
Clonal Lineages and Virulence Factors of Carbapenem Resistant E. coli in Alameda County, California, 2017–2019
 , , , , ,
, , , , , 
Abstract
:1. Introduction
2. Results
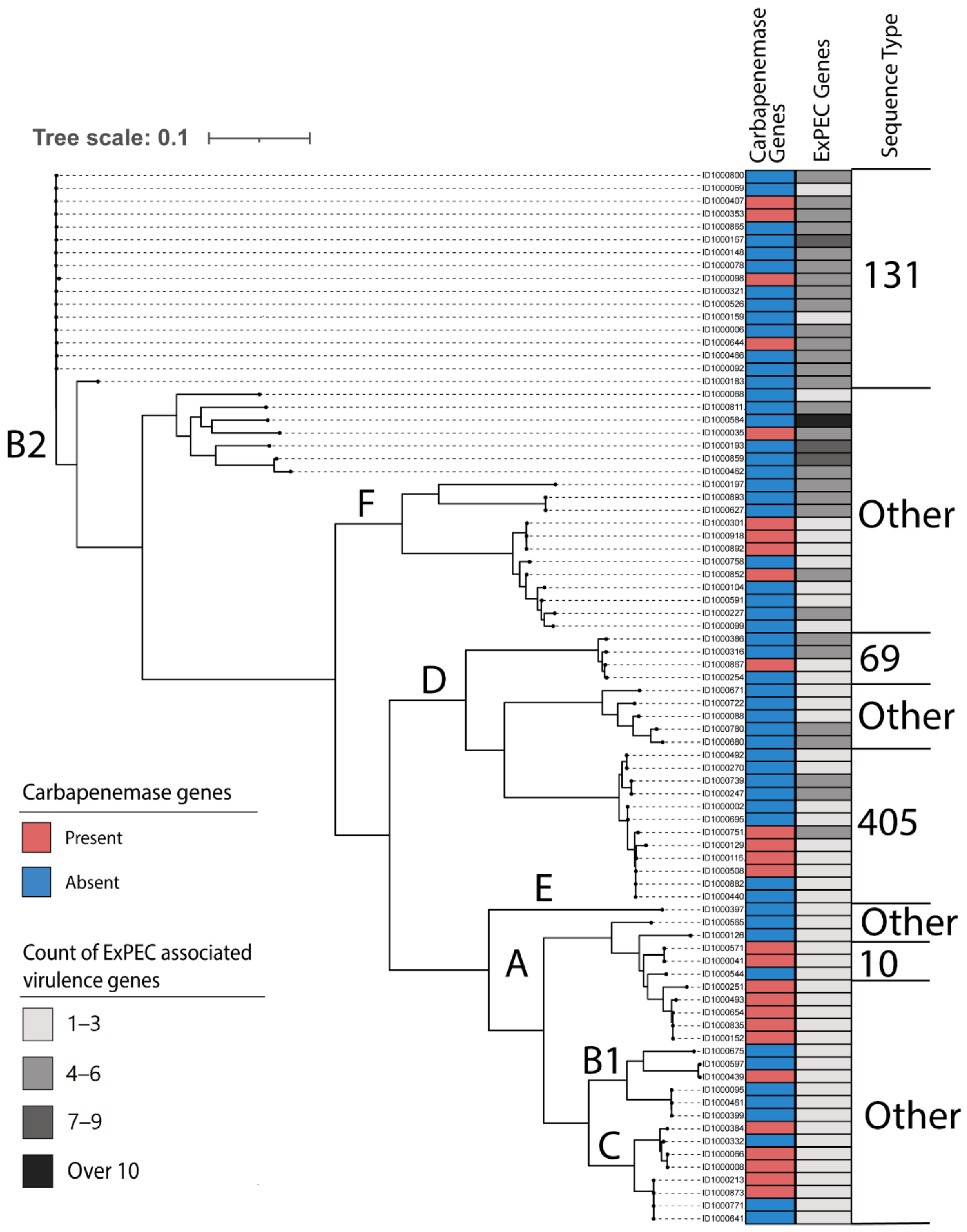
2.1. Characteristics of Clustering, Sequence Types, and Subtypes
2.2. Distribution of Antibiotic Resistance Genes
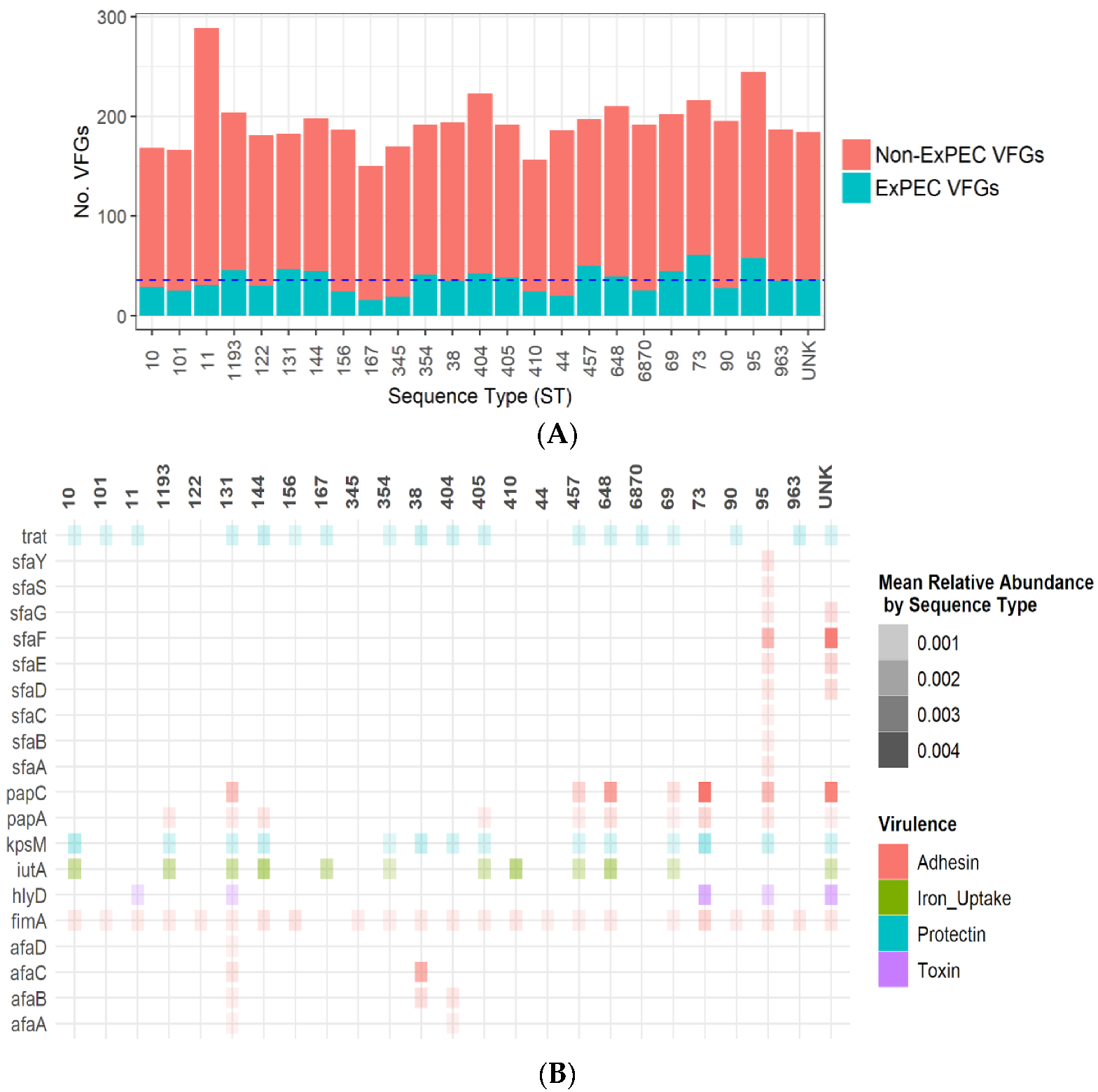
2.3. Distribution of Virulence Genes
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Susceptibility Testing
5.2. Whole Genome Sequencing
5.3. Genotypic Analysis
5.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Logan, L.K.; Weinstein, R.A. The Epidemiology of Carbapenem-Resistant Enterobacteriaceae: The Impact and Evolution of a Global Menace. J. Infect. Dis. 2017, 215, S28–S36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guh, A.Y.; Limbago, B.M.; Kallen, A.J. Epidemiology and Prevention of Carbapenem-Resistant Enterobacteriaceae in the United States. Expert Rev. Anti Infect. Ther. 2014, 12, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Rahal, J.J.; Urban, C.; Horn, D.; Freeman, K.; Segal-Maurer, S.; Maurer, J.; Mariano, N.; Marks, S.; Burns, J.M.; Dominick, D.; et al. Class Restriction of Cephalosporin Use to Control Total Cephalosporin Resistance in Nosocomial Klebsiella. JAMA 1998, 280, 1233–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meletis, G. Carbapenem Resistance: Overview of the Problem and Future Perspectives. Ther. Adv. Infect. Dis. 2016, 3, 15–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- MacKinnon, M.C.; Sargeant, J.M.; Pearl, D.L.; Reid-Smith, R.J.; Carson, C.A.; Parmley, E.J.; McEwen, S.A. Evaluation of the Health and Healthcare System Burden Due to Antimicrobial-Resistant Escherichia coli Infections in Humans: A Systematic Review and Meta-Analysis. Antimicrob. Resist. Infect. Control 2020, 9, 200. [Google Scholar] [CrossRef]
- Johnson, T.J.; Nolan, L.K. Pathogenomics of the Virulence Plasmids of Escherichia coli. Microbiol. Mol. Biol. Rev. 2009, 73, 750–774. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.R.; Johnston, B.; Clabots, C.; Kuskowski, M.A.; Castanheira, M. Escherichia coli Sequence Type ST131 as the Major Cause of Serious Multidrug-Resistant E. Coli Infections in the United States. Clin. Infect. Dis. 2010, 51, 286–294. [Google Scholar] [CrossRef] [Green Version]
- Biran, D.; Ron, E.Z. Extraintestinal Pathogenic Escherichia coli. In Escherichia coli, a Versatile Pathogen; Frankel, G., Ron, E.Z., Eds.; Current Topics in Microbiology and Immunology; Springer International Publishing: Cham, Germany, 2018; pp. 149–161. ISBN 978-3-319-99664-6. [Google Scholar]
