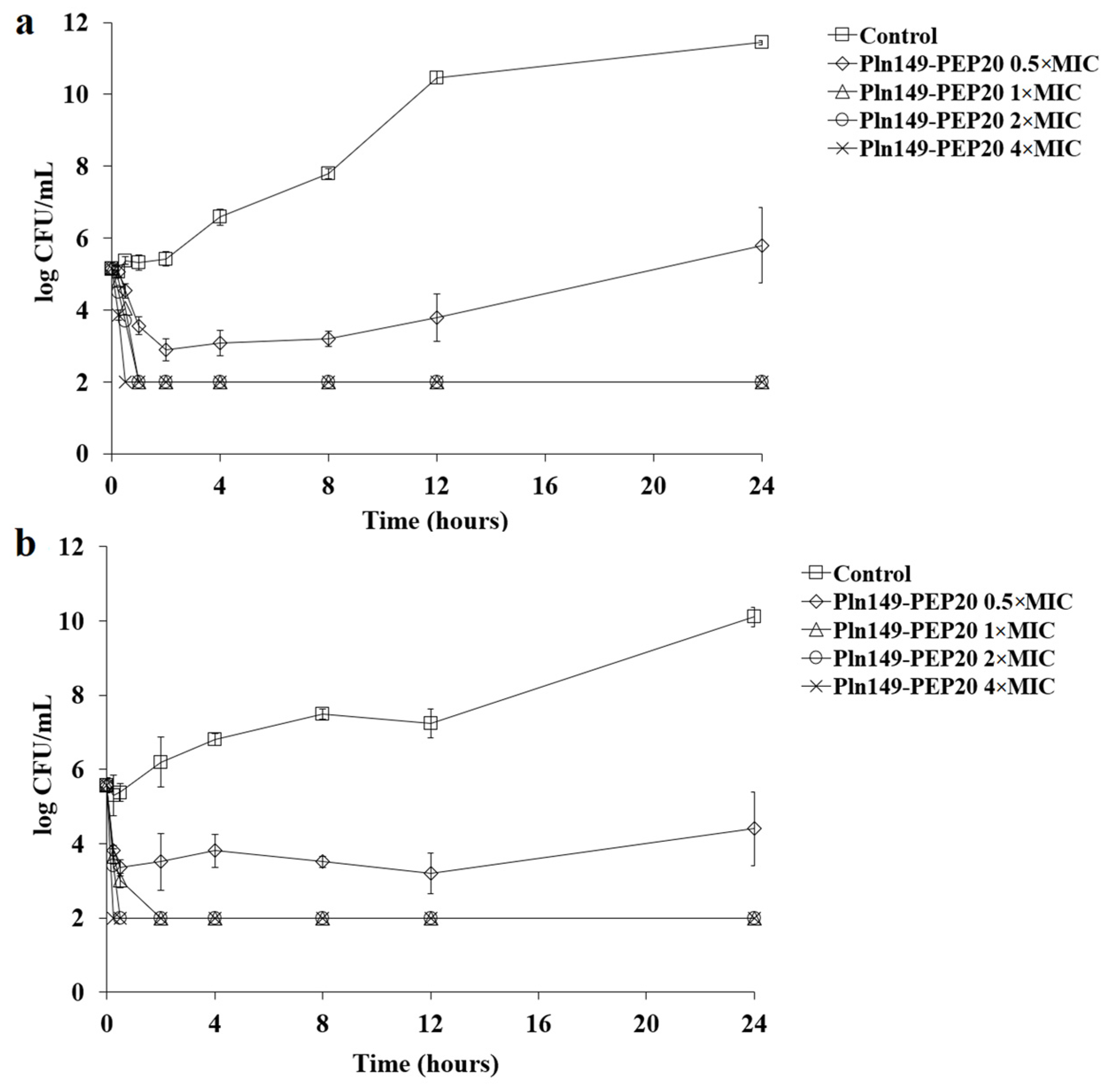
2.2. Time-Kill and Post-Antibiotic Assays
2.3. Cytotoxicity by MTS Assay
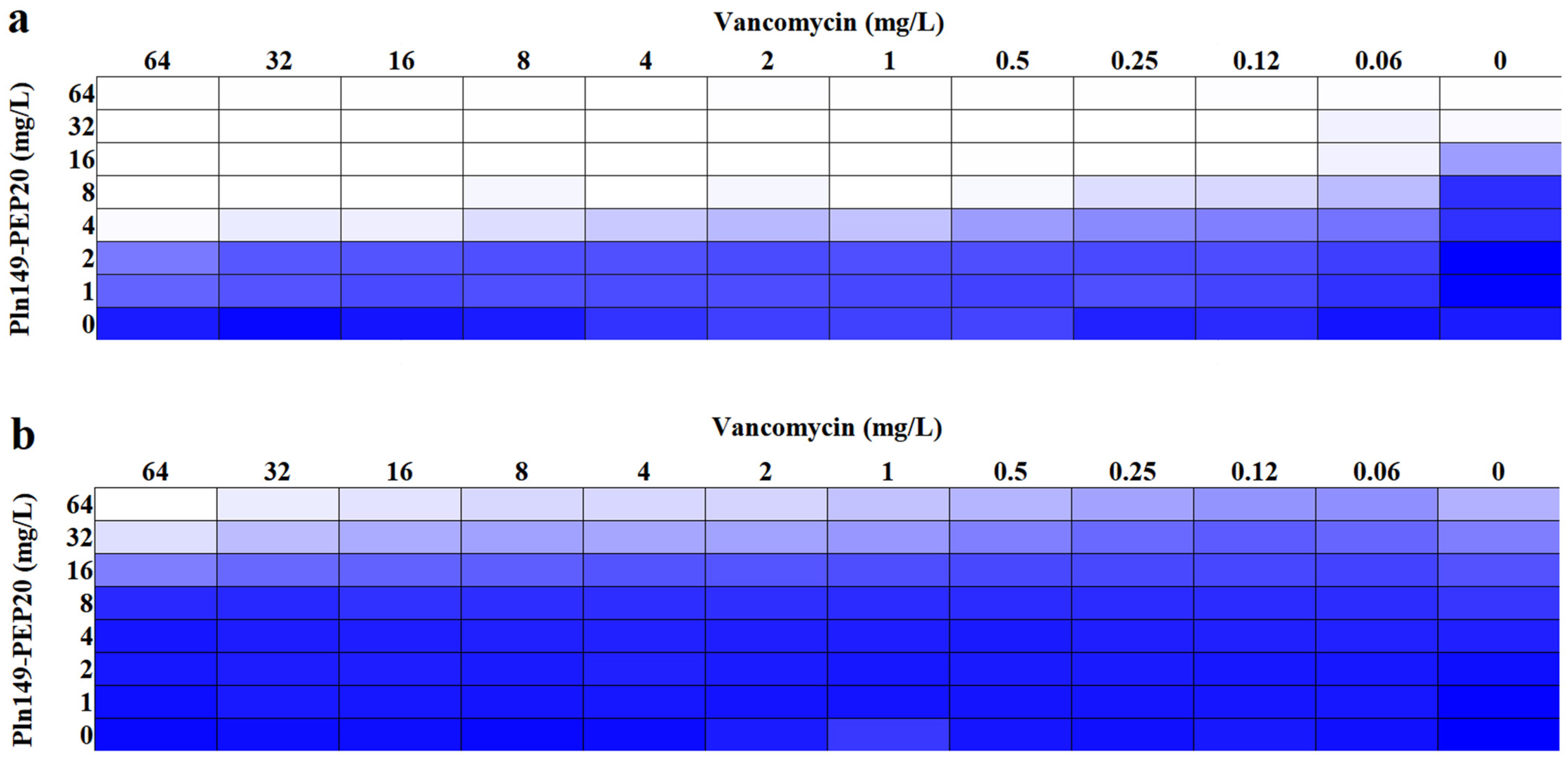
2.4. Synergism Assays
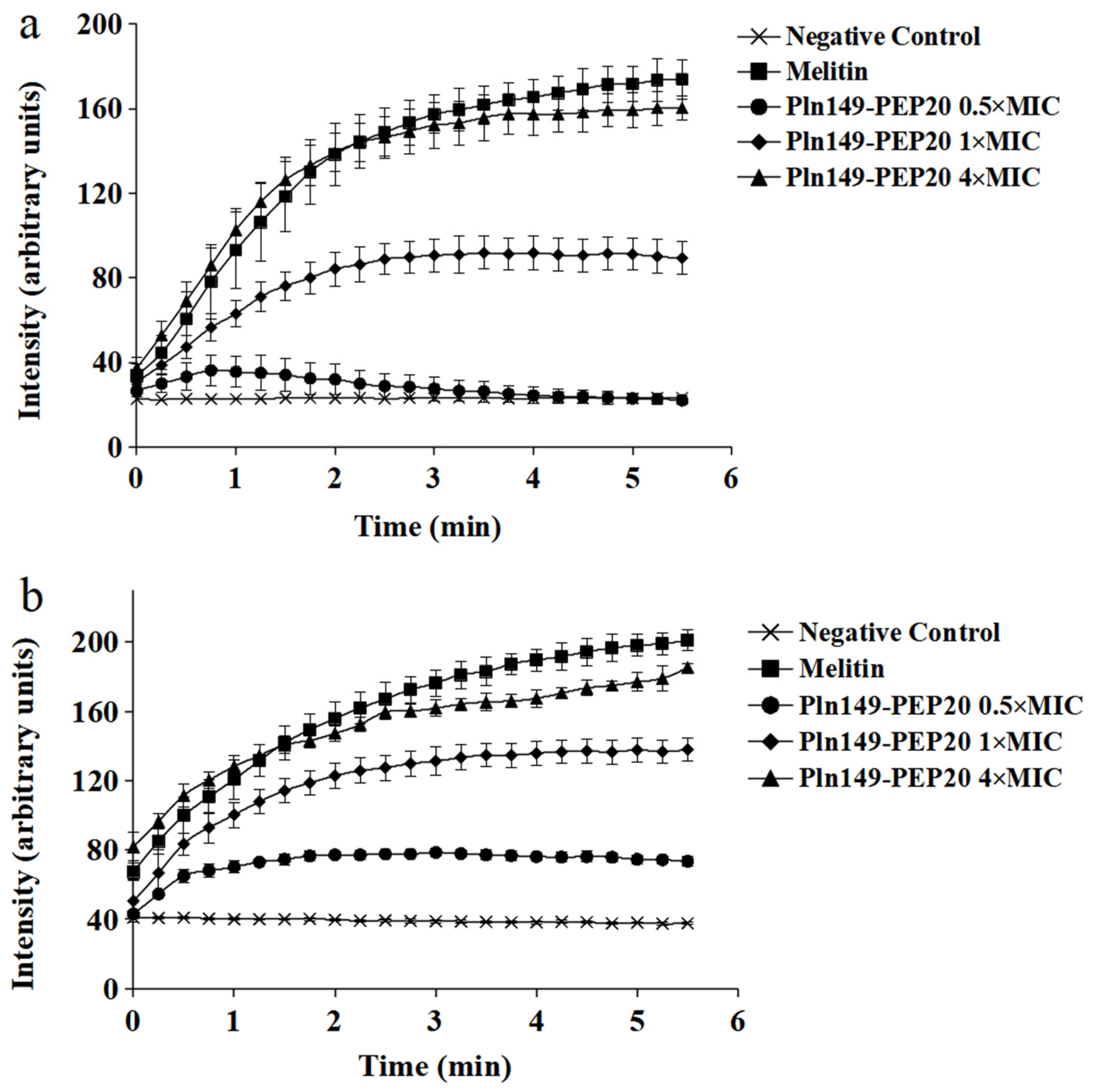
2.5. Membrane Depolarization Assay
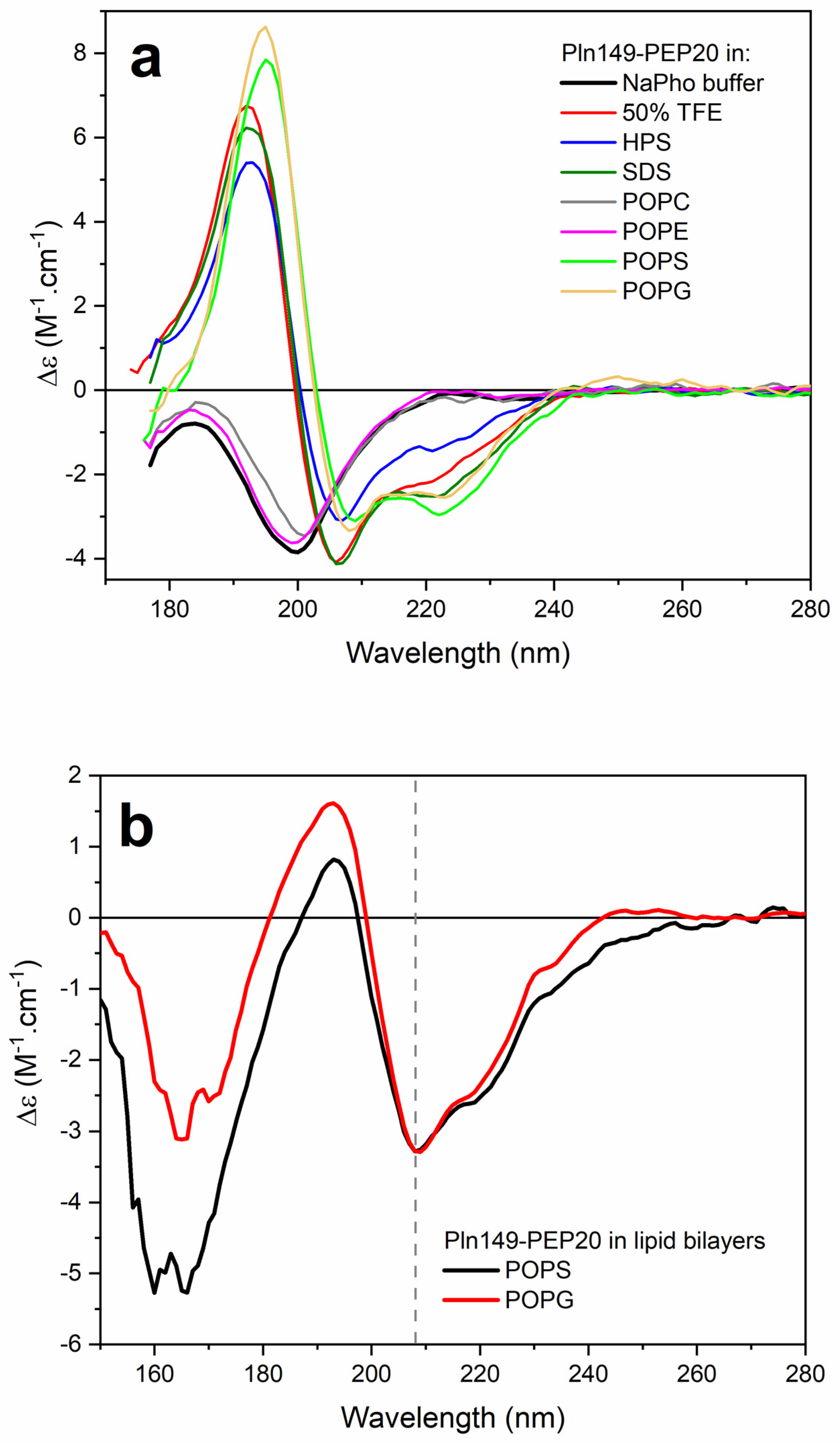
2.6. Circular Dichroism
2.7. Transmission Electron Microscopy (TEM)
2.8. In Vitro Directed Evolution and Genome Analysis
3. Discussion
4. Materials and Methods
4.1. Peptide Synthesis
4.2. Bacterial Strains
4.3. Quantitative Antimicrobial Susceptibility
4.4. Hemolytic Activity
4.5. Time-Kill Assay
4.6. Post-Antibiotic Effect Assay
4.7. Cytotoxicity by MTS Assay
4.8. Synergism Assay
4.9. Membrane Depolarization Assay
4.10. Synchrotron Radiation Circular Dichroism Spectroscopy (SRCD)
4.11. Transmission Electron Microscopy
4.12. In Vitro Directed Evolution
4.13. Genome Sequencing
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
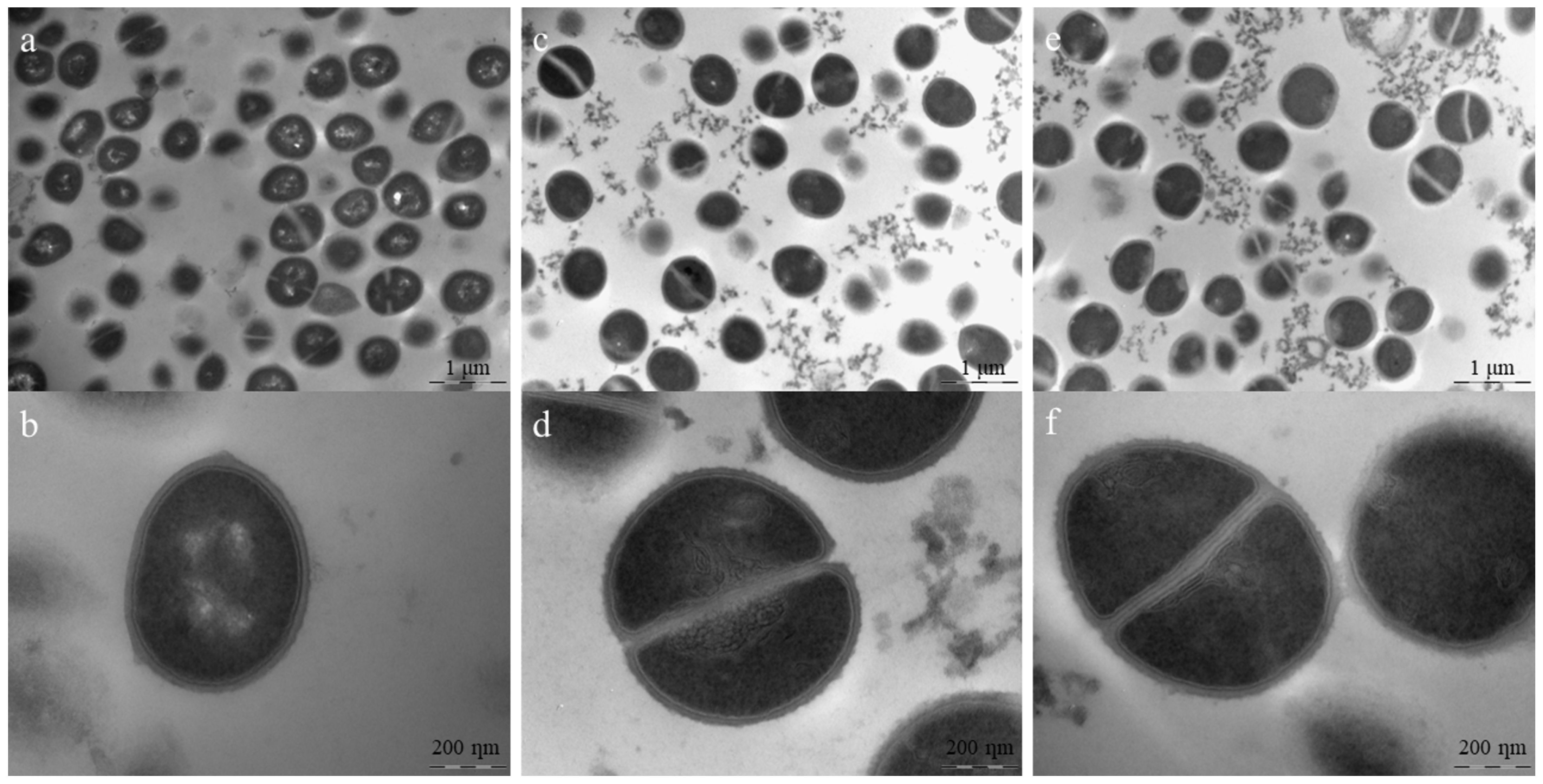{kind=link}
{kind=link}
{kind=link}
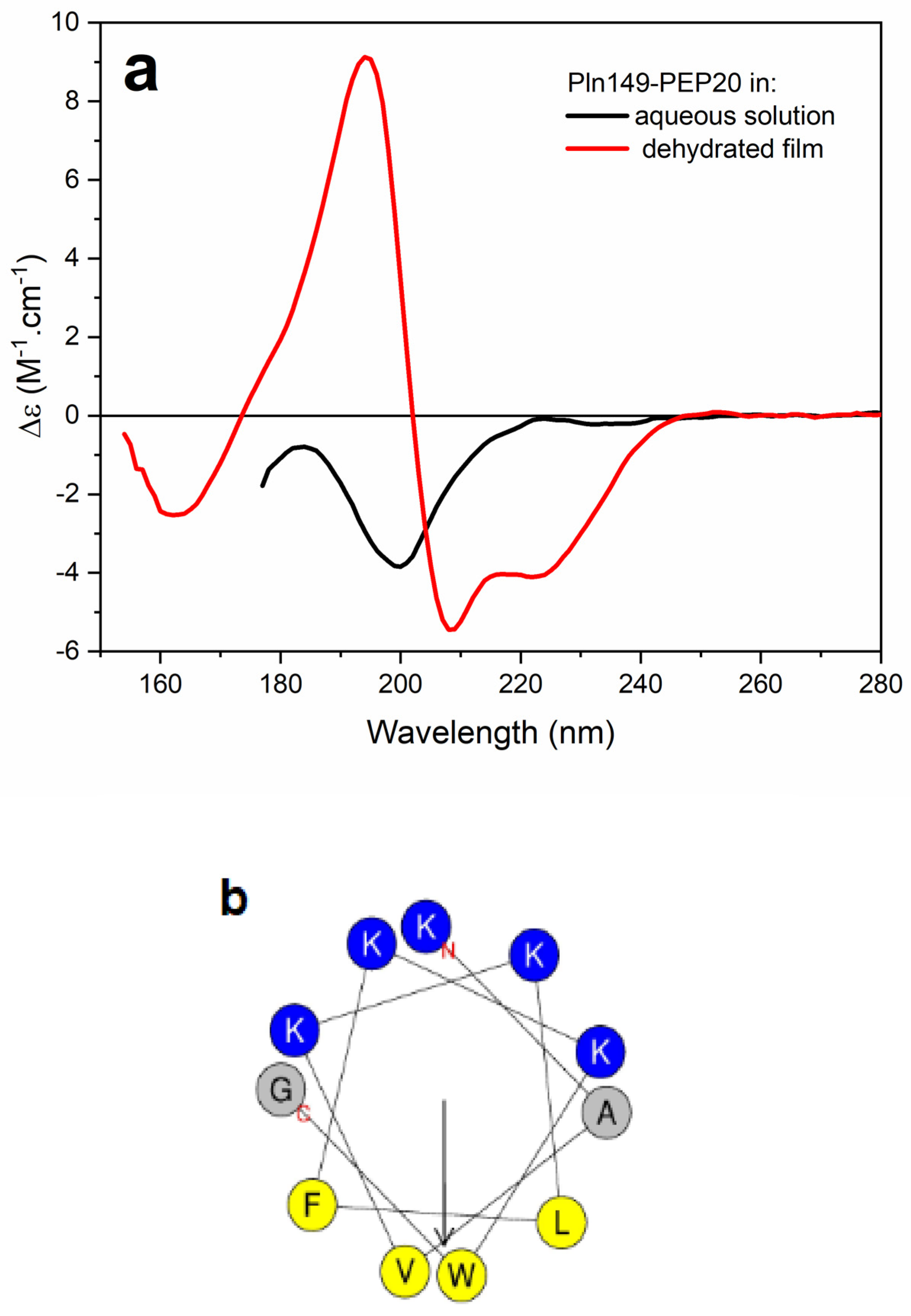{kind=link}
| Species | Strains | Description | MIC (mg/L) | MBC (mg/L) |
|---|---|---|---|---|
| S. epidermidis | ATCC 35984 | Clinical strain, catheter isolate. Strong biofilm former | 2 | 8 |
| ATCC 12228 | 1 | 2 | ||
| S. aureus | ATCC 25923 | Clinical strain. | 8 | 16 |
| SA16 [17] | Clinical strain. MRSA, ST5-SCCmecII | 8 | 16 | |
| SA88 * | Clinical strain. MRSA + h-DNSSA, ST5-SCCmecII | 8 | 16 | |
| SA43 [17] | Clinical strain. MRSA, ST105- SCCmecII, TIG S | 8 | 8 | |
| SA43 B2 [17] | In vitro selected MRSA, ST105- SCCmecII, TIG S | 8 | 8 | |
| SA43 B7 [17] | In vitro selected ST105- MRSA, SCCmecII, TIG R (mepR mutation) | 8 | 8 | |
| Mu50 [52] | MRSA, VISA, ST5 | 2 | >8 | |
| ATCC 8095 | Food isolate. Strong biofilm former | 8 | 16 | |
| E. faecalis | VRE 109 [53] | Clinical strain, ST103, vanA TIG S, VAN R | 32 | 128 |
| VRE 109 42C [53] | In vitro selected. ST103 vanA TIG R, VAN R | 32 | 128 | |
| VRE 80 [53] | Clinical strain. ST103 vanA TIG R, VAN R, | 32 | 128 | |
| V583 | ST 6, vanB. VAN R, CN R | 128 | 256 | |
| RPEfs1 * | Clinical strain. AMP S, CIP R, ERY R, LNZ S, MXF R, TEC S, TIG S, VAN S | 128 | 128 | |
| RPEfs2 * | Clinical strain. AMP S, CIP S, ERY I, LNZ S, MXF S, TEC S, TIG S, VAN S | 128 | 128 | |
| RPEfs3 * | Clinical strain. AMP S, CIP R, ERY R, LNZ S, MXF R, TEC R, TIG S, VAN R | 128 | 128 | |
| RPEfs4 * | Clinical strain. AMP S, CIP R, ERY R, LNZ S, MXF S, TEC S, TIG S, VAN S | 8 | 32 | |
| RPEfs5 * | Clinical strain. AMP S, CIP S, DAP S, LNZ S, NIT S, TEC S, TET R, VAN S | 8 | >32 | |
| ATCC 29212 | Clinical strain, urine isolate | 32 | 64 | |
| E. faecium | VRE 16 [54] | Clinical strain. ST412, vanA. VAN R | 32 | 32 |
| HBSJRP18 [55] | Clinical strain. ST412, DAP supersusceptible (lafB *) | 32 | 64 | |
| HBSJRP18 2.7 [55] | In vitro selected. DAP S | 32 | 64 | |
| HBSJRP18 3.6 [55] | In vitro selected. DAP R (dak *) | 32 | 32 | |
| HBSJRP7 [55] | Clinical strain, muscle biopsy isolate ST896, ermB, msrC, tetL, tetM, vanA. DAP R, LNZ S, TED S, TEC R, VAN R | 16 | 32 | |
| HBSJRP13 [55] | Clinical strain, bronchus alveolar lavage isolate ST896, ermB, msrC, tetL, tetM, vanA. DAP S, LNZ S, TED S, TEC R, VAN R | 16 | 32 | |
| HBSJRP14 [55] | Clinical strain, urine isolate. vanA. DAP S, LNZ S, TED S, TEC R, VAN R | 16 | 64 | |
