
Bacterial Nosocomial Infections: Multidrug Resistance as a Trigger for the Development of Novel Antimicrobials

 , , , , ,
, , , , ,
Abstract
:1. Introduction: Bacterial Nosocomial Infections
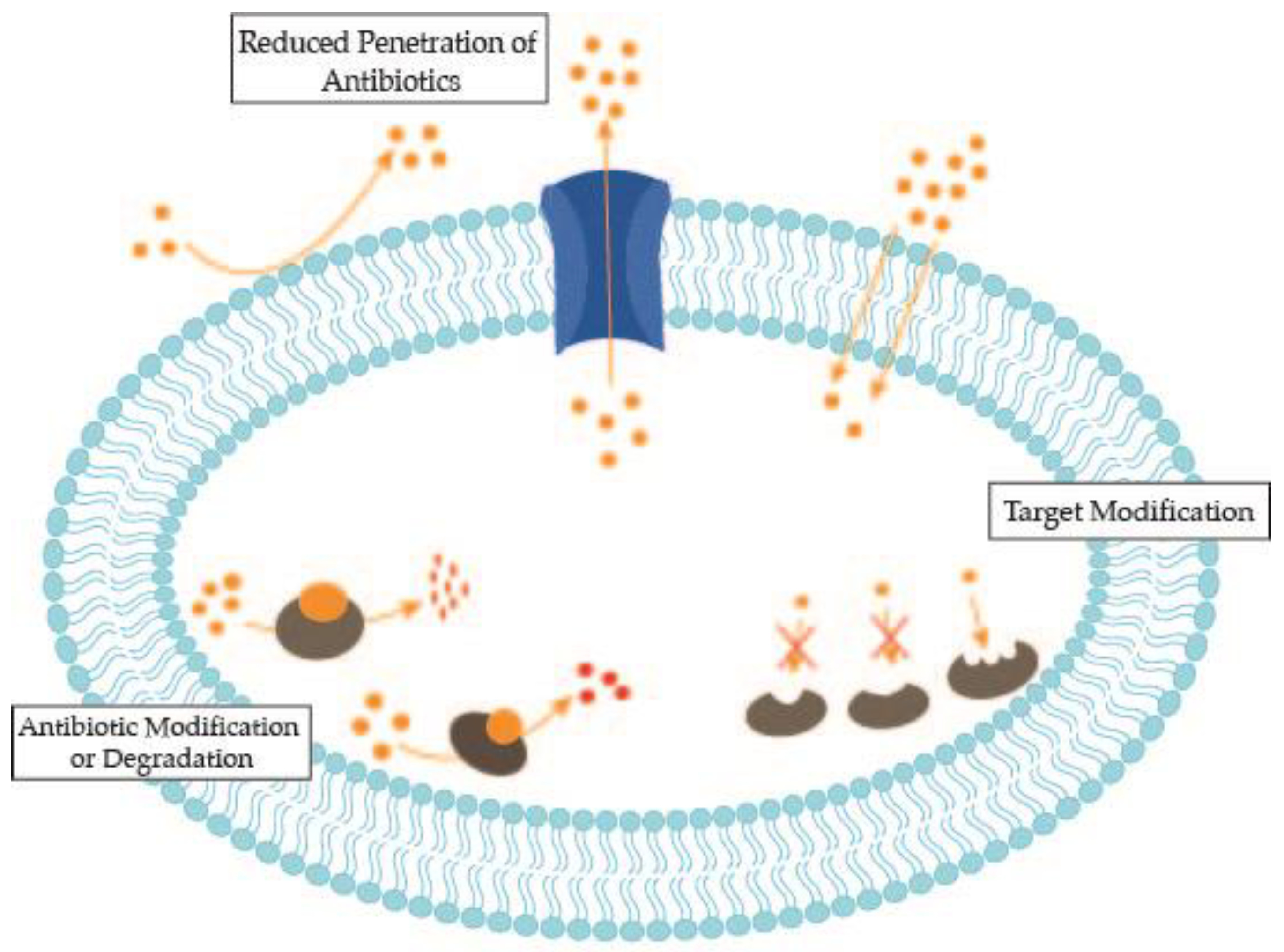
2. Antimicrobial Resistance Mechanisms of Bacterial Nosocomial Pathogens
3. Antimicrobial Alternatives under Investigation
3.1. Repurposing of Existing Drugs
3.2. Metal-Based Complexes
3.3. Antimicrobial Peptides
3.4. Antisense Antimicrobial Therapeutics
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Suetens, C.; Latour, K.; Kärki, T.; Ricchizzi, E.; Kinross, P.; Moro, M.L.; Jans, B.; Hopkins, S.; Hansen, S.; Lyytikäinen, O.; et al. Prevalence of healthcare-associated infections, estimated incidence and composite antimicrobial resistance index in acute care hospitals and long-term care facilities: Results from two european point prevalence surveys, 2016 to 2017. Eurosurveillance 2018, 23, pii1800516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maki, D.G.; Crnich, C.J.; Safdar, N. Nosocomial Infection in the Intensive Care Unit. In Critical Care Medicine: Principles of Diagnosis and Management in the Adult; Elsevier Inc.: Amsterdam, The Netherlands, 2008; pp. 1003–1069. [Google Scholar]
- World Health Organization. Prevention of Hospital-Acquired Infections A Practical Guide, 2nd ed.; Ducel, G., Fabry, J., Nicolle, L., Eds.; World Health Organization: Geneva, Switzerland, 2002. [Google Scholar]
- Stygall, J.; Newman, S. Hospital acquired infection. In Cambridge Handbook of Psychology, Health and Medicine, 2nd ed.; Cambridge University Press: Cambridge, UK, 2014; pp. 736–738. [Google Scholar]
- Centers for Disease Control. Antibiotic Resistance Threats in the United States, 2019; U.S. Department of Health and Human Services, CDC: Atlanta, GA, USA, 2019. [Google Scholar]
- Ayobami, O.; Willrich, N.; Harder, T.; Okeke, I.N.; Eckmanns, T.; Markwart, R. The incidence and prevalence of hospital-acquired (carbapenem-resistant) Acinetobacter baumannii in Europe, Eastern Mediterranean and Africa: A systematic review and meta-analysis. Emerg. Microbes Infect. 2019, 8, 1747–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brink, A.J. Epidemiology of carbapenem-resistant Gram-negative infections globally. Curr. Opin. Infect. Dis. 2019, 32, 609–616. [Google Scholar] [CrossRef]
- Slimings, C.; Riley, T.V. Antibiotics and healthcare facility-associated Clostridioides difficile infection: Systematic review and meta-analysis 2020 update. J. Antimicrob. Chemother. 2021, 76, 1676–1688. [Google Scholar] [CrossRef]
- Spivak, E.S.; Cosgrove, S.E.; Srinivasan, A. Measuring Appropriate Antimicrobial Use: Attempts at Opening the Black Box. Clin. Infect. Dis. 2016, 63, 1–6. [Google Scholar]
- Holmes, A.H.; Moore, L.S.P.; Sundsfjord, A.; Steinbakk, M.; Regmi, S.; Karkey, A.; Guerin, P.J.; Piddock, L.J.V. Understanding the mechanisms and drivers of antimicrobial resistance. Lancet 2016, 387, 176–187. [Google Scholar] [CrossRef]
- Theriot, C.M.; Young, V.B. Interactions between the Gastrointestinal Microbiome and Clostridium difficile. Annu. Rev. Microbiol. 2015, 69, 445–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monnet, D.L.; Harbarth, S. Will coronavirus disease (COVID-19) have an impact on antimicrobial resistance? Eurosurveillance 2020, 25, 1–6. [Google Scholar] [CrossRef]
- Rawson, T.M.; Moore, L.S.P.; Castro-Sanchez, E.; Charani, E.; Davies, F.; Satta, G.; Ellington, M.J.; Holmes, A.H. COVID-19 and the potential long-term impact on antimicrobial resistance. J. Antimicrob. Chemother. 2020, 75, 1681–1684. [Google Scholar] [CrossRef]
- Singh, S.B.; Young, K.; Silver, L.L. What is an “ideal” antibiotic? Discovery challenges and path forward. Biochem. Pharmacol. 2017, 133, 63–73. [Google Scholar] [CrossRef]
- Kaye, K.S.; Fraimow, H.S.; Abrutyn, E. Pathogens resistant to antimicrobial agents: Epidemiology, molecular mechanisms, and clinical management. Infect. Dis. Clin. N. Am. 2000, 14, 293–319. [Google Scholar] [CrossRef]
- Peterson, E.; Kaur, P. Antibiotic resistance mechanisms in bacteria: Relationships between resistance determinants of antibiotic producers, environmental bacteria, and clinical pathogens. Front. Microbiol. 2018, 9, 2928. [Google Scholar] [CrossRef]
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef]
- Rice, L.B. Progress and Challenges in Implementing the Research on ESKAPE Pathogens. Infect. Control Hosp. Epidemiol. 2010, 31, S7–S10. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Singh, D.; Jain, S.; Adhaulia, G.; Barua, S.; Sachan, A.K. Drug repositioning: Achievements, advancements and barriers. IP Int. J. Compr. Adv. Pharmacol. 2019, 4, 11–16. [Google Scholar] [CrossRef]
- Jourdan, J.P.; Bureau, R.; Rochais, C.; Dallemagne, P. Drug repositioning: A brief overview. J. Pharm. Pharmacol. 2020, 72, 1145–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleigh, S.H.; Barton, C.L. Repurposing strategies for therapeutics. Pharm. Med. 2010, 24, 151–159. [Google Scholar] [CrossRef]
- Mazumdar, K.; Asok Kumar, K.; Dutta, N.K. Potential role of the cardiovascular non-antibiotic (helper compound) amlodipine in the treatment of microbial infections: Scope and hope for the future. Int. J. Antimicrob. Agents 2010, 36, 295–302. [Google Scholar] [CrossRef]
- Lagadinou, M.; Onisor, M.O.; Rigas, A.; Musetescu, D.V.; Gkentzi, D.; Assimakopoulos, S.F.; Panos, G.; Marangos, M. Antimicrobial properties on non-antibiotic drugs in the era of increased bacterial resistance. Antibiotics 2020, 9, 107. [Google Scholar] [CrossRef] [Green Version]
- Soo, V.; Kwan, B.; Quezada, H.; Castillo-Juárez, I.; Pérez-Eretza, B.; García-Contreras, S.; Martínez-Vázquez, M.; Wood, T.; García-Contreras, R. Repurposing of Anticancer Drugs for the Treatment of Bacterial Infections. Curr. Top. Med. Chem. 2016, 17, 1157–1176. [Google Scholar] [CrossRef] [Green Version]
- Kwan, B.W.; Chowdhury, N.; Wood, T.K. Combatting bacterial infections by killing persister cells with mitomycin C. Environ. Microbiol. 2015, 17, 4406–4414. [Google Scholar] [CrossRef]
- Chowdhury, N.; Wood, T.L.; Martínez-Vázquez, M.; García-Contreras, R.; Wood, T.K. DNA-crosslinker cisplatin eradicates bacterial persister cells. Biotechnol. Bioeng. 2016, 113, 1984–1992. [Google Scholar] [CrossRef] [PubMed]