- Sarowska, J.; Futoma-Koloch, B.; Jama-Kmiecik, A.; Frej-Madrzak, M.; Ksiazczyk, M.; Bugla-Ploskonska, G.; Choroszy-Krol, I. Virulence Factors, Prevalence and Potential Transmission of Extraintestinal Pathogenic Escherichia coli Isolated from Different Sources: Recent Reports. Gut Pathog. 2019, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Riley, L.W. Pandemic Lineages of Extraintestinal Pathogenic Escherichia coli. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2014, 20, 380–390. [Google Scholar] [CrossRef]
- Manges, A.R.; Geum, H.M.; Guo, A.; Edens, T.J.; Fibke, C.D.; Pitout, J.D.D. Global Extraintestinal Pathogenic Escherichia coli (ExPEC) Lineages. Clin. Microbiol. Rev. 2019, 32, 25. [Google Scholar] [CrossRef]
- Pitout, J.D.D.; DeVinney, R. Escherichia coli ST131: A Multidrug-Resistant Clone Primed for Global Domination. F1000Research 2017, 6, 195. [Google Scholar] [CrossRef] [Green Version]
- Pitout, J.D.; Laupland, K.B. Extended-Spectrum β-Lactamase-Producing Enterobacteriaceae: An Emerging Public-Health Concern. Lancet Infect. Dis. 2008, 8, 159–166. [Google Scholar] [CrossRef]
- Alhashash, F.; Weston, V.; Diggle, M.; McNally, A. Multidrug-Resistant Escherichia coli Bacteremia. Emerg. Infect. Dis. 2013, 19, 1699–1701. [Google Scholar] [CrossRef]
- El Salabi, A.; Walsh, T.R.; Chouchani, C. Extended Spectrum β-Lactamases, Carbapenemases and Mobile Genetic Elements Responsible for Antibiotics Resistance in Gram-Negative Bacteria. Crit. Rev. Microbiol. 2013, 39, 113–122. [Google Scholar] [CrossRef]
- Suay-García, B.; Pérez-Gracia, M.T. Present and Future of Carbapenem-Resistant Enterobacteriaceae (CRE) Infections. Antibiotics 2019, 8, 122. [Google Scholar] [CrossRef] [Green Version]
- Codjoe, F.S.; Donkor, E.S. Carbapenem Resistance: A Review. Med. Sci. 2017, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.R.; Russo, T.A. Extraintestinal Pathogenic Escherichia coli: “The Other Bad E. Coli”. J. Lab. Clin. Med. 2002, 139, 155–162. [Google Scholar] [CrossRef]
- Kaper, J.B.; Nataro, J.P.; Mobley, H.L.T. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004, 2, 123–140. [Google Scholar] [CrossRef]
- Arnold, B.J.; Huang, I.-T.; Hanage, W.P. Horizontal Gene Transfer and Adaptive Evolution in Bacteria. Nat. Rev. Microbiol. 2022, 20, 206–218. [Google Scholar] [CrossRef]
