
The Structures and Binding Modes of Small-Molecule Inhibitors of Pseudomonas aeruginosa Elastase LasB


Abstract
:1. Introduction
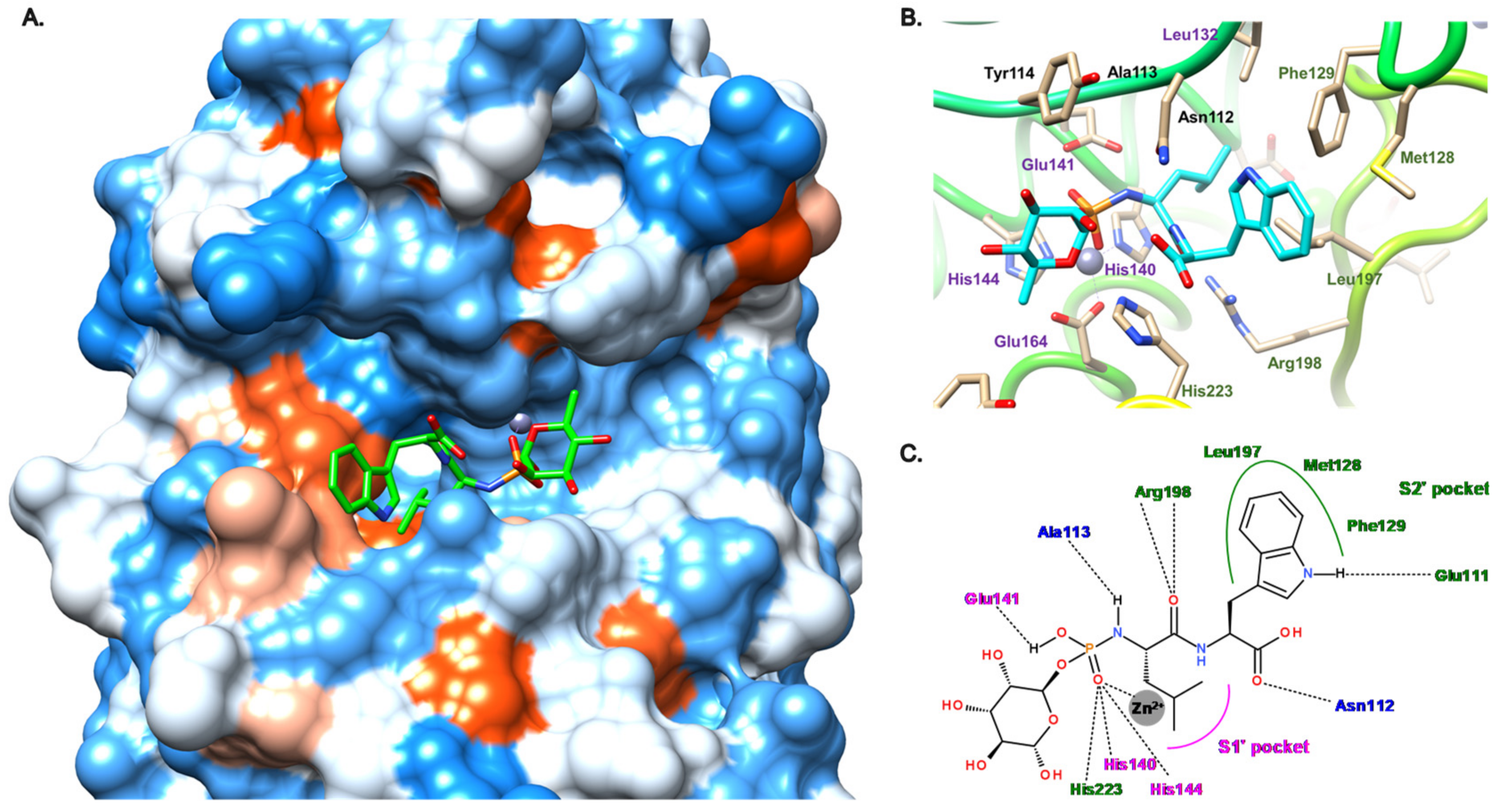
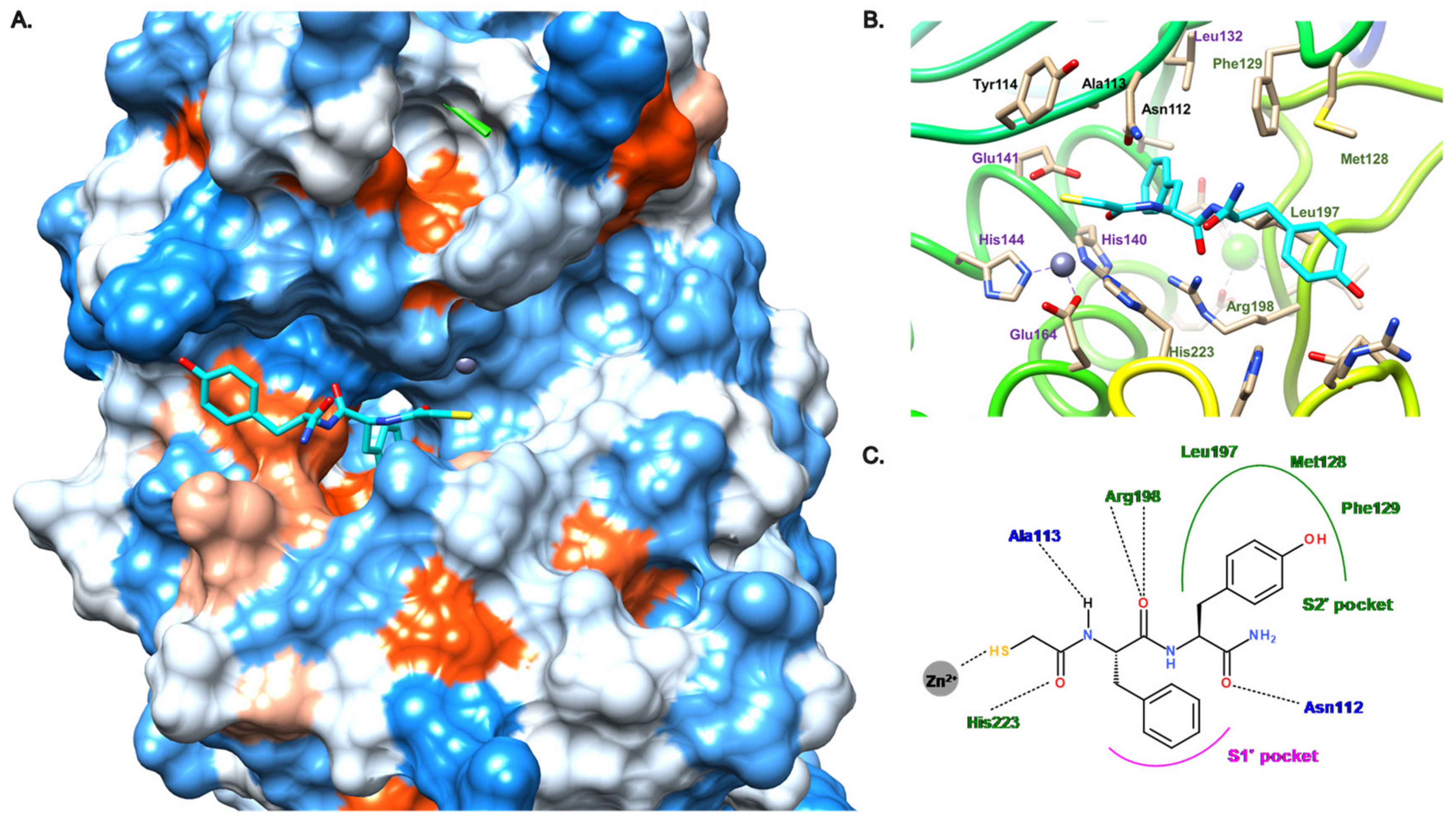
2. Description of the Active Site of LasB