| HBSJRP23 [55] | Clinical strain, urine isolate. DAP S, LNZ S, TED S, TEC S, VAN S | 8 | 8 | |
| HBSJRP11 [55] | Clinical strain, urine isolate DAP S, LNZ S, TED S, TEC S, VAN S | 8 | 16 | |
| ATCC 700221 | Human feces isolate. vanA, VAN R | 8 | 32 |
| Species | Strains | Description | MIC (mg/L) | MBC (mg/L) |
|---|---|---|---|---|
| K. pneumoniae | ATCC 700603 | Clinical strain, urine isolate. blaKPC-, blaSHV-18 + AMP R, ATM R, FOX R, CPD R, CAZ R, CHL R, PIP R, TET R | 16 | >64 |
| ATCC BAA1705 | Clinical strain, urine isolate. blaKPC+ | 16 | >64 | |
| BHKPC50 * | Clinical strain, urine isolate. AK R, AMP R, SAM R, FEP R, FOX R, CAZ R, CRO R, CXM R, CIP R, CL R, ETP R, CN R, IMI R, MEM R, PTZ R, TIG R | 128 | 512 | |
| RPKp01 * | Clinical strain, urine isolate. AK S, AMP S, SAM R, FEP R, FOX R, CAZ R, CRO R, CXM R, CIP R, CL S, ETP R, CN S, IMI R, MEM R, PTZ R, TIG R | 32 | 64 | |
| RPKp02 * | Clinical strain, rectal swab isolate. AK I, AMP R, SAM R, FEP R, FOX R, CAZ R, CRO R, CXM R, CIP R, CL R, ETP R, CN S, IMI R, MEM R, PTZ R, TIG I | 128 | 256 | |
| RPKp09 * | Clinical strain, surgical wound isolate. AK S, AMC R, AMP R, FEP R, FOX R, CAZ R, CRO R, CXM R, CIP S, CL S, ETP S, CN S, IMI R, MEM R, PTZ R, TIG R, | 32 | 64 | |
| RPKp18 * | Clinical strain, blood isolate. AK S, AMP R, SAM R, FEP R, FOX R, CAZ R, CRO R, CXM R, CIP S, CL R, ETP R, CN S, IMI R, MEM R, PTZ R, TIG S | 128 | 256 | |
| NDM-1 (2146) ** | NDM + | 32 | >128 | |
| AMKP4 [18] | Clinical strain. blaKPC-2 CL R, ETP R, IMI R, MEM R, PB R, TIG S | 512 | >512 | |
| AMKP7 [18] | Clinical strain. blaKPC-2 CL S, ETP R, IMI R, MEM R, PB S, TIG S | 256 | 256 | |
| AMKP10 [18] | Clinical strain. ST2306, blaKPC-2, blaCTX-M8, blaSHV-11, tetA, aph(3′)-Ia, catB, aac(6′)Ib-cr, fosA, blaCTX-M15, oqxab, qnrS1, sul1, blaOXA-1, aadA2, dfrA12, mph(A), mgrB AK S, AMP R, SAM R, FEP R, FOX R, CAZ R, CRO R, CXM R, CIP R, CL R, ETP R, CN S, IMI R, MEM R, PB R, TIG S | 512 | >512 | |
| E. coli | ATCC 25922 | 32 | 64 | |
| ATCC 35218 | Canine isolate. TEM-1 + | 32 | 128 | |
| RPEc01 * | Clinical strain, urine isolate. AK R, AMP R, SAM R, FEP R, FOX R, CAZ R, CRO R, CXM R, CIP S, CL S, ETP R, CN R, IMI R, MEM R, PTZ R, TIG S | 32 | 128 | |
| BHKPC10 * | Clinical strain, urine isolate. AK S, AMC R, FEP R, CAZ R, CIP S, CN S, MEM R, NIT S, SXT S. | 32 | 128 | |
| AMEc8 * | Clinical strain, urine isolate. AK S, AMP R, SAM R, FEP R, FOX R, CAZ R, CRO R, CXM R, CIP R, CL S, ETP R, CN R, IMI S, MEM S, PTZ R, TIG I | 16 | 32 | |
| AMEc49 * | Clinical strain, cranial subdural empyema isolate. AK S, AMP R, SAM I, FEP S, FOX S, CAZ S, CRO R, CXM R, CIP S, CL S, MEM S, PTZ S, TIG S | 32 | 64 | |
| AMEc60 * | Clinical strain, tracheal secretion isolate. AK S, AMP R, SAM R, FEP R, FOX S, CAZ R, CRO R, CXM R, CIP R, CL S, MEM S, PTZ S, TIG S | 16 | 64 | |
| A. baumannii | ATCC 19606 | Clinical strain, urine isolate. | 32 | 64 |
| ACI50 [20] | Clinical strain. AK R, SAM R, FEP R, CTX R, CAZ R, CRO R, CIP R, CL R, CN R, IMI R, MEM R, PTZ R PB R, TET I, TIG R, SXT R | 64 | 128 | |
| ACI44 [20] | Clinical strain. AK R, SAM R, FEP R, CTX R, CAZ R, CRO R, CIP R, CL S, CN R, IMI R, MEM R, PTZ R, PB S, TET R, TIG R, SXT R | 128 | 256 | |
| ACI51 [20] | Clinical strain. AK R, SAM R, FEP R, CTX R, CAZ R, CRO R, CIP R, CL R, CN R, IMI R, MEM R, PTZ R PB R, TET I, TIG R, SXT R | 256 | 256 | |