- Brito, I.L. Examining Horizontal Gene Transfer in Microbial Communities. Nat. Rev. Microbiol. 2021, 19, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Bibi, E. MdfA, an Escherichia coli Multidrug Resistance Protein with an Extraordinarily Broad Spectrum of Drug Recognition. J. Bacteriol. 1997, 179, 2274–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezatofighi, S.E.; Mirzarazi, M.; Salehi, M. Virulence Genes and Phylogenetic Groups of Uropathogenic Escherichia coli Isolates from Patients with Urinary Tract Infection and Uninfected Control Subjects: A Case-Control Study. BMC Infect. Dis. 2021, 21, 361. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, R.H.S.; Dias, R.C.B.; Orsi, H.; de Lira, D.R.P.; Vieira, M.A.; dos Santos, L.F.; Ferreira, A.M.; Rall, V.L.M.; Mondelli, A.L.; Gomes, T.A.T.; et al. Characterization of Uropathogenic Escherichia Coli Reveals Hybrid Isolates of Uropathogenic and Diarrheagenic (UPEC/DEC) E. coli. Microorganisms 2022, 10, 645. [Google Scholar] [CrossRef] [PubMed]
- Gultekin, E.O.; Ulger, S.T.; Delialioğlu, N. Distribution of Pathogenicity Island Markers and Virulence Factors Genes of Extraintestinal Pathogenic Escherichia coli Isolates. Jundishapur J. Microbiol. 2022, 15, e121044. [Google Scholar] [CrossRef]
- Koga, V.L.; Tomazetto, G.; Cyoia, P.S.; Neves, M.S.; Vidotto, M.C.; Nakazato, G.; Kobayashi, R.K.T. Molecular Screening of Virulence Genes in Extraintestinal Pathogenic Escherichia coli Isolated from Human Blood Culture in Brazil. BioMed Res. Int. 2014, 2014, e465054. [Google Scholar] [CrossRef] [Green Version]
- Pitout, J.D. Extraintestinal Pathogenic Escherichia coli: An Update on Antimicrobial Resistance, Laboratory Diagnosis and Treatment. Expert Rev. Anti Infect. Ther. 2012, 10, 1165–1176. [Google Scholar] [CrossRef]
- Diard, M.; Garry, L.; Selva, M.; Mosser, T.; Denamur, E.; Matic, I. Pathogenicity-Associated Islands in Extraintestinal Pathogenic Escherichia coli Are Fitness Elements Involved in Intestinal Colonization. J. Bacteriol. 2010, 192, 4885–4893. [Google Scholar] [CrossRef] [Green Version]
- Dobrindt, U.; Chowdary, M.G.; Krumbholz, G.; Hacker, J. Genome Dynamics and Its Impact on Evolution of Escherichia coli. Med. Microbiol. Immunol. 2010, 199, 145–154. [Google Scholar] [CrossRef]
- Johnson, J.R.; Delavari, P.; Kuskowski, M.; Stell, A.L. Phylogenetic Distribution of Extraintestinal Virulence-Associated Traits in Escherichia coli. J. Infect. Dis. 2001, 183, 78–88. [Google Scholar] [CrossRef]
- Ludden, C.; Decano, A.G.; Jamrozy, D.; Pickard, D.; Morris, D.; Parkhill, J.; Peacock, S.J.; Cormican, M.; Downing, T. Genomic Surveillance of Escherichia coli ST131 Identifies Local Expansion and Serial Replacement of Subclones. Microb. Genom. 2020, 6, mgen000352. [Google Scholar] [CrossRef]