3. LasB Inhibitors
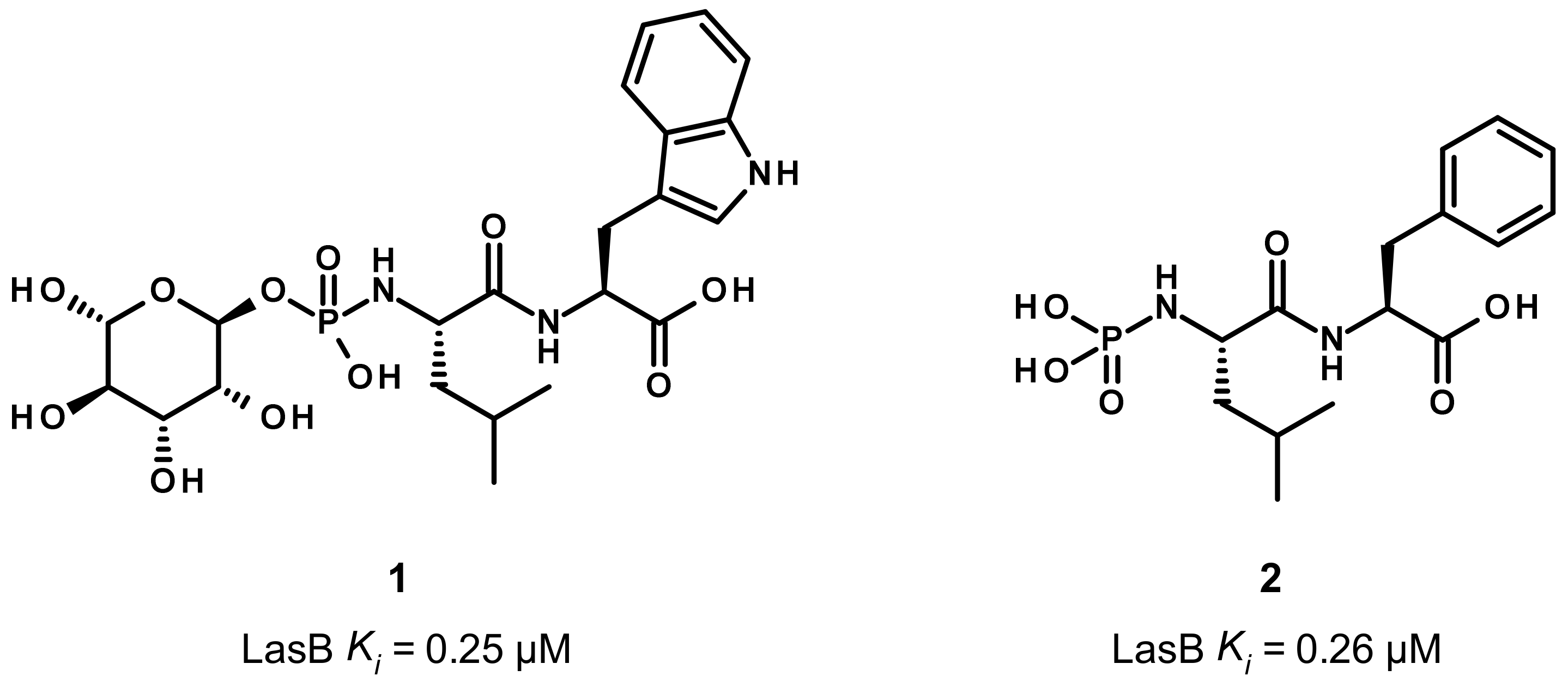
3.1. Phosphoramidate Compounds
3.2. Thiol Compounds
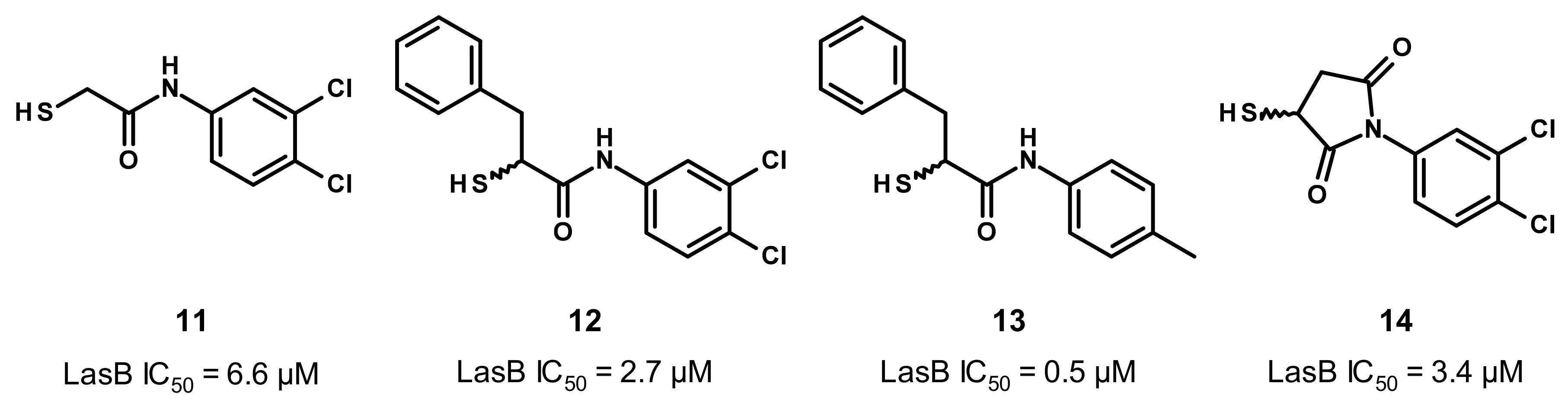
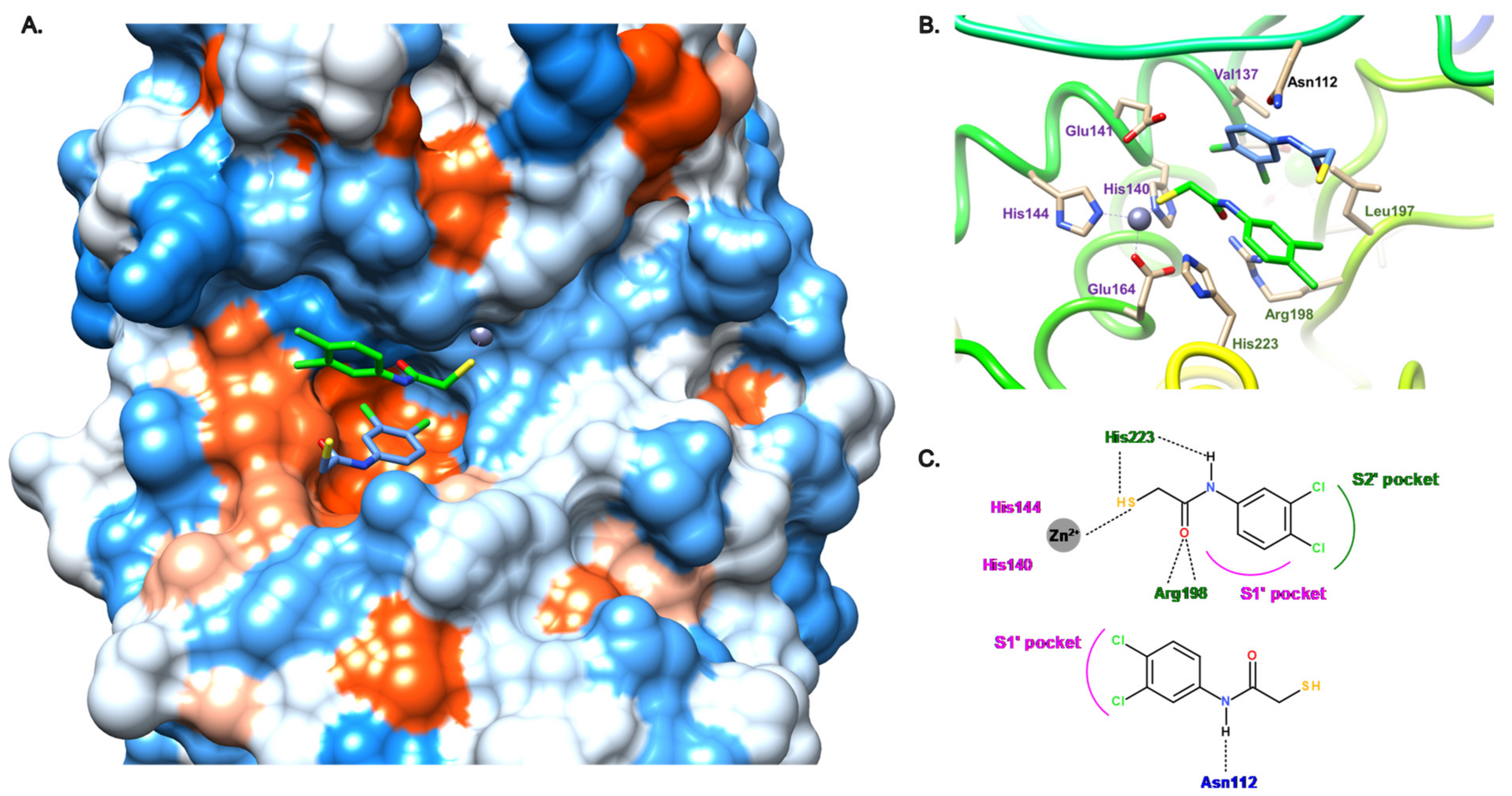
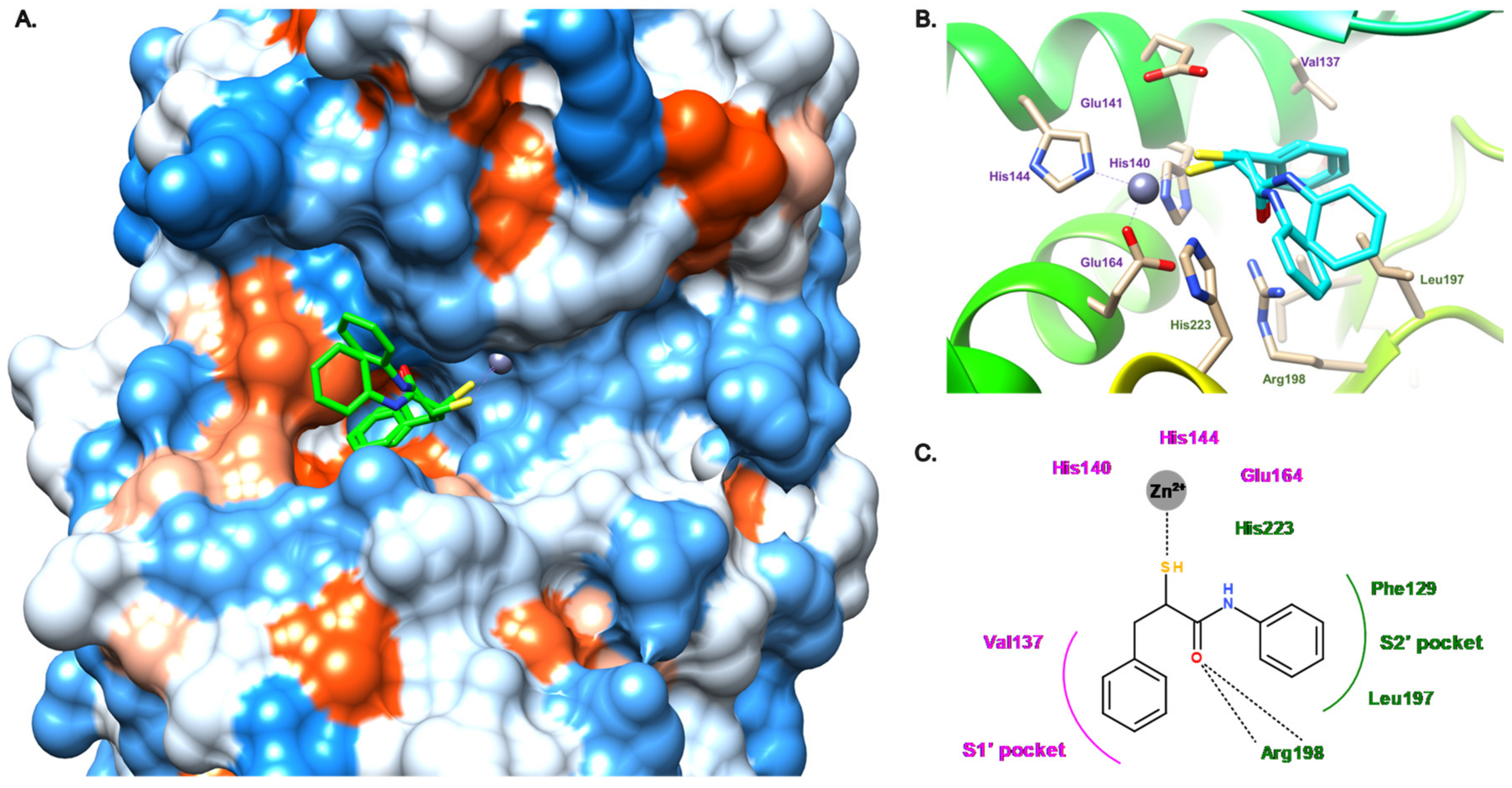