- Goss, C.H.; Kaneko, Y.; Khuu, L.; Anderson, G.D.; Ravishankar, S.; Aitken, M.L.; Lechtzin, N.; Zhou, G.; Czyz, D.M.; McLean, K.; et al. Gallium disrupts bacterial iron metabolism and has therapeutic effects in mice and humans with lung infections. Sci. Transl. Med. 2018, 10, eaat7520. [Google Scholar] [CrossRef] [Green Version]
- Thompson, M.G.; Truong-Le, V.; Alamneh, Y.A.; Black, C.C.; Anderl, J.; Honnold, C.L.; Pavlicek, R.L.; Abu-Taleb, R.; Wise, M.C.; Hall, E.R.; et al. Evaluation of gallium citrate formulations against a multidrug-resistant strain of Klebsiella pneumoniae in a murine wound model of infection. Antimicrob. Agents Chemother. 2015, 59, 6484–6493. [Google Scholar] [CrossRef] [Green Version]
- Antunes, L.C.S.; Imperi, F.; Minandri, F.; Visca, P. In Vitro and In Vivo antimicrobial activities of gallium nitrate against multidrug-resistant acinetobacter baumannii. Antimicrob. Agents Chemother. 2012, 56, 5961–5970. [Google Scholar] [CrossRef] [Green Version]
- Jerwood, S.; Cohen, J. Unexpected antimicrobial effect of statins. J. Antimicrob. Chemother. 2008, 61, 362–364. [Google Scholar] [CrossRef]
- Imperi, F.; Massai, F.; Pillai, C.R.; Longo, F.; Zennaro, E.; Rampioni, G.; Visc, P.; Leoni, L. New life for an old Drug: The anthelmintic drug niclosamide inhibits pseudomonas aeruginosa quorum sensing. Antimicrob. Agents Chemother. 2013, 57, 996–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlmutter, J.I.; Forbes, L.T.; Krysan, D.J.; Ebsworth-Mojica, K.; Colquhoun, J.M.; Wang, J.L.; Dunman, P.M.; Flaherty, D.P. Repurposing the antihistamine terfenadine for antimicrobial activity against staphylococcus aureus. J. Med. Chem. 2014, 57, 8540–8562. [Google Scholar] [CrossRef] [PubMed]
- Prieto, J.M.; Rapún-Araiz, B.; Gil, C.; Penadés, J.R.; Lasa, I.; Latasa, C. Inhibiting the two-component system GraXRS with verteporfin to combat Staphylococcus aureus infections. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Aydin, O.N.; Eyigor, M.; Aydin, N. Antimicrobial activity of ropivacaine and other local anaesthetics. Eur. J. Anaesthesiol. 2001, 18, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Grimmond, T.R.; Brownridge, P. Antimicrobial activity of bupivacaine and pethidine. Anaesth. Intensive Care 1986, 14, 418–420. [Google Scholar] [CrossRef] [Green Version]
- Roder, C.; Thomson, M.J. Auranofin: Repurposing an old drug for a golden new age. Drugs R&D. 2015, 15, 13–20. [Google Scholar]
- Harbut, M.B.; Vilchèze, C.; Luo, X.; Hensler, M.E.; Guo, H.; Yang, B.; Chatterjee, A.K.; Nizet, V.; Jacobs, W.R.; Schultz, P.G.; et al. Auranofin exerts broad-spectrum bactericidal activities by targeting thiol-redox homeostasis. Proc. Natl. Acad. Sci. USA 2015, 112, 4453–4458. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, B.B.; Rajamuthiah, R.; Souza, A.C.R.; Eatemadpour, S.; Rossoni, R.D.; Santos, D.A.; Junqueira, J.C.; Rice, L.B.; Mylonakis, E. Inhibition of bacterial and fungal pathogens by the orphaned drug auranofin. Future Med. Chem. 2016, 8, 117–132. [Google Scholar] [CrossRef] [Green Version]
- De Clercq, E. AMD3100/CXCR4 inhibitor. Front. Immunol. 2015, 6, 276. [Google Scholar] [CrossRef]
- De Clercq, E. The bicyclam AMD3100 story. Nat. Rev. Drug Discov. 2003, 2, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Nagalingam, G.; Ellis, S.; Martinez, E.; Sintchenko, V.; Spain, M.; Rutledge, P.J.; Todd, M.H.; Triccas, J.A. Nontoxic Metal-Cyclam Complexes, a New Class of Compounds with Potency against Drug-Resistant Mycobacterium tuberculosis. J. Med. Chem. 2016, 59, 5917–5921. [Google Scholar] [CrossRef]
- Spain, M.; Wong, J.K.H.; Nagalingam, G.; Batten, J.M.; Hortle, E.; Oehlers, S.H.; Jiang, X.F.; Murage, H.E.; Orford, J.T.; Crisologo, P.; et al. Antitubercular Bis-Substituted Cyclam Derivatives: Structure-Activity Relationships and In Vivo Studies. J. Med. Chem. 2018, 61, 3595–3608. [Google Scholar] [CrossRef]
- Alves, L.G.; Pinheiro, P.F.; Feliciano, J.R.; Dâmaso, D.P.; Leitão, J.H.; Martins, A.M. Synthesis, antimicrobial activity and toxicity to nematodes of cyclam derivatives. Int. J. Antimicrob. Agents 2017, 49, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Alves, L.G.; Portel, J.F.; Sousa, S.A.; Ferreira, O.; Almada, S.; Silva, E.R.; Martins, A.M.; Leitão, J.H. Investigations into the structure/antibacterial activity relationships of cyclam and cyclen derivatives. Antibiotics 2019, 8, 224. [Google Scholar] [CrossRef] [Green Version]
- Allam, A.; Maigre, L.; Alves de Sousa, R.; Dumont, E.; Vergalli, J.; Pagès, J.M.; Artaud, I. New amphiphilic neamine conjugates bearing a metal binding motif active against MDR E. aerogenes Gram-negative bacteria. Eur. J. Med. Chem. 2017, 127, 748–756. [Google Scholar] [CrossRef]
- Younis, W.; Thangamani, S.; Seleem, M. Repurposing Non-Antimicrobial Drugs and Clinical Molecules to Treat Bacterial Infections. Curr. Pharm. Des. 2015, 21, 4106–4111. [Google Scholar] [CrossRef] [Green Version]
- Kamurai, B.; Mombeshora, M.; Mukanganyama, S. Repurposing of Drugs for Antibacterial Activities on Selected ESKAPE Bacteria Staphylococcus aureus and Pseudomonas aeruginosa. Int. J. Microbiol. 2020, 2020, 8885338. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; She, P.; Zhou, L.; Li, S.; Hussain, Z.; Chen, L.; Wu, Y. Drug repurposing: Antimicrobial and antibiofilm effects of penfluridol against Enterococcus faecalis. Microbiologyopen 2021, 10, e1148. [Google Scholar] [CrossRef]
- Thanacoody, H.K.R. Thioridazine: Resurrection as an antimicrobial agent? Br. J. Clin. Pharmacol. 2007, 64, 566–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayaz, M.; Subhan, F.; Ahmed, J.; Khan, A.; Ullah, F.; Ullah, I.; Ali, G.; Syed, N.-H.; Hussain, S. Sertraline enhances the activity of antimicrobial agents against pathogens of clinical relevance. J. Biol. Res. 2015, 22, 1–8. [Google Scholar] [CrossRef] [Green Version]
- May, H.C.; Yu, J.J.; Guentzel, M.N.; Chambers, J.P.; Cap, A.P.; Arulanandam, B.P. Repurposing auranofin, ebselen, and PX-12 as antimicrobial agents targeting the thioredoxin system. Front. Microbiol. 2018, 9, 336. [Google Scholar] [CrossRef]