| ACI40 [20] | Clinical strain. AK R, SAM R, FEP R, CTX R, CAZ R, CRO R, CIP R, CL S, CN R, IMI R, MEM R, PTZ R, PB S, TET R, TIG R, SXT R | 64 | 128 | |
| ACI42 [20] | Clinical strain. AK R, SAM R, FEP R, CTX R, CAZ R, CRO R, CIP R, CL S, CN R, IMI R, MEM R, PTZ R, PB S, TET R, TIG R, SXT R | 64 | 256 | |
| AM83 * | Clinical strain, tracheal secretion isolate. AK R, SAM R, FEP R, CAZ R, CRO R, CL S | 32 | 128 | |
| AM87 * | Clinical strain, urine isolate. AK S, SAM R, FEP R, CAZ R, CRO R, CIP R, CL S | 16 | 16 | |
| P. aeruginosa | ATCC 27853 | Clinical strain, blood isolate. Inducible AmpC | 32 | 128 |
| RPPse09 * | Clinical strain, rectal swab isolate. AK S, FEP S, CAZ S, CIP S, CL S, CN S, IMI R, MEM R, PTZ S | 256 | 512 | |
| RPPse07 * | Clinical strain, urine isolate. AK R, FEP R, CAZ I, CIP R, CL S, CN R, IMI R, MEM R, PTZ I | 64 | 128 | |
| PSE6 [56] | KPC+ | 128 | 128 | |
| PAO1 | 128 | 256 |
Appendix B

References
- Aljeldah, M.M. Antimicrobial Resistance and Its Spread Is a Global Threat. Antibiotics 2022, 11, 1082. [Google Scholar] [CrossRef]
- Reynolds, D.; Burnham, J.P.; Vazquez Guillamet, C.; McCabe, M.; Yuenger, V.; Betthauser, K.; Micek, S.T.; Kollef, M.H. The Threat of Multidrug-Resistant/Extensively Drug-Resistant Gram-Negative Respiratory Infections: Another Pandemic. Eur. Respir. Rev. 2022, 31, 220068. [Google Scholar] [CrossRef]
- De Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial Resistance in ESKAPE Pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Björn, C.; Ekblom, J. Antimicrobial Peptides as Therapeutic Agents: Opportunities and Challenges. Crit. Rev. Biotechnol. 2020, 40, 978–992. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: The Antimicrobial Peptide Database as a Tool for Research and Education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [Green Version]
- Bellotti, D.; Remelli, M. Lights and Shadows on the Therapeutic Use of Antimicrobial Peptides. Molecules 2022, 27, 4584. [Google Scholar] [CrossRef]
- Kristiansen, P.E.; Fimland, G.; Mantzilas, D.; Nissen-meyer, J. Structure and Mode of Action of the Membrane-Permeabilizing Antimicrobial Peptide Pheromone Plantaricin A *. J. Biol. Chem. 2005, 280, 22945–22950. [Google Scholar] [CrossRef] [Green Version]
- Müller, D.M.; Carrasco, M.S.; Simonetta, A.C.; Beltramini, L.M.; Tonarelli, G.G. A Synthetic Analog of Plantaricin 149 Inhibiting Food-Borne Pathogenic Bacteria:Evidence for α-Helical Conformation Involved in Bacteria–Membrane Interaction. J. Pept. Sci. 2007, 13, 171–178. [Google Scholar] [CrossRef]
- de Souza Lopes, J.L.; Hissa, D.C.; Melo, V.M.M.; Beltramini, L.M. Interaction of Antimicrobial Peptide Plantaricin149a and Four Analogs with Lipid Bilayers and Bacterial Membranes. Braz. J. Microbiol. 2013, 44, 1291–1298. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, P.S.; Sousa, V.K.; Donato, M.; Itri, R.; Beltramini, L.M.; Araujo, A.P.U.; Buerck, J.; Wallace, B.A.; Lopes, J.L.S. Unveiling the Binding and Orientation of the Antimicrobial Peptide Plantaricin 149 in Zwitterionic and Negatively Charged Membranes. Eur. Biophys. J. 2019, 48, 621–633. [Google Scholar] [CrossRef]
- Iwamoto, K.; Hayakawa, T.; Murate, M.; Makino, A.; Ito, K.; Fujisawa, T.; Kobayashi, T. Curvature-Dependent Recognition of Ethanolamine Phospholipids by Duramycin and Cinnamycin. Biophys. J. 2007, 93, 1608–1619. [Google Scholar] [CrossRef]