3.2.1. Peptides
3.2.2. Nonpeptidic Small-Molecule Inhibitors
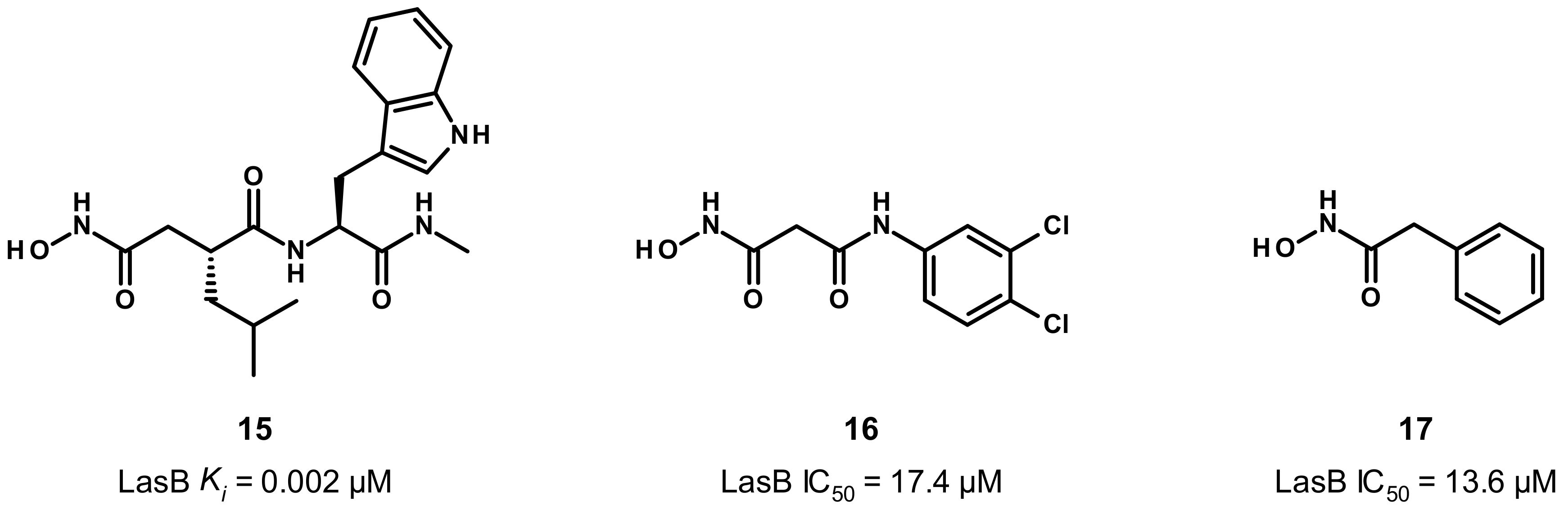
3.3. Hydroxamate Compounds
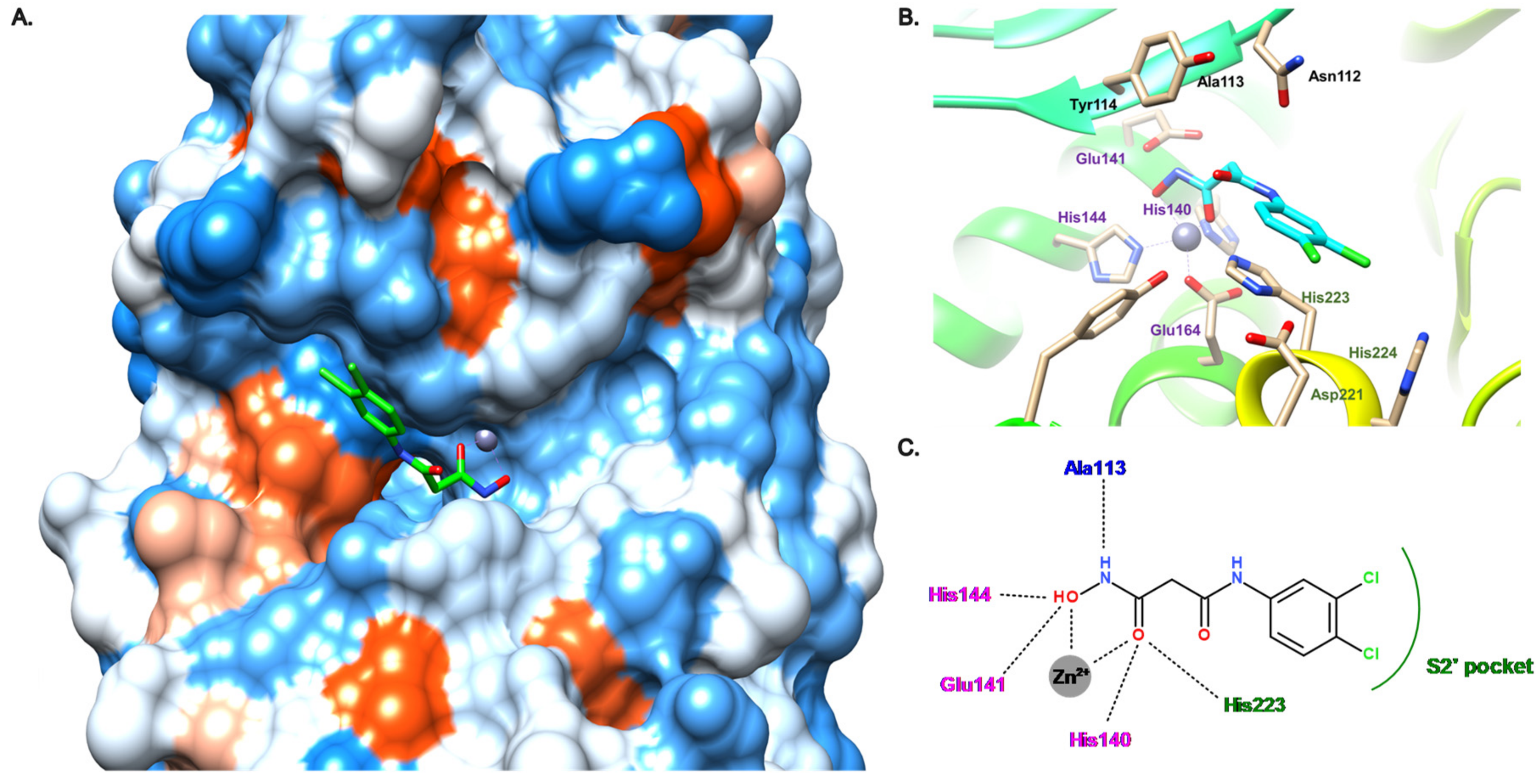
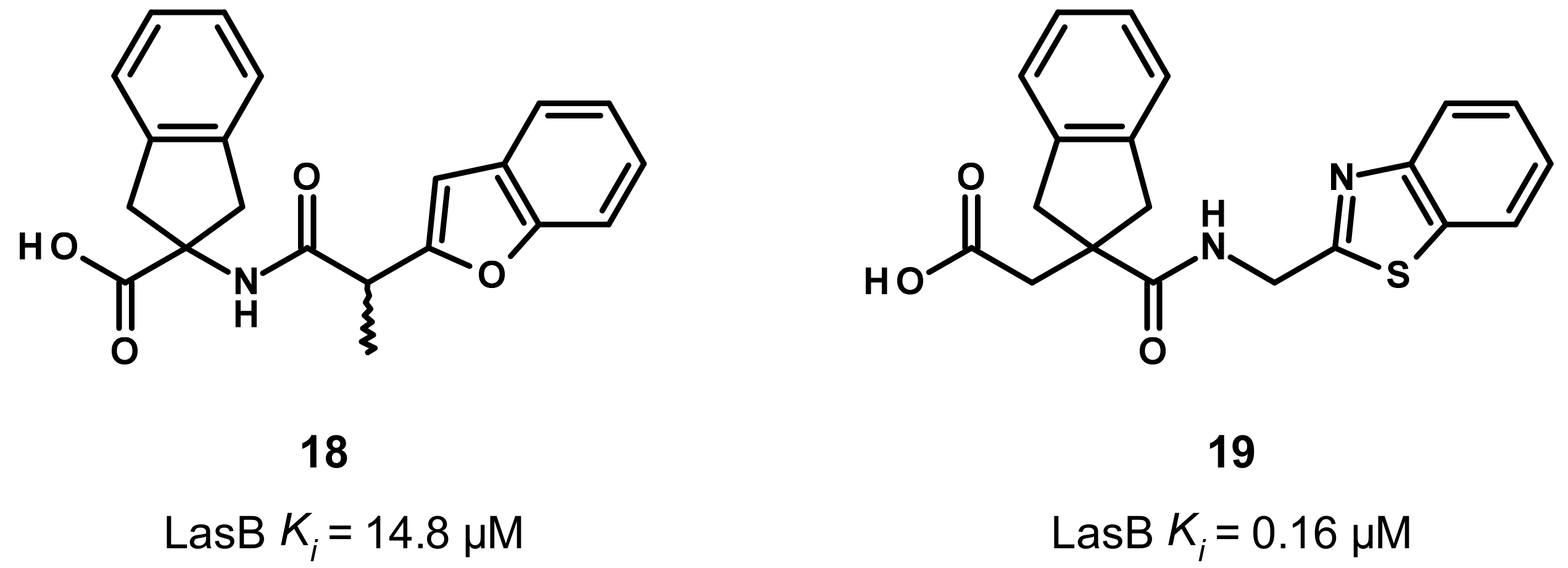