- Shah, P.N.; Marshall-Batty, K.R.; Smolen, J.A.; Tagaev, J.A.; Chen, Q.; Rodesney, C.A.; Le, H.H.; Gordon, V.D.; Greenberg, D.E.; Cannon, C.L. Antimicrobial activity of ibuprofen against cystic fibrosis-associated gram-negative pathogens. Antimicrob. Agents Chemother. 2018, 62, e01574-17. [Google Scholar] [CrossRef] [Green Version]
- Salem-Milani, A.; Balaei-Gajan, E.; Rahimi, S.; Moosavi, Z.; Abdollahi, A.; Zakeri-Milani, P.; Bolourian, M. Antibacterial Effect of Diclofenac Sodium on Enterococcus faecalis. J. Dent. 2013, 10, 16–22. [Google Scholar]
- Lancellotti, P.; Musumeci, L.; Jacques, N.; Servais, L.; Goffin, E.; Pirotte, B.; Oury, C. Antibacterial Activity of Ticagrelor in Conventional Antiplatelet Dosages Against Antibiotic-Resistant Gram-Positive Bacteria. JAMA Cardiol. 2019, 4, 596–599. [Google Scholar] [CrossRef] [Green Version]
- AbdelKhalek, A.; Abutaleb, N.S.; Elmagarmid, K.A.; Seleem, M.N. Repurposing auranofin as an intestinal decolonizing agent for vancomycin-resistant enterococci. Sci. Rep. 2018, 8, 8353. [Google Scholar] [CrossRef] [Green Version]
- Thangamani, S.; Mohammad, H.; Abushahba, M.F.N.; Sobreira, T.J.P.; Hedrick, V.E.; Paul, L.N.; Seleem, M.N. Antibacterial activity and mechanism of action of auranofin against multi-drug resistant bacterial pathogens. Sci. Rep. 2016, 6, 22571. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, S.H.; Murphy, R.A.; Juul-Madsen, K.; Fredborg, M.; Hvam, M.L.; Axelgaard, E.; Skovdal, S.M.; Meyer, R.L.; Sørensen, U.B.S.; Möller, A.; et al. The Immunomodulatory Drug Glatiramer Acetate is Also an Effective Antimicrobial Agent that Kills Gram-negative Bacteria. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.K.; Plott, T. Ciclopirox: A broad-spectrum antifungal with antibacterial and anti-inflammatory properties. Int. J. Dermatol. 2004, 43, 3–8. [Google Scholar] [CrossRef]
- Carlson-Banning, K.M.; Chou, A.; Liu, Z.; Hamill, R.J.; Song, Y.; Zechiedrich, L. Toward Repurposing Ciclopirox as an Antibiotic against Drug-Resistant Acinetobacter baumannii, Escherichia coli, and Klebsiella pneumoniae. PLoS ONE 2013, 8, 69646. [Google Scholar] [CrossRef] [Green Version]
- Domalaon, R.; Okunnu, O.; Zhanel, G.G.; Schweizer, F. Synergistic combinations of anthelmintic salicylanilides oxyclozanide, rafoxanide, and closantel with colistin eradicates multidrug-resistant colistin-resistant Gram-negative bacilli. J. Antibiot. 2019, 72, 605–616. [Google Scholar] [CrossRef]
- Ejim, L.; Farha, M.A.; Falconer, S.B.; Wildenhain, J.; Coombes, B.K.; Tyers, M.; Brown, E.D.; Wright, G.D. Combinations of antibiotics and nonantibiotic drugs enhance antimicrobial efficacy. Nat. Chem. Biol. 2011, 7, 348–350. [Google Scholar] [CrossRef] [PubMed]
- Frei, A.; Zuegg, J.; Elliott, A.G.; Baker, M.; Braese, S.; Brown, C.; Chen, F.; Dowson, C.G.; Dujardin, G.; Jung, N.; et al. Metal complexes as a promising source for new antibiotics. Chem. Sci. 2020, 11, 2627–2639. [Google Scholar] [CrossRef] [Green Version]
- Evans, A.; Kavanagh, K.A. Evaluation of metal-based antimicrobial compounds for the treatment of bacterial pathogens. J. Med. Microbiol. 2021, 70, 001363. [Google Scholar] [CrossRef]
- Claudel, M.; Schwarte, J.V.; Fromm, K.M. New Antimicrobial Strategies Based on Metal Complexes. Chemistry 2020, 2, 849–899. [Google Scholar] [CrossRef]
- Klasen, H.J. Historical review of the use of silver in the treatment of burns. I. Early uses. Burns 2000, 26, 117–130. [Google Scholar] [CrossRef]
- Fox, C.L.; Modak, S.M. Mechanism of silver sulfadiazine action on burn wound infections. Antimicrob. Agents Chemother. 1974, 5, 582–588. [Google Scholar] [CrossRef] [Green Version]
- Yeo, E.D.; Yoon, S.A.; Oh, S.R.; Choi, Y.S.; Lee, Y.K. Degree of the hazards of silver-containing dressings on MRSA-Infected wounds in sprague-dawley and streptozotocin-induced diabetic rats. Wounds 2015, 27, 95–102. [Google Scholar] [PubMed]
- Nunes, J.H.B.; De Paiva, R.E.F.; Cuin, A.; Da Costa Ferreira, A.M.; Lustri, W.R.; Corbi, P.P. Synthesis, spectroscopic characterization, crystallographic studies and antibacterial assays of new copper(II) complexes with sulfathiazole and nimesulide. J. Mol. Struct. 2016, 1112, 14–20. [Google Scholar] [CrossRef]
- Carvalho, M.F.N.N.; Leite, S.; Costa, J.P.; Galvão, A.M.; Leitão, J.H. Ag(I) camphor complexes: Antimicrobial activity by design. J. Inorg. Biochem. 2019, 199, 110791. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.P.; Sousa, S.A.; Galvão, A.M.; Miguel Mata, J.; Leitão, J.H.; Carvalho, M.F.N.N. Key parameters on the antibacterial activity of silver camphor complexes. Antibiotics 2021, 10, 135. [Google Scholar] [CrossRef]
- Kascatan-Nebioglu, A.; Panzner, M.J.; Tessier, C.A.; Cannon, C.L.; Youngs, W.J. N-Heterocyclic carbene-silver complexes: A new class of antibiotics. Coord. Chem. Rev. 2007, 251, 884–895. [Google Scholar] [CrossRef]
- Woo, K.J.; Hye, C.K.; Ki, W.K.; Shin, S.; So, H.K.; Yong, H.P. Antibacterial activity and mechanism of action of the silver ion in Staphylococcus aureus and Escherichia coli. Appl. Environ. Microbiol. 2008, 74, 2171–2178. [Google Scholar]
- Gordon, O.; Slenters, T.V.; Brunetto, P.S.; Villaruz, A.E.; Sturdevant, D.E.; Otto, M.; Landmann, R.; Fromm, K.M. Silver coordination polymers for prevention of implant infection: Thiol interaction, impact on respiratory chain enzymes, and hydroxyl radical induction. Antimicrob. Agents Chemother. 2010, 54, 4208–4218. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, M.; Hara, K.; Kudo, J. Bactericidal actions of a silver ion solution on Escherichia coli, studied by energy-filtering transmission electron microscopy and proteomic analysis. Appl. Environ. Microbiol. 2005, 71, 7589–7593. [Google Scholar] [CrossRef] [Green Version]
- Nomiya, K.; Noguchi, R.; Ohsawa, K.; Tsuda, K.; Oda, M. Synthesis, crystal structure and antimicrobial activities of two isomeric gold(I) complexes with nitrogen-containing heterocycle and triphenylphosphine ligands, [Au(L)(PPh3)] (HL=pyrazole and imidazole). J. Inorg. Biochem. 2000, 78, 363–370. [Google Scholar] [CrossRef]
- Glišić, B.; Djuran, M.I. Gold complexes as antimicrobial agents: An overview of different biological activities in relation to the oxidation state of the gold ion and the ligand structure. Dalton Trans. 2014, 43, 5950–5969. [Google Scholar] [CrossRef]