- Senchyna, F.; Gaur, R.L.; Sandlund, J.; Truong, C.; Tremintin, G.; Kültz, D.; Gomez, C.A.; Tamburini, F.B.; Andermann, T.; Bhatt, A.; et al. Diversity of Resistance Mechanisms in Carbapenem-Resistant Enterobacteriaceae at a Health Care System in Northern California, from 2013 to 2016. Diagn. Microbiol. Infect. Dis. 2019, 93, 250–257. [Google Scholar] [CrossRef]
- Black, C.A.; So, W.; Dallas, S.S.; Gawrys, G.; Benavides, R.; Aguilar, S.; Chen, C.-J.; Shurko, J.F.; Lee, G.C. Predominance of Non-Carbapenemase Producing Carbapenem-Resistant Enterobacterales in South Texas. Front. Microbiol. 2021, 11, 623574. [Google Scholar] [CrossRef]
- Zou, H.; Xiong, S.-J.; Lin, Q.-X.; Wu, M.-L.; Niu, S.-Q.; Huang, S.-F. CP-CRE/Non-CP-CRE Stratification and CRE Resistance Mechanism Determination Help in Better Managing CRE Bacteremia Using Ceftazidime–Avibactam And Aztreonam–Avibactam. Infect. Drug Resist. 2019, 12, 3017–3027. [Google Scholar] [CrossRef] [Green Version]
- Patidar, N.; Vyas, N.; Sharma, S.; Sharma, B. Phenotypic Detection of Carbapenemase Production in Carbapenem-Resistant Enterobacteriaceae by Modified Hodge Test and Modified Strip Carba NP Test. J. Lab. Physicians 2021, 13, 14–21. [Google Scholar] [CrossRef]
- Seyedjavadi, S.S.; Goudarzi, M.; Sabzehali, F. Relation between BlaTEM, BlaSHV and BlaCTX-M Genes and Acute Urinary Tract Infections. J. Acute Dis. 2016, 5, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Braz, V.S.; Melchior, K.; Moreira, C.G. Escherichia coli as a Multifaceted Pathogenic and Versatile Bacterium. Front. Cell. Infect. Microbiol. 2020, 10, 548492. [Google Scholar] [CrossRef]
- Bevan, E.R.; Jones, A.M.; Hawkey, P.M. Global Epidemiology of CTX-M β-Lactamases: Temporal and Geographical Shifts in Genotype. J. Antimicrob. Chemother. 2017, 72, 2145–2155. [Google Scholar] [CrossRef] [Green Version]
- Petty, N.K.; Zakour, N.L.B.; Stanton-Cook, M.; Skippington, E.; Totsika, M.; Forde, B.M.; Phan, M.-D.; Moriel, D.G.; Peters, K.M.; Davies, M.; et al. Global Dissemination of a Multidrug Resistant Escherichia coli Clone. Proc. Natl. Acad. Sci. USA 2014, 111, 5694–5699. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Sun, Q.; Chen, S.; Ding, S.; Zhang, R.; Zhu, K. Genomic and Phenotypic Analysis of Persistent Carbapenem-Resistant Klebsiella pneumoniae Isolates from a 5-Year Hospitalized Patient. Microb. Drug Resist. 2020, 27, 1117–1125. [Google Scholar] [CrossRef]