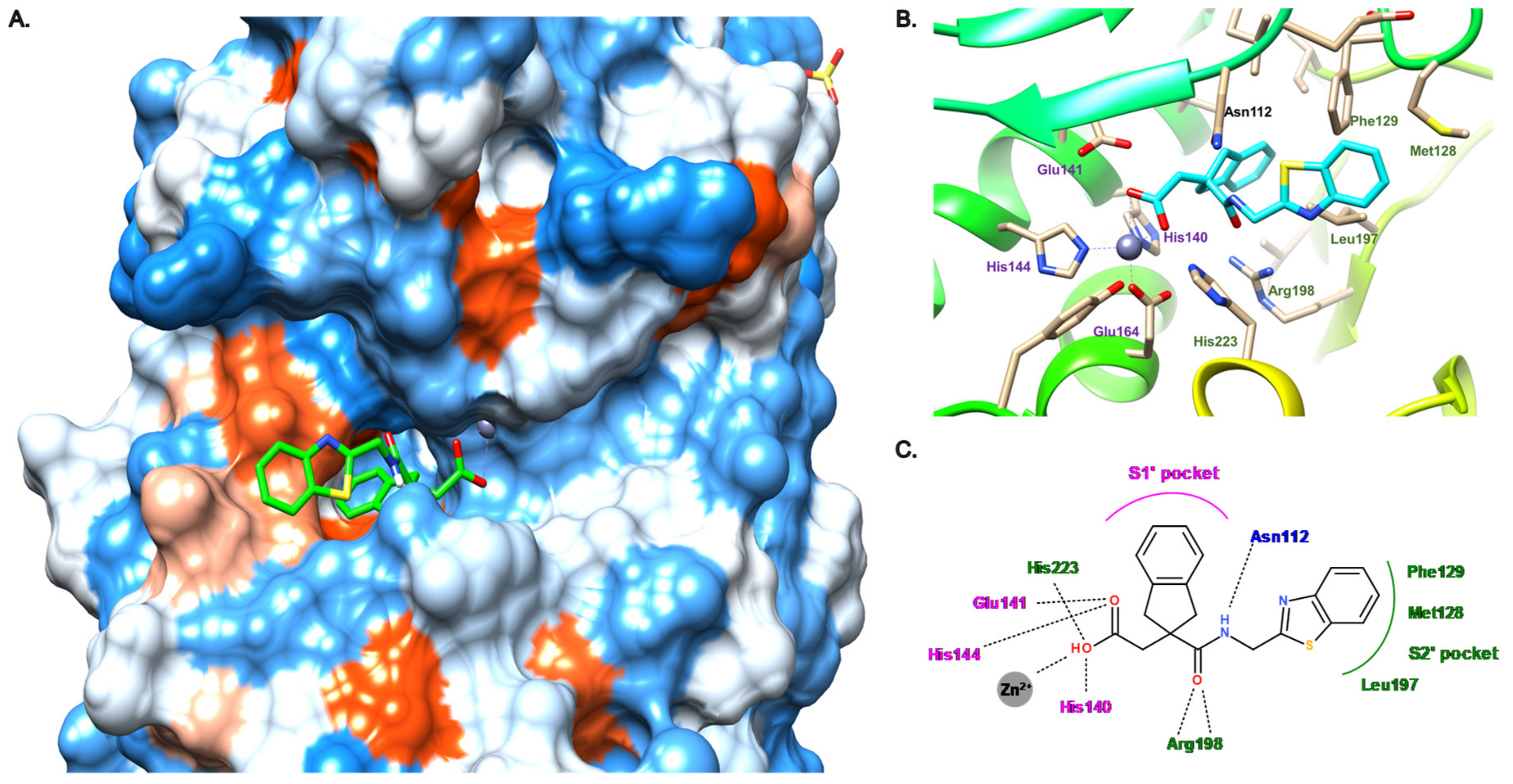
3.4. Carboxylate Compounds
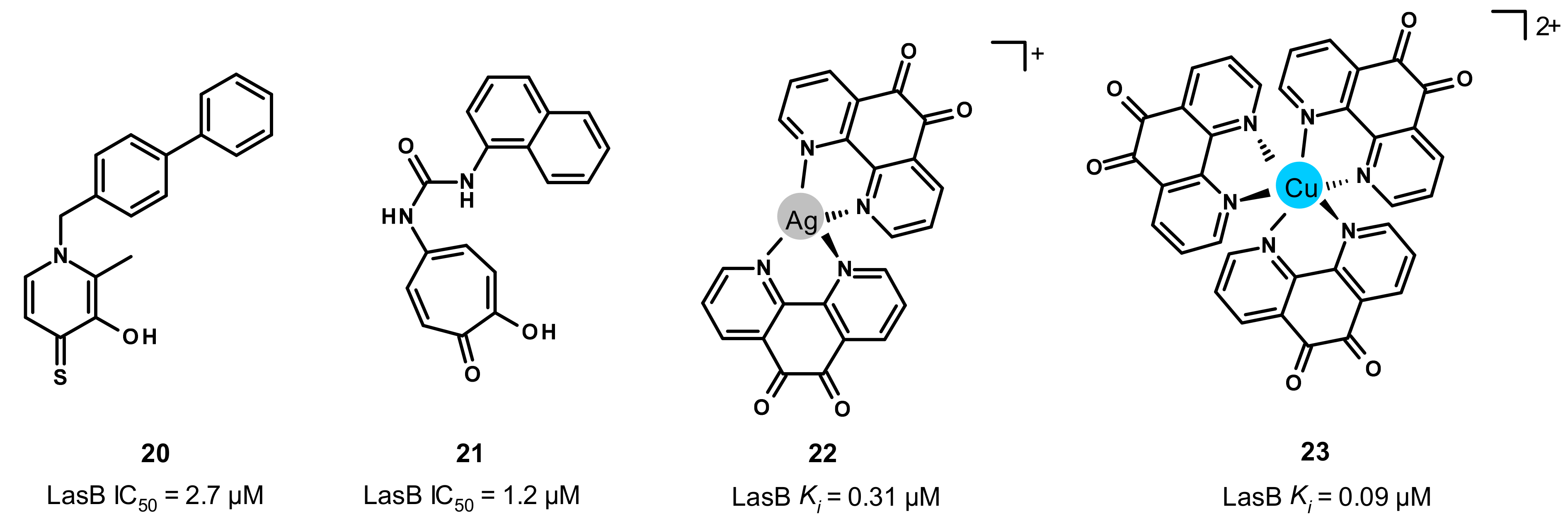
3.5. Other Compounds
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moradali, M.F.; Ghods, S.; Rehm, B.H.A. Pseudomonas aeruginosa Lifestyle: A Paradigm for Adaptation, Survival, and Persistence. Front. Cell. Infect. Microbiol. 2017, 7, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed. Available online: https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 21 July 2022).