- Stenger-Smith, J.R.; Mascharak, P.K. Gold Drugs with {Au(PPh3)}+ Moiety: Advantages and Medicinal Applications. ChemMedChem 2020, 15, 2136–2145. [Google Scholar] [CrossRef] [PubMed]
- Hussaini, S.Y.; Haque, R.A.; Razali, M.R. Recent progress in silver(I)-, gold(I)/(III)- and palladium(II)-N-heterocyclic carbene complexes: A review towards biological perspectives. J. Organomet. Chem. 2019, 882, 96–111. [Google Scholar] [CrossRef]
- Ray, S.; Mohan, R.; Singh, J.K.; Samantaray, M.K.; Shaikh, M.M.; Panda, D.; Ghosh, P. Anticancer and antimicrobial metallopharmaceutical agents based on palladium, gold, and silver N-heterocyclic carbene complexes. J. Am. Chem. Soc. 2007, 129, 15042–15053. [Google Scholar] [CrossRef]
- Novelli, F.; Recine, M.; Sparatore, F.; Juliano, C. Gold(I) complexes as antimicrobial agents. Farmaco 1999, 54, 232–236. [Google Scholar] [CrossRef]
- Doǧan, Ö.; Kaloǧlu, N.; Demir, S.; Özdemir, I.; Günal, S.; Özdemir, I. Synthesis and antimicrobial activity of novel gold(I) N-heterocyclic carbene complexes. Mon. Chem. 2013, 144, 313–319. [Google Scholar] [CrossRef]
- Eiter, L.C.; Hall, N.W.; Day, C.S.; Saluta, G.; Kucera, G.L.; Bierbach, U. Gold(I) analogues of a platinum-acridine antitumor agent are only moderately cytotoxic but show potent activity against Mycobacterium tuberculosis. J. Med. Chem. 2009, 52, 6519–6522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiekink, E.R.T. Gold derivatives for the treatment of cancer. Crit. Rev. Oncol. Hematol. 2002, 42, 225–248. [Google Scholar] [CrossRef]
- Parish, R.V.; Howe, B.P.; Wright, J.P.; Mack, J.; Pritchard, R.G.; Buckley, R.G.; Elsome, A.M.; Fricker, S.P. Chemical and Biological Studies of Dichloro(2-((dimethylamino)methyl)phenyl)gold(III). Inorg. Chem. 1996, 35, 1659–1666. [Google Scholar] [CrossRef]
- Parish, R.V.; Mack, J.; Hargreaves, L.; Wright, J.P.; Buckley, R.G.; Elsome, A.M.; Fricker, S.P.; Theobald, B.R.C. Chemical and biological reactions of diacetato[2-(dimethylaminoniethyl)-phenyl]gold(III), [Au(O2CMe)2(dmamp)]. J. Chem. Soc. Dalton Trans. 1996, 69–74. [Google Scholar] [CrossRef]
- Al-Khodir, F.A.I.; Refat, M.S. Synthesis, structural characterization and biological studies of some nalidixic acid-metal complexes: Metalloantibiotic complexes of some divalent and trivalent metal ions. J. Mol. Struct. 2015, 1094, 22–35. [Google Scholar] [CrossRef]
- Sousa, S.A.; Leitão, J.H.; Silva, R.A.L.; Belo, D.; Santos, I.C.; Guerreiro, J.F.; Martins, M.; Fontinha, D.; Prudêncio, M.; Almeida, M.; et al. On the path to gold: Monoanionic Au bisdithiolate complexes with antimicrobial and antitumor activities. J. Inorg. Biochem. 2020, 202, 110904. [Google Scholar] [CrossRef] [PubMed]
- Kharat, A.N.; Foroutannejad, S.; Khavasi, H.R. Biological evaluation of 4′-(2-thienyl)-2, 2′; 6′, 2″-terpyridine-1,1″-diium chloride tetrachloridoaurate (III): The effect of countercations. Synth. React. Inorg. Met. Nano Metal Chem. 2012, 42, 752–757. [Google Scholar] [CrossRef]
- Fontinha, D.; Sousa, S.A.; Morais, T.S.; Prudêncio, M.; Leitão, J.H.; Le Gal, Y.; Lorcy, D.; Silva, R.A.L.; Velho, M.F.G.; Velho, M.F.G.; et al. Gold(iii) bis(dithiolene) complexes: From molecular conductors to prospective anticancer, antimicrobial and antiplasmodial agents. Metallomics 2020, 12, 974–987. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Karge, B.; Misgeld, R.; Prokop, A.; Franke, R.; Brönstrup, M.; Ott, I. Gold(I) NHC Complexes: Antiproliferative Activity, Cellular Uptake, Inhibition of Mammalian and Bacterial Thioredoxin Reductases, and Gram-Positive Directed Antibacterial Effects. Chem. A Eur. J. 2017, 23, 1869–1880. [Google Scholar] [CrossRef] [PubMed]
- Ritz, D.; Beckwith, J. Roles of thiol-redox pathways in bacteria. Annu. Rev. Microbiol. 2001, 55, 21–48. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Uziel, O.; Borovok, I.; Schreiber, R.; Cohen, G.; Aharonowitz, Y. Transcriptional Regulation of the Staphylococcus aureus Thioredoxin and Thioredoxin Reductase Genes in Response to Oxygen and Disulfide Stress. J. Bacteriol. 2004, 186, 326–334. [Google Scholar] [CrossRef] [Green Version]
- Marzo, T.; Cirri, D.; Pollini, S.; Prato, M.; Fallani, S.; Cassetta, M.I.; Novelli, A.; Rossolini, G.M.; Messori, L. Auranofin and its Analogues Show Potent Antimicrobial Activity against Multidrug-Resistant Pathogens: Structure–Activity Relationships. ChemMedChem 2018, 13, 2448–2454. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Anis, R.; Hwang, E.; Ovalle, R.; Varela-Ramírez, A.; Aguilera, R.J.; Contel, M. Group 11 metal compounds with tripodal bis(imidazole) thioether ligands. applications as catalysts in the oxidation of alkenes and as antimicrobial agents. Molecules 2011, 16, 6701–6720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, B.C.; Henderson, W.; Andy Hor, T.S.; Nicholson, B.K. Synthesis and characterization of new trimetallic complexes with {Pt2Au(μ-S)2}n+ (n = 2, 3) cores containing C, N and N, N donor ligands. Inorg. Chim. Acta 2013, 394, 146–151. [Google Scholar] [CrossRef]
- Wuerth, K.; Hancock, R.E.W. New insights into cathelicidin modulation of adaptive immunity. Eur. J. Immunol. 2011, 41, 2817–2819. [Google Scholar] [CrossRef]
- Hancock, R.E.W. Cationic peptides: Effectors in innate immunity and novel antimicrobials. Lancet Infect. Dis. 2001, 1, 156–164. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Beisswenger, C.; Bals, R. Functions of Antimicrobial Peptides in Host Defense and Immunity. Curr. Protein Pept. Sci. 2005, 6, 255–264. [Google Scholar] [CrossRef]
- Zhang, L.; Gallo, R.L. Antimicrobial peptides. Curr. Biol. 2016, 26, R14–R19. [Google Scholar] [CrossRef]
- Berglund, N.A.; Piggot, T.J.; Jefferies, D.; Sessions, R.B.; Bond, P.J.; Khalid, S. Interaction of the Antimicrobial Peptide Polymyxin B1 with Both Membranes of E. coli: A Molecular Dynamics Study. PLoS Comput. Biol. 2015, 11, e1004180. [Google Scholar] [CrossRef] [Green Version]
- Gagnon, M.G.; Roy, R.N.; Lomakin, I.B.; Florin, T.; Mankin, A.S.; Steitz, T.A. Structures of proline-rich peptides bound to the ribosome reveal a common mechanism of protein synthesis inhibition. Nucleic Acids Res. 2016, 44, 2439–2450. [Google Scholar] [CrossRef] [Green Version]