- Fernandez, D.I.; Sani, M.-A.; Miles, A.J.; Wallace, B.A.; Separovic, F. Membrane Defects Enhance the Interaction of Antimicrobial Peptides, Aurein 1.2 versus Caerin 1.1. Biochim. Biophys. Acta (BBA)-Biomembr. 2013, 1828, 1863–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shai, Y. Mode of Action of Membrane Active Antimicrobial Peptides. Pept. Sci. Orig. Res. Biomol. 2002, 66, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical Relevance of the ESKAPE Pathogens. Expert Rev. Anti-Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef]
- WHO. Global Priority List of Antibiotic-Resistant Batceria to Guide Research, Discovery, and Development of New Antibiotics; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Tran, T.B.; Velkov, T.; Nation, R.L.; Forrest, A.; Tsuji, B.T.; Bergen, P.J.; Li, J. Pharmacokinetics/Pharmacodynamics of Colistin and Polymyxin B: Are We There Yet? Int. J. Antimicrob. Agents 2016, 48, 592–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabul, A.N.G.; Avaca-Crusca, J.S.; Van Tyne, D.; Gilmore, M.S.; Camargo, I.L.B.C. Resistance in In Vitro Selected Tigecycline-Resistant Methicillin-Resistant Staphylococcus aureus Sequence Type 5 Is Driven by Mutations in MepR and MepA Genes. Microb. Drug Resist. 2018, 24, 519–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Souza, R.C.; Dabul, A.N.G.; dos Santos Boralli, C.M.; Zuvanov, L.; da Cunha Camargo, I.L.B. Dissemination of BlaKPC-2 in an NTEKPC by an IncX5 Plasmid. Plasmid 2019, 106, 102446. [Google Scholar] [CrossRef]
- Garbacz, K. Anticancer Activity of Lactic Acid Bacteria. Semin. Cancer Biol. 2022, 86, 356–366. [Google Scholar] [CrossRef]
- de Melo Carrasco, L.D.; Dabul, A.N.G.; dos Santos Boralli, C.M.; Righetto, G.M.; Silva e Carvalho, I.; Dornelas, J.V.; Martins da Mata, C.P.S.; de Araújo, C.A.; Leite, E.M.M.; Lincopan, N.; et al. Polymyxin Resistance Among XDR ST1 Carbapenem-Resistant Acinetobacter Baumannii Clone Expanding in a Teaching Hospital. Front. Microbiol. 2021, 12, 622704. [Google Scholar] [CrossRef]
- Li, Q.; Cebrián, R.; Montalbán-López, M.; Ren, H.; Wu, W.; Kuipers, O.P. Outer-Membrane-Acting Peptides and Lipid II-Targeting Antibiotics Cooperatively Kill Gram-Negative Pathogens. Commun. Biol. 2021, 4, 31. [Google Scholar] [CrossRef]
- Kumagai, P.S.; DeMarco, R.; Lopes, J.L.S. Advantages of Synchrotron Radiation Circular Dichroism Spectroscopy to Study Intrinsically Disordered Proteins. Eur. Biophys. J. 2017, 46, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Bürck, J.; Wadhwani, P.; Fanghänel, S.; Ulrich, A.S. Oriented Circular Dichroism: A Method to Characterize Membrane-Active Peptides in Oriented Lipid Bilayers. Acc. Chem. Res. 2016, 49, 184–192. [Google Scholar] [CrossRef]
- Wenzel, M.; Dekker, M.P.; Wang, B.; Burggraaf, M.J.; Bitter, W.; van Weering, J.R.T.; Hamoen, L.W. A Flat Embedding Method for Transmission Electron Microscopy Reveals an Unknown Mechanism of Tetracycline. Commun. Biol. 2021, 4, 306. [Google Scholar] [CrossRef]
- Müller, A.; Klöckner, A.; Schneider, T. Targeting a Cell Wall Biosynthesis Hot Spot. Nat. Prod. Rep. 2017, 34, 909–932. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.-B.; Wenzel, M. A How-To Guide for Mode of Action Analysis of Antimicrobial Peptides. Front. Cell. Infect. Microbiol. 2020, 10, 540898. [Google Scholar] [CrossRef] [PubMed]