- Livermore, D.M.; Day, M.; Cleary, P.; Hopkins, K.L.; Toleman, M.A.; Wareham, D.W.; Wiuff, C.; Doumith, M.; Woodford, N. OXA-1 β-Lactamase and Non-Susceptibility to Penicillin/β-Lactamase Inhibitor Combinations among ESBL-Producing Escherichia coli. J. Antimicrob. Chemother. 2019, 74, 326–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alousi, S.; Salloum, T.; Arabaghian, H.; Matar, G.M.; Araj, G.F.; Tokajian, S.T. Genomic Characterization of MDR Escherichia coli Harboring BlaOXA-48 on the IncL/M-Type Plasmid Isolated from Blood Stream Infection. BioMed Res. Int. 2018, 2018, 3036143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielaszewska, M.; Dobrindt, U.; Gärtner, J.; Gallitz, I.; Hacker, J.; Karch, H.; Müller, D.; Schubert, S.; Alexander Schmidt, M.; Sorsa, L.J.; et al. Aspects of Genome Plasticity in Pathogenic Escherichia coli. Int. J. Med. Microbiol. 2007, 297, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.A.; Shin, S.; Chung, J.W.; Kim, K.J.; Elliott, S.; Wang, Y.; Kim, K.S. Outer Membrane Protein A and Cytotoxic Necrotizing Factor-1 Use Diverse Signaling Mechanisms for Escherichia coli K1 Invasion of Human Brain Microvascular Endothelial Cells. Microb. Pathog. 2003, 35, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.R.; Johnston, B.D.; Porter, S.; Thuras, P.; Aziz, M.; Price, L.B. Accessory Traits and Phylogenetic Background Predict Escherichia coli Extraintestinal Virulence Better Than Does Ecological Source. J. Infect. Dis. 2018, 219, 121–132. [Google Scholar] [CrossRef]
- Hung, W.-T.; Cheng, M.-F.; Tseng, F.-C.; Chen, Y.-S.; Shin-Jung Lee, S.; Chang, T.-H.; Lin, H.-H.; Hung, C.-H.; Wang, J.-L. Bloodstream Infection with Extended-Spectrum Beta-Lactamase-Producing Escherichia coli: The Role of Virulence Genes. J. Microbiol. Immunol. Infect. Wei Mian Yu Gan Ran Za Zhi 2019, 52, 947–955. [Google Scholar] [CrossRef]
- Bingen, E.; Picard, B.; Brahimi, N.; Mathy, S.; Desjardins, P.; Elion, J.; Denamur, E. Phylogenetic Analysis of Escherichia coli Strains Causing Neonatal Meningitis Suggests Horizontal Gene Transfer from a Predominant Pool of Highly Virulent B2 Group Strains. J. Infect. Dis. 1998, 177, 642–650. [Google Scholar] [CrossRef] [Green Version]
- Boyd, E.F.; Hartl, D.L. Chromosomal Regions Specific to Pathogenic Isolates of Escherichia coli Have a Phylogenetically Clustered Distribution. J. Bacteriol. 1998, 180, 1159–1165. [Google Scholar] [CrossRef] [Green Version]