- ECDC European Centre for Disease Prevention and Control. Antimicrobial Resistance in the EU/EEA (EARS-Net). In Annual Epidemiological Report 2019; ECDC European Centre for Disease Prevention and Control: Stockholm, Sweden, 2019. [Google Scholar]
- U.S. Department of Health and Human Services; CDC. CDC Antibiotic Resistance Threats in the United States, 2019; U.S. Department of Health and Human Services, CDC: Atlanta, GA, USA, 2019. [Google Scholar]
- Jurado-Martín, I.; Sainz-Mejías, M.; McClean, S. Pseudomonas aeruginosa: An Audacious Pathogen with an Adaptable Arsenal of Virulence Factors. Int. J. Mol. Sci. 2021, 22, 3128. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, S.; Bouffartigues, E.; Bodilis, J.; Maillot, O.; Lesouhaitier, O.; Feuilloley, M.G.J.; Orange, N.; Dufour, A.; Cornelis, P. Structure, function and regulation of Pseudomonas aeruginosa porins. FEMS Microbiol. Rev. 2017, 41, 698–722. [Google Scholar] [CrossRef] [PubMed]
- Sampedro, I.; Parales, R.E.; Krell, T.; Hill, J.E. Pseudomonas chemotaxis. FEMS Microbiol. Rev. 2015, 39, 17–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thi, M.T.T.; Wibowo, D.; Rehm, B.H. Pseudomonas aeruginosa Biofilms. Int. J. Mol. Sci. 2020, 21, E8671. [Google Scholar] [CrossRef]
- Ciofu, O.; Tolker-Nielsen, T. Tolerance and Resistance of Pseudomonas aeruginosa Biofilms to Antimicrobial Agents—How P. aeruginosa Can Escape Antibiotics. Front. Microbiol. 2019, 10, 913. [Google Scholar] [CrossRef] [Green Version]
- Lebrun, I.; Marques-Porto, R.; Pereira, A.S.; Pereira, A.; Perpetuo, E.A. Bacterial Toxins: An Overview on Bacterial Proteases and their Action as Virulence Factors. Mini Rev. Med. Chem. 2009, 9, 820–828. [Google Scholar] [CrossRef]
- Frees, D.; Brøndsted, L.; Ingmer, H. Bacterial Proteases and Virulence. In Regulated Proteolysis in Microorganisms; Subcellular Biochemistry; Dougan, D., Ed.; Springer: Dordrecht, The Netherlands, 2013; Volume 66, pp. 161–192. ISBN 978-94-007-5939-8. [Google Scholar]
- Escaich, S. Antivirulence as a new antibacterial approach for chemotherapy. Curr. Opin. Chem. Biol. 2008, 12, 400–408. [Google Scholar] [CrossRef]
- Rasko, D.A.; Sperandio, V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 2010, 9, 117–128. [Google Scholar] [CrossRef]
- Dickey, S.W.; Cheung, G.Y.C.; Otto, M. Different drugs for bad bugs: Antivirulence strategies in the age of antibiotic resistance. Nat. Rev. Drug Discov. 2017, 16, 457–471. [Google Scholar] [CrossRef]
- Mühlen, S.; Dersch, P. Anti-virulence Strategies to Target Bacterial Infections. In How to Overcome the Antibiotic Crisis; Current Topics in Microbiology and Immunology; Springer: Cham, Switzeland, 2015; Volume 398, pp. 147–183. [Google Scholar] [CrossRef]
- Wagner, S.; Sommer, R.; Hinsberger, S.; Lu, C.; Hartmann, R.W.; Empting, M.; Titz, A. Novel Strategies for the Treatment of Pseudomonas aeruginosa Infections. J. Med. Chem. 2016, 59, 5929–5969. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Mullally, C.; Haese, E.; Kibble, E.; McCluskey, N.; Mikucki, E.; Thai, V.; Stubbs, K.; Sarkar-Tyson, M.; Kahler, C. Anti-Virulence Therapeutic Approaches for Neisseria gonorrhoeae. Antibiotics 2021, 10, 103. [Google Scholar] [CrossRef] [PubMed]
- Denis, K.; Le Bris, M.; Le Guennec, L.; Barnier, J.-P.; Faure, C.; Gouge, A.; Bouzinba-Ségard, H.; Jamet, A.; Euphrasie, D.; Durel, B.; et al. Targeting Type IV pili as an antivirulence strategy against invasive meningococcal disease. Nat. Microbiol. 2019, 4, 972–984. [Google Scholar] [CrossRef] [PubMed]
- Kassegne, K.; Hu, W.; Ojcius, D.; Sun, D.; Ge, Y.; Zhao, J.; Yang, X.F.; Li, L.; Yan, J. Identification of Collagenase as a Critical Virulence Factor for Invasiveness and Transmission of Pathogenic Leptospira Species. J. Infect. Dis. 2014, 209, 1105–1115. [Google Scholar] [CrossRef] [Green Version]
- Schönauer, E.; Kany, A.M.; Haupenthal, J.; Hüsecken, K.; Hoppe, I.J.; Voos, K.; Yahiaoui, S.; Elsässer, B.; Ducho, C.; Brandstetter, H.; et al. Discovery of a Potent Inhibitor Class with High Selectivity toward Clostridial Collagenases. J. Am. Chem. Soc. 2017, 139, 12696–12703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstantinović, J.; Yahiaoui, S.; Alhayek, A.; Haupenthal, J.; Schönauer, E.; Andreas, A.; Kany, A.M.; Mueller, R.; Koehnke, J.; Berger, F.K.; et al. N-Aryl-3-mercaptosuccinimides as Antivirulence Agents Targeting Pseudomonas aeruginosa Elastase and Clostridium Collagenases. J. Med. Chem. 2020, 63, 8359–8368. [Google Scholar] [CrossRef] [PubMed]
- Voos, K.; Schönauer, E.; Alhayek, A.; Haupenthal, J.; Andreas, A.; Müller, R.; Hartmann, R.W.; Brandstetter, H.; Hirsch, A.K.H.; Ducho, C. Phosphonate as a Stable Zinc-Binding Group for “Pathoblocker” Inhibitors of Clostridial Collagenase H (ColH). ChemMedChem 2021, 16, 1257–1267. [Google Scholar] [CrossRef]