- Saravolatz, L.D.; Pawlak, J.; Martin, H.; Saravolatz, S.; Johnson, L.; Wold, H.; Husbyn, M.; Olsen, W.M. Postantibiotic Effect and Postantibiotic Sub-MIC Effect of LTX-109 and Mupirocin on Staphylococcus aureus Blood Isolates. Lett. Appl. Microbiol. 2017, 65, 410–413. [Google Scholar] [CrossRef]
- Alves, A.C.; Ribeiro, D.; Nunes, C.; Reis, S. Biophysics in Cancer: The Relevance of Drug-Membrane Interaction Studies. Biochim. Biophys. Acta (BBA)-Biomembr. 2016, 1858, 2231–2244. [Google Scholar] [CrossRef] [PubMed]
- Awouafack, M.D.; McGaw, L.J.; Gottfried, S.; Mbouangouere, R.; Tane, P.; Spiteller, M.; Eloff, J.N. Antimicrobial Activity and Cytotoxicity of the Ethanol Extract, Fractions and Eight Compounds Isolated from Eriosema Robustum (Fabaceae). BMC Complement. Altern. Med. 2013, 13, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholar, E. Vancomycin. In xPharm: The Comprehensive Pharmacology Reference; Elsevier: Amsterdam, The Netherlands, 2007; pp. 1–6. [Google Scholar]
- Dik, D.A.; Fisher, J.F.; Mobashery, S. Cell-Wall Recycling of the Gram-Negative Bacteria and the Nexus to Antibiotic Resistance. Chem. Rev. 2018, 118, 5952–5984. [Google Scholar] [CrossRef]
- Azam, M.W.; Khan, A.U. Updates on the Pathogenicity Status of Pseudomonas Aeruginosa. Drug Discov. Today 2019, 24, 350–359. [Google Scholar] [CrossRef]
- Olaitan, A.O.; Morand, S.; Rolain, J.M. Mechanisms of Polymyxin Resistance: Acquired and Intrinsic Resistance in Bacteria. Front. Microbiol. 2014, 5, 643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keane, F.M.; Loughman, A.; Valtulina, V.; Brennan, M.; Speziale, P.; Foster, T.J. Fibrinogen and Elastin Bind to the Same Region within the A Domain of Fibronectin Binding Protein A, an MSCRAMM of Staphylococcus aureus. Mol. Microbiol. 2007, 63, 711–723. [Google Scholar] [CrossRef]
- Rudkin, J.K.; Edwards, A.M.; Bowden, M.G.; Brown, E.L.; Pozzi, C.; Waters, E.M.; Chan, W.C.; Williams, P.; O’Gara, J.P.; Massey, R.C. Methicillin Resistance Reduces the Virulence of Healthcare-Associated Methicillin-Resistant Staphylococcus aureus by Interfering with the agr Quorum Sensing System. J. Infect. Dis. 2012, 205, 798–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taglialegna, A.; Varela, M.C.; Rosato, R.R.; Rosato, A.E. VraSR and Virulence Trait Modulation during Daptomycin Resistance in Methicillin-Resistant Staphylococcus aureus Infection. mSphere 2019, 4, e00557-18. [Google Scholar] [CrossRef] [Green Version]
- Tocchetti, A.; Iorio, M.; Hamid, Z.; Armirotti, A.; Reggiani, A.; Donadio, S. Understanding the Mechanism of Action of NAI-112, a Lanthipeptide with Potent Antinociceptive Activity. Molecules 2021, 26, 6764. [Google Scholar] [CrossRef] [PubMed]
- Graf, A.; Lewis, R.J.; Fuchs, S.; Pagels, M.; Engelmann, S.; Riedel, K.; Pané-Farré, J. The Hidden Lipoproteome of Staphylococcus aureus. Int. J. Med. Microbiol. 2018, 308, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.; Goerke, C.; Fritz, M.; Schäfer, T.; Ohlsen, K.; Liebeke, M.; Lalk, M.; Wolz, C. Role of the (p)PpGpp Synthase RSH, a RelA/SpoT Homolog, in Stringent Response and Virulence of Staphylococcus aureus. Infect. Immun. 2010, 78, 1873–1883. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.L.; Roberts, C.; Disz, T.; Vonstein, V.; Hwang, K.; Overbeek, R.; Olson, P.D.; Projan, S.J.; Dunman, P.M. Characterization of the Staphylococcus aureus Heat Shock, Cold Shock, Stringent, and SOS Responses and Their Effects on Log-Phase MRNA Turnover. J. Bacteriol. 2006, 188, 6739–6756. [Google Scholar] [CrossRef] [Green Version]