- Lecointre, G.; Rachdi, L.; Darlu, P.; Denamur, E. Escherichia coli Molecular Phylogeny Using the Incongruence Length Difference Test. Mol. Biol. Evol. 1998, 15, 1685–1695. [Google Scholar] [CrossRef] [Green Version]
- Spurbeck, R.R.; Dinh, P.C.; Walk, S.T.; Stapleton, A.E.; Hooton, T.M.; Nolan, L.K.; Kim, K.S.; Johnson, J.R.; Mobley, H.L.T. Escherichia coli Isolates That Carry Vat, FyuA, ChuA, and YfcV Efficiently Colonize the Urinary Tract. Infect. Immun. 2012, 80, 4115–4122. [Google Scholar] [CrossRef]
- Huang, S.H.; Wan, Z.S.; Chen, Y.H.; Jong, A.Y.; Kim, K.S. Further Characterization of Escherichia coli Brain Microvascular Endothelial Cell Invasion Gene IbeA by Deletion, Complementation, and Protein Expression. J. Infect. Dis. 2001, 183, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Logue, C.M.; Doetkott, C.; Mangiamele, P.; Wannemuehler, Y.M.; Johnson, T.J.; Tivendale, K.A.; Li, G.; Sherwood, J.S.; Nolan, L.K. Genotypic and Phenotypic Traits That Distinguish Neonatal Meningitis-Associated Escherichia coli from Fecal E. coli Isolates of Healthy Human Hosts. Appl. Environ. Microbiol. 2012, 78, 5824–5830. [Google Scholar] [CrossRef] [Green Version]
- Wijetunge, D.S.S.; Gongati, S.; DebRoy, C.; Kim, K.S.; Couraud, P.O.; Romero, I.A.; Weksler, B.; Kariyawasam, S. Characterizing the Pathotype of Neonatal Meningitis Causing Escherichia coli (NMEC). BMC Microbiol. 2015, 15, 211. [Google Scholar] [CrossRef] [Green Version]
- Le Gall, T.; Clermont, O.; Gouriou, S.; Picard, B.; Nassif, X.; Denamur, E.; Tenaillon, O. Extraintestinal Virulence Is a Coincidental By-Product of Commensalism in B2 Phylogenetic Group Escherichia coli Strains. Mol. Biol. Evol. 2007, 24, 2373–2384. [Google Scholar] [CrossRef] [Green Version]
- Jerse, A.E.; Kaper, J.B. The Eae Gene of Enteropathogenic Escherichia Coli Encodes a 94-Kilodalton Membrane Protein, the Expression of Which Is Influenced by the EAF Plasmid. Infect. Immun. 1991, 59, 4302–4309. [Google Scholar] [CrossRef] [Green Version]
- Sheikh, A.; Luo, Q.; Roy, K.; Shabaan, S.; Kumar, P.; Qadri, F.; Fleckenstein, J.M. Contribution of the Highly Conserved EaeH Surface Protein to Enterotoxigenic Escherichia coli Pathogenesis. Infect. Immun. 2014, 82, 3657–3666. [Google Scholar] [CrossRef] [Green Version]
- Riley, L.W. Distinguishing Pathovars from Nonpathovars: Escherichia coli. Microbiol. Spectr. 2020, 8, 1–23. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving Bacterial Genome Assemblies from Short and Long Sequencing Reads. PLOS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Beghain, J.; Bridier-Nahmias, A.; Le Nagard, H.; Denamur, E.; Clermont, O. ClermonTyping: An Easy-to-Use and Accurate in Silico Method for Escherichia Genus Strain Phylotyping. Microb. Genom. 2018, 4, e000192. [Google Scholar] [CrossRef]