- Alhayek, A.; Khan, E.S.; Schönauer, E.; Däinghaus, T.; Shafiei, R.; Voos, K.; Han, M.K.L.; Ducho, C.; Posselt, G.; Wessler, S.; et al. Inhibition of Collagenase Q1 of Bacillus cereus as a Novel Antivirulence Strategy for the Treatment of Skin-Wound Infections. Adv. Ther. 2022, 5, 2100222. [Google Scholar] [CrossRef]
- Ford, C.A.; Hurford, I.M.; Cassat, J.E. Antivirulence Strategies for the Treatment of Staphylococcus aureus Infections: A Mini Review. Front. Microbiol. 2020, 11, 632706. [Google Scholar] [CrossRef]
- Sully, E.K.; Malachowa, N.; Elmore, B.O.; Alexander, S.M.; Femling, J.K.; Gray, B.M.; DeLeo, F.; Otto, M.; Cheung, A.L.; Edwards, B.S.; et al. Selective Chemical Inhibition of agr Quorum Sensing in Staphylococcus aureus Promotes Host Defense with Minimal Impact on Resistance. PLoS Pathog. 2014, 10, e1004174. [Google Scholar] [CrossRef]
- Duplantier, M.; Lohou, E.; Sonnet, P. Quorum Sensing Inhibitors to Quench P. aeruginosa Pathogenicity. Pharmaceuticals 2021, 14, 1262. [Google Scholar] [CrossRef] [PubMed]
- Galdino, A.C.M.; Branquinha, M.H.; Santos, A.L.S.; Viganor, L. Pseudomonas aeruginosa and Its Arsenal of Proteases: Weapons to Battle the Host. In Pathophysiological Aspects of Proteases; Chakraborti, S., Dhalla, N., Eds.; Springer: Singapore, 2017; pp. 381–397. [Google Scholar] [CrossRef] [Green Version]
- Everett, M.J.; Davies, D.T. Pseudomonas aeruginosa elastase (LasB) as a therapeutic target. Drug Discov. Today 2021, 26, 2108–2123. [Google Scholar] [CrossRef] [PubMed]
- Gi, M.; Jeong, J.; Lee, K.; Lee, K.-M.; Toyofuku, M.; Yong, D.E.; Yoon, S.S.; Choi, J.Y. A Drug-Repositioning Screening Identifies Pentetic Acid as a Potential Therapeutic Agent for Suppressing the Elastase-Mediated Virulence of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2014, 58, 7205–7214. [Google Scholar] [CrossRef] [Green Version]
- Kany, A.M.; Sikandar, A.; Haupenthal, J.; Yahiaoui, S.; Maurer, C.K.; Proschak, E.; Koehnke, J.; Hartmann, R.W. Binding Mode Characterization and Early in vivo Evaluation of Fragment-Like Thiols as Inhibitors of the Virulence Factor LasB from Pseudomonas aeruginosa. ACS Infect. Dis. 2018, 4, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Kaya, C.; Walter, I.; Yahiaoui, S.; Sikandar, A.; Alhayek, A.; Konstantinović, J.; Kany, A.M.; Haupenthal, J.; Köhnke, J.; Hartmann, R.W.; et al. Substrate-Inspired Fragment Merging and Growing Affords Efficacious LasB Inhibitors. Angew. Chem. Int. Ed. 2022, 61, e202112295. [Google Scholar] [CrossRef]
- Kaya, C.; Walter, I.; Alhayek, A.; Shafiei, R.; Jézéquel, G.; Andreas, A.; Konstantinović, J.; Schönauer, E.; Sikandar, A.; Haupenthal, J.; et al. Structure-Based Design of α-Substituted Mercaptoacetamides as Inhibitors of the Virulence Factor LasB from Pseudomonas aeruginosa. ACS Infect. Dis. 2022, 8, 1010–1021. [Google Scholar] [CrossRef]
- Galdino, A.C.M.; De Oliveira, M.P.; Ramalho, T.C.; De Castro, A.A.; Branquinha, M.H.; Santos, A.L.S. Anti-Virulence Strategy against the Multidrug-Resistant Bacterial Pathogen Pseudomonas aeruginosa: Pseudolysin (Elastase B) as a Potential Druggable Target. Curr. Protein Pept. Sci. 2019, 20, 471–487. [Google Scholar] [CrossRef]
- Thayer, M.M.; Flaherty, K.M.; McKay, D.B. Three-dimensional structure of the elastase of Pseudomonas aeruginosa at 1.5-A resolution. J. Biol. Chem. 1991, 266, 2864–2871. [Google Scholar] [CrossRef]
- Velázquez-Libera, J.L.; Murillo-López, J.A.; de la Torre, A.F.; Caballero, J. Structural Requirements of N-alpha-Mercaptoacetyl Dipeptide (NAMdP) Inhibitors of Pseudomonas aeruginosa Virulence Factor LasB: 3D-QSAR, Molecular Docking, and Interaction Fingerprint Studies. Int. J. Mol. Sci. 2019, 20, E6133. [Google Scholar] [CrossRef] [Green Version]
- Poncz, L.; Gerken, T.A.; Dearborn, D.G.; Grobelny, D.; Galardy, R.E. Inhibition of the elastase of Pseudomonas aeruginosa by N.alpha.-phosphoryl dipeptides and kinetics of spontaneous hydrolysis of the inhibitors. Biochemistry 1984, 23, 2766–2772. [Google Scholar] [CrossRef]
- Kessler, E.; Israel, M.; Landshman, N.; Chechick, A.; Blumberg, S. In Vitro Inhibition of Pseudomonas aeruginosa Elastase by Metal-Chelating Peptide Derivatives. Infect. Immun. 1982, 38, 716–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cathcart, G.R.; Gilmore, B.F.; Greer, B.; Harriott, P.; Walker, B. Inhibitor profiling of the Pseudomonas aeruginosa virulence factor LasB using N-alpha mercaptoamide template-based inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 6230–6232. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, G.R.A.; Quinn, D.; Greer, B.; Harriott, P.; Lynas, J.F.; Gilmore, B.F.; Walker, B. Novel Inhibitors of the Pseudomonas aeruginosa Virulence Factor LasB: A Potential Therapeutic Approach for the Attenuation of Virulence Mechanisms in Pseudomonal Infection. Antimicrob. Agents Chemother. 2011, 55, 2670–2678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, F.R.; Paterson, C.A.; Gray, R.D.; Wells, J.T. Inhibition of Pseudomonas aeruginosa elastase and Pseudomonas keratitis using a thiol-based peptide. Antimicrob. Agents Chemother. 1990, 34, 2065–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishino, N.; Powers, J.C. Pseudomonas Aeruginosa Elastase. Development of a New Substrate, Inhibitors, and an Affinity Ligand. J. Biol. Chem. 1980, 255, 3482–3486. [Google Scholar] [CrossRef]