- Horvatek, P.; Salzer, A.; Hanna, A.M.F.; Gratani, F.L.; Keinhörster, D.; Korn, N.; Borisova, M.; Mayer, C.; Rejman, D.; Mäder, U.; et al. Inducible Expression of (Pp)PGpp Synthetases in Staphylococcus aureus Is Associated with Activation of Stress Response Genes. PLoS Genet. 2020, 16, e1009282. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Trisolini, L.; Gambacorta, N.; Gorgoglione, R.; Montaruli, M.; Laera, L.; Colella, F.; Volpicella, M.; De Grassi, A.; Pierri, C.L. FAD/NADH Dependent Oxidoreductases: From Different Amino Acid Sequences to Similar Protein Shapes for Playing an Ancient Function. J. Clin. Med. 2019, 8, 2117. [Google Scholar] [CrossRef] [Green Version]
- Quistgaard, E.M.; Löw, C.; Guettou, F.; Nordlund, P. Understanding Transport by the Major Facilitator Superfamily (MFS): Structures Pave the Way. Nat. Rev. Mol. Cell Biol. 2016, 17, 123–132. [Google Scholar] [CrossRef]
- Loehfelm, T.W.; Luke, N.R.; Campagnari, A.A. Identification and Characterization of an Acinetobacter Baumannii Biofilm-Associated Protein. J. Bacteriol. 2008, 190, 1036–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The Conserved Domain Database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, C.-T.; Wu, C.-C.; Lee, J.-C.; Chen, S.-L.; Morris-Natschke, S.L.; Hsieh, P.-W.; Wu, Y.-C. Cytotoxic N-(Fluorenyl-9-Methoxycarbonyl) (Fmoc)-Dipeptides: Structure–Activity Relationships and Synergistic Studies. Eur. J. Med. Chem. 2010, 45, 2494–2502. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kizhakkedathu, J.; Straus, S. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- DABUL, A.N.G.; CAMARGO, I.L.B.C. Molecular Characterization of Methicillin-Resistant Staphylococcus aureus Resistant to Tigecycline and Daptomycin Isolated in a Hospital in Brazil. Epidemiol. Infect. 2014, 142, 479–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, M.; Ohta, T.; Uchiyama, I.; Baba, T.; Yuzawa, H.; Kobayashi, I.; Cui, L.; Oguchi, A.; Aoki, K.; Nagai, Y.; et al. Whole Genome Sequencing of Meticillin-Resistant Staphylococcus aureus. Lancet 2001, 357, 1225–1240. [Google Scholar] [CrossRef] [PubMed]
- Dabul, A.N.G.; Avaca-Crusca, J.S.; Navais, R.B.; Merlo, T.P.; Van Tyne, D.; Gilmore, M.S.; Camargo, I.L.B. da C. Molecular Basis for the Emergence of a New Hospital Endemic Tigecycline-Resistant Enterococcus Faecalis ST103 Lineage. Infect. Genet. Evol. 2019, 67, 23–32. [Google Scholar] [CrossRef]
- de Mello, S.S.; Van Tyne, D.; Dabul, A.N.G.; Gilmore, M.S.; Camargo, I.L.B.C. High-Quality Draft Genome Sequence of the Multidrug-Resistant Clinical Isolate Enterococcus Faecium VRE16. Genome Announc. 2016, 4, e00992-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mello, S.S.; Van Tyne, D.; Lebreton, F.; Silva, S.Q.; Nogueira, M.C.L.; Gilmore, M.S.; Camargo, I.L.B.C. A Mutation in the Glycosyltransferase Gene LafB Causes Daptomycin Hypersusceptibility in Enterococcus Faecium. J. Antimicrob. Chemother. 2020, 75, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Galetti, R.; Andrade, L.N.; Chandler, M.; de Mello Varani, A.; Darini, A.L.C. New Small Plasmid Harboring Bla KPC-2 in Pseudomonas Aeruginosa. Antimicrob. Agents Chemother. 2016, 60, 3211–3214. [Google Scholar] [CrossRef] [Green Version]