- Florin, T.; Maracci, C.; Graf, M.; Karki, P.; Klepacki, D.; Berninghausen, O.; Beckmann, R.; Vázquez-Laslop, N.; Wilson, D.N.; Rodnina, M.V.; et al. An antimicrobial peptide that inhibits translation by trapping release factors on the ribosome. Nat. Struct. Mol. Biol. 2017, 24, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Hale, J.D.F.; Hancock, R.E.W. Alternative mechanisms of action of cationic antimicrobial peptides on bacteria. Expert Rev. Anti Infect. Ther. 2007, 5, 951–959. [Google Scholar] [CrossRef]
- Lazzaro, B.P.; Zasloff, M.; Rolff, J. Antimicrobial peptides: Application informed by evolution. Science 2020, 368, eaau5480. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, C.L.; Rozek, A.; Patrzykat, A.; Hancock, R.E.W. Structure and Mechanism of Action of an Indolicidin Peptide Derivative with Improved Activity against Gram-positive Bacteria. J. Biol. Chem. 2001, 276, 24015–24022. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.-P.; Zhou, L.-F.; Li, N.-N.; Chen, Y.-Q.; Li, B.-C.; Cai, Y.-F.; Zhang, S.-Q. Lipopolysaccharide neutralization by the antibacterial peptide CM4. Eur. J. Pharmacol. 2008, 596, 160–165. [Google Scholar] [CrossRef]
- Paranjape, S.M.; Lauer, T.W.; Montelaro, R.C.; Mietzner, T.A.; Vij, N. Modulation of proinflammatory activity by the engineered cationic antimicrobial peptide WLBU-2. F1000Research 2013, 2, 36. [Google Scholar] [CrossRef]
- Melvin, J.A.; Lashua, L.P.; Kiedrowski, M.R.; Yang, G.; Deslouches, B.; Montelaro, R.C.; Bomberger, J.M. Simultaneous Antibiofilm and Antiviral Activities of an Engineered Antimicrobial Peptide during Virus-Bacterium Coinfection. mSphere 2016, 1, e00083-16. [Google Scholar] [CrossRef] [Green Version]
- Lashua, L.P.; Melvin, J.A.; Deslouches, B.; Pilewski, J.M.; Montelaro, R.C.; Bomberger, J.M. Engineered cationic antimicrobial peptide (eCAP) prevents Pseudomonas aeruginosa biofilm growth on airway epithelial cells. J. Antimicrob. Chemother. 2016, 71, 2200–2207. [Google Scholar] [CrossRef] [Green Version]
- De Zoysa, G.H.; Cameron, A.J.; Hegde, V.V.; Raghothama, S.; Sarojini, V. Antimicrobial Peptides with Potential for Biofilm Eradication: Synthesis and Structure Activity Relationship Studies of Battacin Peptides. J. Med. Chem. 2015, 58, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, J.; Montville, T.J.; Nes, I.F.; Chikindas, M.L. Bacteriocins: Safe, natural antimicrobials for food preservation. Int. J. Food Microbiol. 2001, 71, 1–20. [Google Scholar] [CrossRef]
- Shin, J.M.; Gwak, J.W.; Kamarajan, P.; Fenno, J.C.; Rickard, A.H.; Kapila, Y.L. Biomedical applications of nisin. J. Appl. Microbiol. 2016, 120, 1449–1465. [Google Scholar] [CrossRef] [Green Version]
- Blay, G.L.; Lacroix, C.; Zihler, A.; Fliss, I. In vitro inhibition activity of nisin A, nisin Z, pediocin PA-1 and antibiotics against common intestinal bacteria. Lett. Appl. Microbiol. 2007, 45, 252–257. [Google Scholar] [CrossRef]
- Lee, G.; Bae, H. Anti-Inflammatory Applications of Melittin, a Major Component of Bee Venom: Detailed Mechanism of Action and Adverse Effects. Molecules 2016, 21, 616. [Google Scholar] [CrossRef]
- Son, D.J.; Lee, J.W.; Lee, Y.H.; Song, H.S.; Lee, C.K.; Hong, J.T. Therapeutic application of anti-arthritis, pain-releasing, and anti-cancer effects of bee venom and its constituent compounds. Pharmacol. Ther. 2007, 115, 246–270. [Google Scholar] [CrossRef]
- Choi, J.H.; Jang, A.Y.; Lin, S.; Lim, S.; Kim, D.; Park, K.; Han, S.-M.; Yeo, J.-H.; Seo, H.S. Melittin, a honeybee venom_derived antimicrobial peptide, may target methicillin_resistant Staphylococcus aureus. Mol. Med. Rep. 2015, 12, 6483–6490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Bogaart, G.; Guzmán, J.V.; Mika, J.T.; Poolman, B. On the mechanism of pore formation by melittin. J. Biol. Chem. 2008, 283, 33854–33857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, J.M.; Rajasekaran, A.K. Gramicidin A: A New Mission for an Old Antibiotic. J. Kidney Cancer VHL 2015, 2, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, J.H.; Crawford, S.A.; Kelly, C.C.; Patterson, J.E. In vitro activity of daptomycin against vancomycin-resistant enterococci of various Van types and comparison of susceptibility testing methods. Antimicrob. Agents Chemother. 2003, 47, 3760–3763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, S.C.J.; Zasowski, E.J.; Trinh, T.D.; Lagnf, A.M.; Bhatia, S.; Sabagha, N.; Abdul-Mutakabbir, J.C.; Alosaimy, S.; Mynatt, R.P.; Davis, S.L.; et al. Daptomycin Plus β-Lactam Combination Therapy for Methicillin-resistant Staphylococcus aureus Bloodstream Infections: A Retrospective, Comparative Cohort Study. Clin. Infect. Dis. 2020, 71, 1–10. [Google Scholar] [CrossRef]
- Berditsch, M.; Afonin, S.; Reuster, J.; Lux, H.; Schkolin, K.; Babii, O.; Radchenko, D.S.; Abdullah, I.; William, N.; Middel, V.; et al. Supreme activity of gramicidin S against resistant, persistent and biofilm cells of staphylococci and enterococci. Sci. Rep. 2019, 9, 17938. [Google Scholar] [CrossRef] [PubMed]
- Zavascki, A.P.; Goldani, L.Z.; Li, J.; Nation, R.L. Polymyxin B for the treatment of multidrug-resistant pathogens: A critical review. J. Antimicrob. Chemother. 2007, 60, 1206–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falagas, M.E.; Kasiakou, S.K. Toxicity of polymyxins: A systematic review of the evidence from old and recent studies. Crit. Care 2006, 10, R27. [Google Scholar] [CrossRef] [Green Version]
- Cisneros, J.M.; Rosso-Fernández, C.M.; Roca-Oporto, C.; De Pascale, G.; Jiménez-Jorge, S.; Fernández-Hinojosa, E.; Matthaiou, D.K.; Ramírez, P.; Díaz-Miguel, R.O.; Estella, A.; et al. Colistin versus meropenem in the empirical treatment of ventilator-associated pneumonia (Magic Bullet study): An investigator-driven, open-label, randomized, noninferiority controlled trial. Crit. Care 2019, 23, 383. [Google Scholar] [CrossRef] [Green Version]