- Clermont, O.; Dixit, O.V.A.; Vangchhia, B.; Condamine, B.; Dion, S.; Bridier-Nahmias, A.; Denamur, E.; Gordon, D. Characterization and Rapid Identification of Phylogroup G in Escherichia coli, a Lineage with High Virulence and Antibiotic Resistance Potential. Environ. Microbiol. 2019, 21, 3107–3117. [Google Scholar] [CrossRef]
- Jolley, K.A.; Maiden, M.C. BIGSdb: Scalable Analysis of Bacterial Genome Variation at the Population Level. BMC Bioinform. 2010, 11, 595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roer, L.; Tchesnokova, V.; Allesøe, R.; Muradova, M.; Chattopadhyay, S.; Ahrenfeldt, J.; Thomsen, M.C.F.; Lund, O.; Hansen, F.; Hammerum, A.M.; et al. Development of a Web Tool for Escherichia Coli Subtyping Based on FimH Alleles. J. Clin. Microbiol. 2017, 55, 2538–2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, T. Tseemann/Abricate 2021. Available online: https://github.com/tseemann/abricate (accessed on 27 August 2022).
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for Predictions of Phenotypes from Genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A Reference Database for Bacterial Virulence Factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Snippy: Fast Bacterial Variant Calling from NGS Reads, 2022. Available online: https://github.com/tseemann/snippy (accessed on 27 August 2022).
- Seemann, T. Snp-Dists: Pairwise SNP Distance Matrix from a FASTA Sequence Alignment 2022. Available online: https://github.com/tseemann/snp-dists (accessed on 1 August 2022).
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL): An Online Tool for Phylogenetic Tree Display and Annotation. Bioinforma. Oxf. Engl. 2007, 23, 127–128. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Springer International Publishing: Imprint; Springer: Cham, Germany, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Wickham, H.; François, R.; Henry, L.; Müller, K. Dplyr: A Grammar of Data Manipulation. R Package Version 1.0.5. 2021. Available online: https://dplyr.tidyverse.org (accessed on 1 August 2022).
- Wickham, H. Stringr: Simple, Consistent Wrappers for Common String Operations. R Package Version 1.4.0. 2019. Available online: https://stringr.tidyverse.org (accessed on 1 August 2022).
- Wickham, H. Tidyr: Tidy Messy Data. R Package Version 1.1.2. 2020. Available online: https://github.com/tidyverse/tidyr (accessed on 27 August 2022).