- CLSI. Methods for Determining Bactericidal Activity of Antimicrobial Agents, Approved Guideline, 1st ed.; NCCLS document M26-A; CLSI: Wayne, PA, USA, 1999; ISBN 1-56238-384-1. [Google Scholar]
- CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, 10th ed.; CLSI Standard M07; CLSI: Wayne, PA, USA, 2018; ISBN 1562389874. [Google Scholar]
- Castro, M.S.; Ferreira, T.C.G.; Cilli, E.M.; Crusca, E.; Mendes-Giannini, M.J.S.; Sebben, A.; Ricart, C.A.O.; Sousa, M.V.; Fontes, W. Hylin A1, the First Cytolytic Peptide Isolated from the Arboreal South American Frog Hypsiboas Albopunctatus (“spotted Treefrog”). Peptides 2009, 30, 291–296. [Google Scholar] [CrossRef]
- Indrayanto, G.; Putra, G.S.; Suhud, F. Validation of In-Vitro Bioassay Methods: Application in Herbal Drug Research. Excip. Relat. Methodol. 2021, 46, 273–307. [Google Scholar]
- Jahnsen, R.D.; Haney, E.F.; Franzyk, H.; Hancock, R.E.W. Characterization of a Proteolytically Stable Multifunctional Host Defense Peptidomimetic. Chem. Biol. 2013, 20, 1286–1295. [Google Scholar] [CrossRef] [Green Version]
- Bürck, J.; Roth, S.; Wadhwani, P.; Afonin, S.; Kanithasen, N.; Strandberg, E.; Ulrich, A.S. Conformation and Membrane Orientation of Amphiphilic Helical Peptides by Oriented Circular Dichroism. Biophys. J. 2008, 95, 3872–3881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miles, A.J.; Wallace, B.A. CDtoolX, a Downloadable Software Package for Processing and Analyses of Circular Dichroism Spectroscopic Data. Protein Sci. 2018, 27, 1717–1722. [Google Scholar] [CrossRef]








| Peptide | Sequence | Minimal Inhibitory Concentration (mg/L) | HC50 (mg/L) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| S. epidermidis ATCC 35984 | S. aureus ATCC 25923 | E. faecalis ATCC 29212 | E. faecium ATCC 700221 | K. pneumoniae ATCC 700603 | E. coli ATCC 25922 | A. baumannii ATCC 19606 | P. aeruginosa ATCC 27583 | |||
| Fmoc-Pln149 | Fmoc -YSLQMGATAIKQVKKLFKKKGG | 32 | 256 | 128 | 64 | 256 | 128 | 32 | 256 | 4 |
| Fmoc-Pln149(6-22) | Fmoc -GATAIKQVKKLFKKKGG | 128 | 512 | 512 | 128 | 512 | 512 | 64 | 512 | >128 |
| Pln149-PEP20 | Fmoc -KAVKKLFKKWG | 4 | 16 | 32 | 16 | 32 | 64 | 32 | 64 | >512 |
| Bacterial Strains | PAE of the Treatments (h ± s.d.) | |
|---|---|---|
| Pln149-PEP20 0.5 × MIC | Pln149-PEP20 1 × MIC | |
| S. aureus ATCC 25923 (MIC = 8 mg/L) | 4.0 ± 1.0 | 5.5 ± 2.0 |
| S. aureus SA43 [17] (MIC = 8 mg/L) | 4.0 ± 0.5 | 5.0 ± 1.0 |
| A. baumannii ATCC 19606 (MIC = 32 mg/L) | 3.5 ± 0.5 | 4.5 ± 0.5 |
| A. baumannii ACI50 [18] (MIC = 64 mg/L) | N.O. | 2.5 ± 0.5 |
| CIM50 (mg/L) | SI (THP-1) | SI (HFF-1) | |
|---|---|---|---|
| Gram-positives | 8 | 8 | 7 |
| Gram-negatives | 32 | 2 | 2 |
| Antibiotics | Antibiotics MIC (mg/L) | Combination (mg/L) | FIC Index | |
|---|---|---|---|---|
| MICANTIBIOTIC | MICPln149-PEP20 | |||
| Ciprofloxacin | 1 | 0.06 | 8 | 2 |
| Tobramycin | 4 | 4 | 16 | 1.5 |
| Polymyxin B | 1 | 0.25 | 2 | 0.312 |
| Vancomycin | >64 | 2 | 4 | 0.133 |
| Ampicillin | >64 | 16 | 4 | 0.375 |
| Bacterial Strains | Main Phenotype | Pln149-PEP20 MIC (mg/L) | Vancomycin MIC (mg/L) | Combination (mg/L) | FIC Index | |
|---|---|---|---|---|---|---|
| MICPln149-PEP20 | MICVancomycin | |||||