- Konai, M.M.; Pakrudheen, I.; Barman, S.; Sharma, N.; Tabbasum, K.; Garg, P.; Haldar, J. Cyclam-based antibacterial molecules eradicate Gram-negative superbugs with potent efficacy against human corneal infection. Chem. Commun. 2020, 56, 2147–2150. [Google Scholar] [CrossRef] [PubMed]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Kosikowska, P.; Lesner, A. Antimicrobial peptides (AMPs) as drug candidates: A patent review (2003–2015). Expert Opin. Ther. Pat. 2016, 26, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.-L.; Marshall, J.C.; Dellinger, R.P.; Simonson, S.G.; Guntupalli, K.; Levy, M.M.; Singer, M.; Malik, R. Talactoferrin in Severe Sepsis: Results From the Phase II/III Oral tAlactoferrin in Severe sepsIS Trial. Crit. Care Med. 2015, 43, 1832–1838. [Google Scholar] [CrossRef]
- Wach, A.; Dembowsky, K.; Dale, G.E. Pharmacokinetics and Safety of Intravenous Murepavadin Infusion in Healthy Adult Subjects Administered Single and Multiple Ascending Doses. Antimicrob. Agents Chemother. 2018, 62, e02355-17. [Google Scholar] [CrossRef] [Green Version]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dijksteel, G.S.; Ulrich, M.M.W.; Middelkoop, E.; Boekema, B.K.H.L. Review: Lessons Learned From Clinical Trials Using Antimicrobial Peptides (AMPs). Front. Microbiol. 2021, 12, 287. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, A.C.; Janson, H.; Wold, H.; Fugelli, A.; Andersson, K.; Håkangård, C.; Olsson, P.; Olsen, W.M. LTX-109 Is a Novel Agent for Nasal Decolonization of Methicillin-Resistant and -Sensitive Staphylococcus aureus. Antimicrob. Agents Chemother. 2015, 59, 145–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Hong, Y.; Xi, Y.; Zou, Y.; Gao, J.; Du, J. Synthesis, Self-Assembly, and Biomedical Applications of Antimicrobial Peptide–Polymer Conjugates. Biomacromolecules 2018, 19, 1701–1720. [Google Scholar] [CrossRef] [PubMed]
- Ugurlu, T.; Turkoglu, M.; Gurer, U.S.; Akarsu, B.G. Colonic delivery of compression coated nisin tablets using pectin/HPMC polymer mixture. Eur. J. Pharm. Biopharm. 2007, 67, 202–210. [Google Scholar] [CrossRef]
- Yüksel, E.; Karakeçili, A. Antibacterial activity on electrospun poly(lactide-co-glycolide) based membranes via Magainin II grafting. Mater. Sci. Eng. C 2014, 45, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-Y.; Chang, H.-Y.; Lu, J.-K.; Huang, Y.-C.; Harroun, S.G.; Tseng, Y.-T.; Li, Y.-J.; Huang, C.-C.; Chang, H.-T. Self-Assembly of Antimicrobial Peptides on Gold Nanodots: Against Multidrug-Resistant Bacteria and Wound-Healing Application. Adv. Funct. Mater. 2015, 25, 7189–7199. [Google Scholar] [CrossRef]
- Sharma, D.; Choudhary, M.; Vashistt, J.; Shrivastava, R.; Bisht, G.S. Cationic antimicrobial peptide and its poly-N-substituted glycine congener: Antibacterial and antibiofilm potential against A. baumannii. Biochem. Biophys. Res. Commun. 2019, 518, 472–478. [Google Scholar] [CrossRef]
- Kim, H.; Jang, J.H.; Kim, S.C.; Cho, J.H. Development of a novel hybrid antimicrobial peptide for targeted killing of Pseudomonas aeruginosa. Eur. J. Med. Chem. 2020, 185, 111814. [Google Scholar] [CrossRef] [PubMed]
- Mohid, S.A.; Ghorai, A.; Ilyas, H.; Mroue, K.H.; Narayanan, G.; Sarkar, A.; Ray, S.K.; Biswas, K.; Bera, A.K.; Malmsten, M.; et al. Application of tungsten disulfide quantum dot-conjugated antimicrobial peptides in bio-imaging and antimicrobial therapy. Colloids Surf. B Biointerfaces 2019, 176, 360–370. [Google Scholar] [CrossRef]
- Lenhard, J.R.; Nation, R.L.; Tsuji, B.T. Synergistic combinations of polymyxins. Int. J. Antimicrob. Agents 2016, 48, 607–613. [Google Scholar] [CrossRef] [Green Version]
- Otvos, L., Jr.; Ostorhazi, E.; Szabo, D.; Zumbrun, S.D.; Miller, L.L.; Halasohoris, S.A.; Desai, P.D.; Int Veldt, S.M.; Kraus, C.N. Synergy Between Proline-Rich Antimicrobial Peptides and Small Molecule Antibiotics against Selected Gram-Negative Pathogens in vitro and in vivo. Front. Chem. 2018, 6, 309. [Google Scholar] [CrossRef] [PubMed]
- Brophy, J.A.; Voigt, C.A. Antisense transcription as a tool to tune gene expression. Mol. Syst. Biol. 2016, 12, 854. [Google Scholar] [CrossRef] [PubMed]
- Pita, T.; Feliciano, J.R.; Leitão, J.H. Extracellular RNAs in bacterial infections: From emerging key players on host-pathogen interactions to exploitable biomarkers and therapeutic targets. Int. J. Mol. Sci. 2020, 21, 9634. [Google Scholar] [CrossRef]
- Millar, J.A.; Raghavan, R. Modulation of Bacterial Fitness and Virulence Through Antisense RNAs. Front. Cell. Infect. Microbiol. 2021, 10, 596277. [Google Scholar] [CrossRef]
- Sully, E.K.; Geller, B.L. Antisense antimicrobial therapeutics. Curr. Opin. Microbiol. 2016, 33, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Jani, S.; Ramirez, M.S.; Tolmasky, M.E. Silencing Antibiotic Resistance with Antisense Oligonucleotides. Biomedicines 2021, 9, 416. [Google Scholar] [CrossRef]
- Hegarty, J.P.; Stewart, D.B. Advances in therapeutic bacterial antisense biotechnology. Appl. Microbiol. Biotechnol. 2018, 102, 1055–1065. [Google Scholar] [CrossRef]
- Wesolowski, D.; Alonso, D.; Altman, S. Combined effect of a peptide–morpholino oligonucleotide conjugate and a cell-penetrating peptide as an antibiotic. Proc. Natl. Acad. Sci. USA 2013, 110, 8686–8689. [Google Scholar] [CrossRef] [Green Version]
- Patenge, N.; Pappesch, R.; Krawack, F.; Walda, C.; Mraheil, M.A.; Jacob, A.; Hain, T.; Kreikemeyer, B. Inhibition of Growth and Gene Expression by PNA-peptide Conjugates in Streptococcus pyogenes. Mol. Ther. Nucleic Acids 2013, 2, e132. [Google Scholar] [CrossRef] [PubMed]