- Xie, Y. Knitr: A General-Purpose Package for Dynamic Report Generation in R. R Package Version 1.30. Available online: https://yihui.org/knitr/ (accessed on 1 August 2022).
- Zhu, H. KableExtra: Construct Complex Table with “kable” and Pipe Syntax. R Package Version 1.3.1. 2020. Available online: https://haozhu233.github.io/kableExtra/ (accessed on 1 August 2022).
- Kaplan, J. FastDummies: Fast Creation of Dummy (Binary) Columns and Rows from Categorical Variables. R Package Version 1.6.3. 2020. Available online: https://github.com/jacobkap/fastDummies (accessed on 1 August 2022).



{kind=link}
{kind=link}
| Phylogroup | fimH Type | Sequence Type | Prevalence (n = 82) |
|---|---|---|---|
| A | fimH27 | ST10 | 1 |
| fimH54 | ST10 | 2 | |
| ST44 | 1 | ||
| ST UNK1 a | 1 | ||
| fimH Absent | ST167 | 5 | |
| B1 | fimH31 | ST345 | 3 |
| fimH38 | ST156 | 2 | |
| fimH191 | ST101 | 1 | |
| B2 | fimH10 | ST73 | 1 |
| fimH18 | ST95 | 1 | |
| fimH21 | ST122 | 1 | |
| fimH27 | ST404 | 1 | |
| fimH30 | ST131 | 16 | |
| fimH41 | ST131 | 1 | |
| fimH54 | ST144 | 1 | |
| fimH64 | ST1193 | 1 | |
| fimH120 | ST UNK2 b | 1 | |
| C | fimH24 | ST410 | 4 |
| fimH142 | ST90 | 4 | |
| D | fimH5 | ST38 | 1 |
| fimH26 | ST963 | 1 | |
| fimH27 | ST69 | 4 | |
| ST405 | 12 | ||
| fimH54 | ST38 | 1 | |
| fimH65 | ST38 | 1 | |
| ST UNK3 c | 1 | ||
| E | fimH82 | ST11 | 1 |
| F | fimH5 | ST648 | 1 |
| fimH27 | ST648 | 2 | |
| fimH171 | ST648 | 1 | |
| fimH145 | ST457 | 1 | |
| fimH58 | ST354 | 2 | |
| fimH Absent | ST6870 | 3 | |
| fimH Absent | ST UNK4 d/5 e | 2 |
| Ambler Class | Carbapenemase Gene | Sequence Type | Isolates (n = 82) |
|---|---|---|---|
| A | blaKPC-2 | ST131 | 1 |
| B | blaNDM-1 | ST131 | 2 |
| ST90 | 2 | ||
| blaNDM-5 | ST167 | 5 | |
| ST405 | 4 | ||
| ST6870 | 3 | ||
| ST10 | 2 | ||
| ST69 | 1 | ||
| ST73 | 1 | ||
| D | blaOXA-181 | ST410 | 3 |
| ST156 | 1 | ||
| STUNK5 * | 1 | ||
| blaOXA-48 | ST131 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Slown, S.; Walas, N.; Amato, H.K.; Lloyd, T.; Varghese, V.; Bender, M.; Pandori, M.; Graham, J. Clonal Lineages and Virulence Factors of Carbapenem Resistant E. coli in Alameda County, California, 2017–2019. Antibiotics 2022, 11, 1794. https://doi.org/10.3390/antibiotics11121794
Slown S, Walas N, Amato HK, Lloyd T, Varghese V, Bender M, Pandori M, Graham J. Clonal Lineages and Virulence Factors of Carbapenem Resistant E. coli in Alameda County, California, 2017–2019. Antibiotics. 2022; 11(12):1794. https://doi.org/10.3390/antibiotics11121794
Chicago/Turabian StyleSlown, Samuel, Nikolina Walas, Heather K. Amato, Tyler Lloyd, Vici Varghese, Monica Bender, Mark Pandori, and Jay Graham. 2022. "Clonal Lineages and Virulence Factors of Carbapenem Resistant E. coli in Alameda County, California, 2017–2019" Antibiotics 11, no. 12: 1794. https://doi.org/10.3390/antibiotics11121794