| K. pneumoniae ATCC 700603 | 64 | >64 | 8 | 2 | 0.133 | |
| K. pneumoniae AMKP7 [18] | KPC+, CL S | 512 | 128 | 128 | 16 | 0.375 |
| K. pneumoniae AMKP4 [18] | KPC+, CL R | 512 | >64 | 256 | 64 | 0.75 |
| K. pneumoniae AMKP10 [18] | KPC+, CL R | 512 | >64 | 512 | >64 | 2 |
| A. baumannii ATCC 19606 | 32 | >64 | 4 | 2 | 0.133 | |
| A. baumannii ACI40 [20] | CL S | 64 | >64 | 8 | 2 | 0.133 |
| A. baumannii ACI50 [20] | CL R | 64 | >64 | 2 | 16 | 0.093 |
| E. coli ATCC 25922 | 32 | >64 | 4 | 2 | 0.133 | |
| P. aeruginosa ATCC 27853 | Inducible AmpC | 32 | >64 | 32 | >64 | 2 |
| Replicate | Sequencing Coverage * | N50 | Mutation | Change | Altered Protein |
|---|---|---|---|---|---|
| A | 153× | 19,562 | G > A | Ala111Thr | Hypothetical protein |
| C > T | Ser164Phe | Bifunctional synthetase (p)ppGpp/guanosine-3′-5′-bis(diphosphate)-3′-pyrophosphohydrolase | |||
| A > T | Gln152His | DUF1672 domain protein | |||
| T > A | Ser194Thr | FAD-containing oxidoreductase | |||
| B | 173× | 14,266 | 394_395Ins ** | Frameshift mutation | Fibronectin binding precursor (fnbA) |
| C > A | Leu171Ile | Major facilitator superfamily | |||
| 5_6Ins *** | Frameshift mutation | Major facilitator superfamily | |||
| C | 138× | 12,146 | T > C | Leu893Ser | Clumping factor A (ClfA) |
| A > T | Gln152His | DUF1672 domain protein |
| Replicate | Sequencing Coverage * | N50 | Mutation | Change | Altered Protein |
|---|---|---|---|---|---|
| A | 201× | 14,063 | G > C | Ser126Thr | Ig-like domain-containing protein |
| A > G | Thr128Ala | ||||
| CT > GC | Thr137Ser | ||||
| A > T | Thr139Ser | ||||
| T > AACC | Asn ** | ||||
| AAAT > GGTG | Asn146Val | ||||
| A > C | Lys150Thr | ||||
| TT > CA | Ile154Thr | ||||
| C > A | Pro162Thr | ||||
| GGA > TTG | Gly164Leu | ||||
| CT > GC | Ala168Gly | ||||
| TT > CA | Ile169Thr | ||||
| GAT > TCA | Asp171Ser | ||||
| 517_518Ins *** | *** | ||||
| T > CACA | Thr **** | ||||
| GACA > TGTG | Thr176Val | ||||
| TA > GG | Asn180Asp | ||||
| A > G | Thr265Ala | ||||
| B | 190× | 8,576 | T > AACC | Asn ***** | Ig-like domain-containing protein |
| AAAT > GGTG | Asn243Val | ||||
| A > C | Lys247Thr | ||||
| TT > CA | Ile251Thr | ||||
| C > A | Pro259Thr | ||||
| C | 141× | 7,603 | TA > GG | Asn164Asp | Ig-like domain-containing protein |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Righetto, G.M.; Lopes, J.L.d.S.; Bispo, P.J.M.; André, C.; Souza, J.M.; Andricopulo, A.D.; Beltramini, L.M.; Camargo, I.L.B.d.C. Antimicrobial Activity of an Fmoc-Plantaricin 149 Derivative Peptide against Multidrug-Resistant Bacteria. Antibiotics 2023, 12, 391. https://doi.org/10.3390/antibiotics12020391
Righetto GM, Lopes JLdS, Bispo PJM, André C, Souza JM, Andricopulo AD, Beltramini LM, Camargo ILBdC. Antimicrobial Activity of an Fmoc-Plantaricin 149 Derivative Peptide against Multidrug-Resistant Bacteria. Antibiotics. 2023; 12(2):391. https://doi.org/10.3390/antibiotics12020391
Chicago/Turabian StyleRighetto, Gabriela Marinho, José Luiz de Souza Lopes, Paulo José Martins Bispo, Camille André, Julia Medeiros Souza, Adriano Defini Andricopulo, Leila Maria Beltramini, and Ilana Lopes Baratella da Cunha Camargo. 2023. "Antimicrobial Activity of an Fmoc-Plantaricin 149 Derivative Peptide against Multidrug-Resistant Bacteria" Antibiotics 12, no. 2: 391. https://doi.org/10.3390/antibiotics12020391