- Barkowsky, G.; Lemster, A.-L.; Pappesch, R.; Jacob, A.; Krüger, S.; Schröder, A.; Kreikemeyer, B.; Patenge, N. Influence of Different Cell-Penetrating Peptides on the Antimicrobial Efficiency of PNAs in Streptococcus pyogenes. Mol. Ther. Nucleic Acids 2019, 18, 444–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Równicki, M.; Pieńko, T.; Czarnecki, J.; Kolanowska, M.; Bartosik, D.; Trylska, J. Artificial Activation of Escherichia coli mazEF and hipBA Toxin–Antitoxin Systems by Antisense Peptide Nucleic Acids as an Antibacterial Strategy. Front. Microbiol. 2018, 9, 2870. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.; He, Y.; Xia, Y.; Wang, H.; Wang, L.; Gao, R.; Zhang, M. Inhibiting the growth of methicillin-resistant Staphylococcus aureus in vitro with antisense peptide nucleic acid conjugates targeting the ftsZ gene. Int. J. Infect. Dis. 2015, 30, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Oh, E.; Zhang, Q.; Jeon, B. Target optimization for peptide nucleic acid (PNA)-mediated antisense inhibition of the CmeABC multidrug efflux pump in Campylobacter jejuni. J. Antimicrob. Chemother. 2014, 69, 375–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, B.; Zhang, Q. Sensitization of Campylobacter jejuni to fluoroquinolone and macrolide antibiotics by antisense inhibition of the CmeABC multidrug efflux transporter. J. Antimicrob. Chemother. 2009, 63, 946–948. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, Y.; Ling, Z.; Zhang, C.; Fu, M.; Wang, Y.; Wang, S.; Zhang, S.; Shen, Z. Peptide nucleic acid restores colistin susceptibility through modulation of MCR-1 expression in Escherichia coli. J. Antimicrob. Chemother. 2020, 75, 2059–2065. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Meng, J.; Jia, M.; Ma, X.; He, G.; Yu, J.; Wang, R.; Bai, H.; Hou, Z.; Luo, X. oprM as a new target for reversion of multidrug resistance in Pseudomonas aeruginosa by antisense phosphorothioate oligodeoxynucleotides. FEMS Immunol. Med. Microbiol. 2010, 60, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Al Husseini, L.B.; Maleki, A.; Al Marjani, M.F. Antisense mqsR-PNA as a putative target to the eradication of Pseudomonas aeruginosa persisters. New Microbes New Infect. 2021, 41, 100868. [Google Scholar] [CrossRef]
- Pereira, S.; Santos, R.S.; Moreira, L.; Guimarães, N.M.; Braeckmans, K.; De Smedt, S.C.; Azevedo, N.F. Delivery of Oligonucleotides into Bacteria by Fusogenic Liposomes. Methods Mol. Biol. 2021, 2246, 87–96. [Google Scholar]
- Kauss, T.; Arpin, C.; Bientz, L.; Vinh Nguyen, P.; Vialet, B.; Benizri, S.; Barthélémy, P. Lipid oligonucleotides as a new strategy for tackling the antibiotic resistance. Sci. Rep. 2020, 10, 1054. [Google Scholar] [CrossRef]
- Eller, K.A.; Aunins, T.R.; Courtney, C.M.; Campos, J.K.; Otoupal, P.B.; Erickson, K.E.; Madinger, N.E.; Chatterjee, A. Facile accelerated specific therapeutic (FAST) platform develops antisense therapies to counter multidrug-resistant bacteria. Commun. Biol. 2021, 4, 331. [Google Scholar] [CrossRef] [PubMed]
- Aunins, T.R.; Erickson, K.E.; Chatterjee, A. Transcriptome-based design of antisense inhibitors potentiates carbapenem efficacy in CRE Escherichia coli. Proc. Natl. Acad. Sci. USA 2020, 117, 30699–30709. [Google Scholar] [CrossRef]
- Geller, B.L.; Deere, J.; Tilley, L.; Iversen, P.L. Antisense phosphorodiamidate morpholino oligomer inhibits viability of Escherichia coli in pure culture and in mouse peritonitis. J. Antimicrob. Chemother. 2005, 55, 983–988. [Google Scholar] [CrossRef] [Green Version]
- Castillo, J.I.; Równicki, M.; Wojciechowska, M.; Trylska, J. Antimicrobial synergy between mRNA targeted peptide nucleic acid and antibiotics in E. coli. Bioorg. Med. Chem. Lett. 2018, 28, 3094–3098. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.M.; Bonke, G.; Hogendorf, W.F.J.; Björkling, F.; Nielsen, J.; Kongstad, K.T.; Zabicka, D.; Tomczak, M.; Urbas, M.; Nielsen, P.E.; et al. Microwave-assisted solid-phase synthesis of antisense acpP peptide nucleic acid-peptide conjugates active against colistin- and tigecycline-resistant E. coli and K. pneumoniae. Eur. J. Med. Chem. 2019, 168, 134–145. [Google Scholar] [CrossRef]
- Yavari, N.; Goltermann, L.; Nielsen, P.E. Uptake, Stability, and Activity of Antisense Anti-acpP PNA-Peptide Conjugates in Escherichia coli and the Role of SbmA. ACS Chem. Biol. 2021, 16, 471–479. [Google Scholar] [CrossRef]
- Sully, E.K.; Geller, B.L.; Li, L.; Moody, C.M.; Bailey, S.M.; Moore, A.L.; Wong, M.; Nordmann, P.; Daly, S.M.; Sturge, C.R.; et al. Peptide-conjugated phosphorodiamidate morpholino oligomer (PPMO) restores carbapenem susceptibility to NDM-1-positive pathogens in vitro and in vivo. J. Antimicrob. Chemother. 2017, 72, 782–790. [Google Scholar] [PubMed]
- Geller, B.L.; Marshall-Batty, K.; Schnell, F.J.; McKnight, M.M.; Iversen, P.L.; Greenberg, D.E. Gene-silencing antisense oligomers inhibit acinetobacter growth in vitro and in vivo. J. Infect. Dis. 2013, 208, 1553–1560. [Google Scholar] [CrossRef] [Green Version]
- Lopez, C.; Arivett, B.A.; Actis, L.A.; Tolmasky, M.E. Inhibition of AAC(6′)-Ib-Mediated Resistance to Amikacin in Acinetobacter baumannii by an Antisense Peptide-Conjugated 2′,4′-Bridged Nucleic Acid-NC-DNA Hybrid Oligomer. Antimicrob. Agents Chemother. 2015, 59, 5798–5803. [Google Scholar] [CrossRef] [Green Version]
- Soler Bistué, A.J.C.; Martín, F.A.; Vozza, N.; Ha, H.; Joaquín, J.C.; Zorreguieta, A.; Tolmasky, M.E. Inhibition of aac(6′)-Ib-mediated amikacin resistance by nuclease-resistant external guide sequences in bacteria. Proc. Natl. Acad. Sci. USA 2009, 106, 13230–13235. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Guitián, M.; Vázquez-Ucha, J.C.; Álvarez-Fraga, L.; Conde-Pérez, K.; Bou, G.; Poza, M.; Beceiro, A. Antisense inhibition of lpxB gene expression in Acinetobacter baumannii by peptide–PNA conjugates and synergy with colistin. J. Antimicrob. Chemother. 2020, 75, 51–59. [Google Scholar] [CrossRef]
- Sarno, R.; Ha, H.; Weinsetel, N.; Tolmasky, M.E. Inhibition of aminoglycoside 6′-N-acetyltransferase type Ib-mediated amikacin resistance by antisense oligodeoxynucleotides. Antimicrob. Agents Chemother. 2003, 47, 3296–3304. [Google Scholar] [CrossRef] [Green Version]
- Arivett, B.A.; Fiester, S.E.; Ream, D.C.; Centrón, D.; Ramírez, M.S.; Tolmasky, M.E.; Actis, L.A. Draft Genome of the Multidrug-Resistant Acinetobacter baumannii Strain A155 Clinical Isolate. Genome Announc. 2015, 3, e00212-15. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, M.S.; Xie, G.; Marshall, S.H.; Hujer, K.M.; Chain, P.S.G.; Bonomo, R.A.; Tolmasky, M.E. Multidrug-resistant (MDR) Klebsiella pneumoniae clinical isolates: A zone of high heterogeneity (HHZ) as a tool for epidemiological studies. Clin. Microbiol. Infect. 2012, 18, E254–E258. [Google Scholar] [CrossRef] [Green Version]
- Howard, J.J.; Sturge, C.R.; Moustafa, D.A.; Daly, S.M.; Marshall-Batty, K.R.; Felder, C.F.; Zamora, D.; Yabe-Gill, M.; Labandeira-Rey, M.; Bailey, S.M.; et al. Inhibition of Pseudomonas aeruginosa by Peptide-Conjugated Phosphorodiamidate Morpholino Oligomers. Antimicrob. Agents Chemother. 2017, 61, e01938-16. [Google Scholar] [CrossRef] [Green Version]