- Zhu, J.; Cai, X.; Harris, T.L.; Gooyit, M.; Wood, M.; Lardy, M.; Janda, K.D. Disarming Pseudomonas aeruginosa Virulence Factor LasB by Leveraging a Caenorhabditis elegans Infection Model. Chem. Biol. 2015, 22, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Grobelny, D.; Poncz, L.; Galardy, R.E. Inhibition of human skin fibroblast collagenase, thermolysin, and Pseudomonas aeruginosa elastase by peptide hydroxamic acids. Biochemistry 1992, 31, 7152–7154. [Google Scholar] [CrossRef]
- Holmes, M.A.; Matthews, B.W. Binding of hydroxamic acid inhibitors to crystalline thermolysin suggests a pentacoordinate zinc intermediate in catalysis. Biochemistry 1981, 20, 6912–6920. [Google Scholar] [CrossRef]
- Kany, A.M.; Sikandar, A.; Yahiaoui, S.; Haupenthal, J.; Walter, I.; Empting, M.; Koehnke, J.; Hartmann, R.W. Tackling Pseudomonas aeruginosa Virulence by a Hydroxamic Acid-Based LasB Inhibitor. ACS Chem. Biol. 2018, 13, 2449–2455. [Google Scholar] [CrossRef]
- Garner, A.L.; Struss, A.K.; Fullagar, J.L.; Agrawal, A.; Moreno, A.Y.; Cohen, S.M.; Janda, K.D. 3-Hydroxy-1-alkyl-2-methylpyridine-4(1H)-thiones: Inhibition of the Pseudomonas aeruginosa Virulence Factor LasB. ACS Med. Chem. Lett. 2012, 3, 668–672. [Google Scholar] [CrossRef]
- Leiris, S.; Davies, D.T.; Sprynski, N.; Castandet, J.; Beyria, L.; Bodnarchuk, M.S.; Sutton, J.M.; Mullins, T.M.G.; Jones, M.W.; Forrest, A.K.; et al. Virtual Screening Approach to Identifying a Novel and Tractable Series of Pseudomonas aeruginosa Elastase Inhibitors. ACS Med. Chem. Lett. 2021, 12, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Fullagar, J.L.; Garner, A.L.; Struss, A.K.; Day, J.A.; Martin, D.P.; Yu, J.; Cai, X.; Janda, K.D.; Cohen, S.M. Antagonism of a zinc metalloprotease using a unique metal-chelating scaffold: Tropolones as inhibitors of P. aeruginosa elastase. Chem. Commun. 2013, 49, 3197–3199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galdino, A.C.M.; Viganor, L.; de Castro, A.A.; da Cunha, E.F.F.; Mello, T.P.; Mattos, L.M.; Pereira, M.D.; Hunt, M.C.; O’Shaughnessy, M.; Howe, O.; et al. Disarming Pseudomonas aeruginosa Virulence by the Inhibitory Action of 1,10-Phenanthroline-5,6-Dione-Based Compounds: Elastase B (LasB) as a Chemotherapeutic Target. Front. Microbiol. 2019, 10, 1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santajit, S.; Kong-Ngoen, T.; Chongsa-Nguan, M.; Boonyuen, U.; Pumirat, P.; Sookrung, N.; Chaicumpa, W.; Indrawattana, N. Human Single-Chain Antibodies That Neutralize Elastolytic Activity of Pseudomonas aeruginosa LasB. Pathogens 2021, 10, 765. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Sarma, D.K.; Shubham, S.; Kumawat, M.; Verma, V.; Nina, P.B.; Jp, D.; Kumar, S.; Singh, B.; Tiwari, R.R. Futuristic Non-antibiotic Therapies to Combat Antibiotic Resistance: A Review. Front. Microbiol. 2021, 12, 609459. [Google Scholar] [CrossRef] [PubMed]
- Mindt, B.C.; DiGiandomenico, A. Microbiome Modulation as a Novel Strategy to Treat and Prevent Respiratory Infections. Antibiotics 2022, 11, 474. [Google Scholar] [CrossRef] [PubMed]
- Plé, C.; Tam, H.-K.; Da Cruz, A.V.; Compagne, N.; Jiménez-Castellanos, J.-C.; Müller, R.T.; Pradel, E.; Foong, W.E.; Malloci, G.; Ballée, A.; et al. Pyridylpiperazine-based allosteric inhibitors of RND-type multidrug efflux pumps. Nat. Commun. 2022, 13, 115. [Google Scholar] [CrossRef]
- Yuan, Y.; Rosado-Lugo, J.D.; Zhang, Y.; Datta, P.; Sun, Y.; Cao, Y.; Banerjee, A.; Parhi, A.K. Evaluation of Heterocyclic Carboxamides as Potential Efflux Pump Inhibitors in Pseudomonas aeruginosa. Antibiotics 2021, 11, 30. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
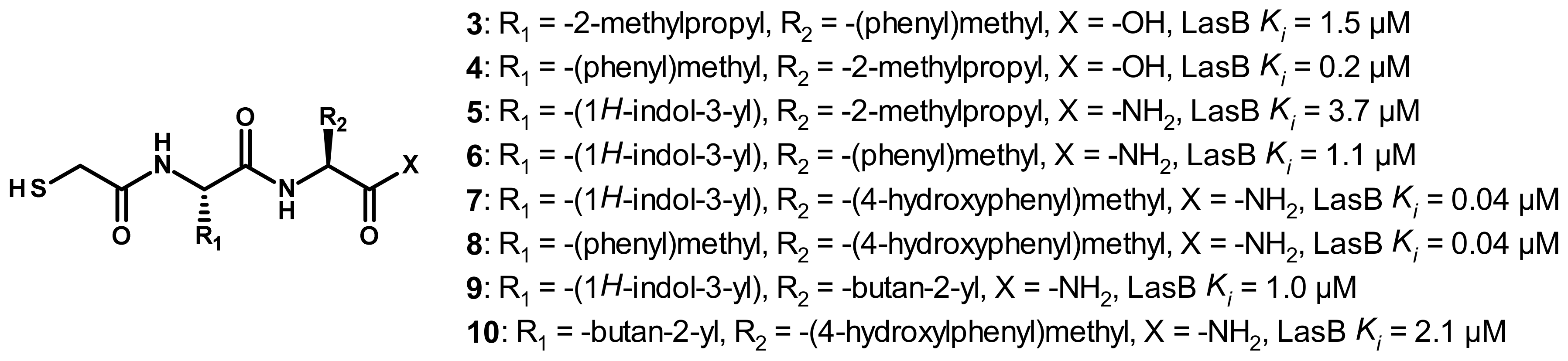{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camberlein, V.; Jézéquel, G.; Haupenthal, J.; Hirsch, A.K.H. The Structures and Binding Modes of Small-Molecule Inhibitors of Pseudomonas aeruginosa Elastase LasB. Antibiotics 2022, 11, 1060. https://doi.org/10.3390/antibiotics11081060
Camberlein V, Jézéquel G, Haupenthal J, Hirsch AKH. The Structures and Binding Modes of Small-Molecule Inhibitors of Pseudomonas aeruginosa Elastase LasB. Antibiotics. 2022; 11(8):1060. https://doi.org/10.3390/antibiotics11081060
Chicago/Turabian StyleCamberlein, Virgyl, Gwenaëlle Jézéquel, Jörg Haupenthal, and Anna K. H. Hirsch. 2022. "The Structures and Binding Modes of Small-Molecule Inhibitors of Pseudomonas aeruginosa Elastase LasB" Antibiotics 11, no. 8: 1060. https://doi.org/10.3390/antibiotics11081060