- Montagner, G.; Bezzerri, V.; Cabrini, G.; Fabbri, E.; Borgatti, M.; Lampronti, I.; Finotti, A.; Nielsen, P.E.; Gambari, R. An antisense peptide nucleic acid against Pseudomonas aeruginosa inhibiting bacterial-induced inflammatory responses in the cystic fibrosis IB3-1 cellular model system. Int. J. Biol. Macromol. 2017, 99, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Tekintas, Y.; Demir-Dora, D.; Erac, B.; Erac, Y.; Yilmaz, O.; Aydemir, S.S.; Kocagoz, Z.T.; Hosgor-Limoncu, M. Silencing acpP gene via antisense oligonucleotide-niosome complex in clinical Pseudomonas aeruginosa isolates. Res. Microbiol. 2021, 172, 103834. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, D.E.; Marshall-Batty, K.R.; Brinster, L.R.; Zarember, K.A.; Shaw, P.A.; Mellbye, B.L.; Iversen, P.L.; Holland, S.M.; Geller, B.L. Antisense phosphorodiamidate morpholino oligomers targeted to an essential gene inhibit Burkholderia cepacia complex. J. Infect. Dis. 2010, 201, 1822–1830. [Google Scholar] [CrossRef] [Green Version]
- Sawyer, A.J.; Wesolowski, D.; Gandotra, N.; Stojadinovic, A.; Izadjoo, M.; Altman, S.; Kyriakides, T.R. A peptide-morpholino oligomer conjugate targeting Staphylococcus aureus gyrA mRNA improves healing in an infected mouse cutaneous wound model. Int. J. Pharm. 2013, 453, 651–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, J.; Da, F.; Ma, X.; Wang, N.; Wang, Y.; Zhang, H.; Li, M.; Zhou, Y.; Xue, X.; Hou, Z.; et al. Antisense growth inhibition of methicillin-resistant Staphylococcus aureus by locked nucleic acid conjugated with cell-penetrating peptide as a novel FtsZ inhibitor. Antimicrob. Agents Chemother. 2015, 59, 914–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goh, S.; Loeffler, A.; Lloyd, D.H.; Nair, S.P.; Good, L. Oxacillin sensitization of methicillin-resistant Staphylococcus aureus and methicillin-resistant Staphylococcus pseudintermedius by antisense peptide nucleic acids in vitro. BMC Microbiol. 2015, 15, 262. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; He, G.; Wang, H.; Jia, M.; Ma, X.; Da, F.; Wang, N.; Hou, Z.; Xue, X.; Li, M.; et al. Reversion of antibiotic resistance by inhibiting mecA in clinical methicillin-resistant Staphylococci by antisense phosphorothioate oligonucleotide. J. Antibiot. 2015, 68, 158–164. [Google Scholar] [CrossRef]
- Bai, H.; Sang, G.; You, Y.; Xue, X.; Zhou, Y.; Hou, Z.; Meng, J.; Luo, X. Targeting RNA polymerase primary σ70 as a therapeutic strategy against methicillin-resistant Staphylococcus aureus by antisense peptide nucleic acid. PLoS ONE 2012, 7, e29886. [Google Scholar] [CrossRef]
- Abushahba, M.F.N.; Mohammad, H.; Thangamani, S.; Hussein, A.A.A.; Seleem, M.N. Impact of different cell penetrating peptides on the efficacy of antisense therapeutics for targeting intracellular pathogens. Sci. Rep. 2016, 6, 20832. [Google Scholar] [CrossRef]
- Rajasekaran, P.; Alexander, J.C.; Seleem, M.N.; Jain, N.; Sriranganathan, N.; Wattam, A.R.; Setubal, J.C.; Boyle, S.M. Peptide nucleic acids inhibit growth of Brucella suis in pure culture and in infected murine macrophages. Int. J. Antimicrob. Agents 2013, 41, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Matthews, H.; Hanison, J.; Nirmalan, N. ‘Omics’-informed drug and biomarker discovery: Opportunities, challenges and future perspectives. Proteomes 2016, 4, 28. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Drug | Primary Medical Indication | Antimicrobial Activity | Mechanism of Action | Reference |
|---|---|---|---|---|
| Mitomycin C | Anticancer | E. coli, S. aureus, P. aeruginosa | Crosslinking DNA. Eradicates cells in biofilms. | [26] |
| Cisplatin | Anticancer | E. coli, S. aureus, P. aeruginosa | Crosslinking DNA. Effective against biofilm and planktonic cells of P. aeruginosa. | [27] |
| Gallium | Anticancer | P. aeruginosa (CF patients isolates), K. pneumoniae, A. baumannii | P. aeruginosa: Disrupts iron metabolism and increases oxidant sensitivity. K. pneumoniae: reduces CFUs and biofilm. A. baumannii: disrupts the iron metabolism. | [28,29,30] |
| 5-fluoro-2′-deoxyuridine | Anticancer | MRSA, VRSA, MSSA, VISA, E. faecium; Enterecoccus faecalis | Inhibits thymidylate synthase and impairs nucleic acids’ metabolism and structure. | [47] |
| Curcumin | Anticancer | P. aeruginosa, S. aureus | Causes nucleic acid and protein leakage, indicating impairment of the membrane. | [48] |
| Simvastatin | Statin | MSSA, MRSA, VRE, VSE | Targets HMG-CoA reductase in eukaryotes. However, no mechanism described for prokaryotes. | [31] |
| Niclosamide | Anthelmintic | P. aeruginosa | Inhibits the QS response and the production of acyl-homoserine lactone signal molecules. Inhibits the transcription of genes related to adhesion and biofilm formation. | [32] |
| Terfenadine | Antihistamine | S. aureus, E. faecium, E. faecalis | Type II topoisomerase inhibitor. Targets both DNA gyrase and topoisomerase IV. Antimicrobial activity against planktonic, biofilm and small-colony variant (scv) forms of S. aureus. | [33] |
| Verteporfin | Photosensitizer | S. aureus | Inhibits the GraXRS-dependent promoter. Sensitizes bacteria against PMN cells. | [34] |
| Lidocaine | Anesthetic | P. aeruginosa, E. coli, S. aureus | No mechanism described. | [35] |
| Prilocaine | Anesthetic | P. aeruginosa, E. coli, S. aureus | No mechanism described. | [35] |
| Bupivacaine | Anesthetic | MRSA, S. aureus, E. coli | No mechanism described. | [36] |
| Pethidine | Opioid | MRSA, S. aureus, E. coli, P. aeruginosa | No mechanism described. | [36] |
| Penfluridol | Antipsychotic | E. faecalis | Eradicates biofilm, probably inhibiting the QS system and the second messenger c-di-GMP. | [49] |
| Thioridazine | Antipsychotic | MRSA, Enterococcus species | MRSA: Inhibits bacterial efflux pumps. Inhibits replication of phagocytosed MRSA or causing ultrastructural changes in the cell envelope and consequent lysis after phagocytosis. | [50] |
| Sertraline | Antidepressant | S. aureus, E. coli, P. aeruginosa | Hypothesized to inhibit efflux pumps in bacteria once it is a serotonin reuptake inhibitor in humans. However, further studies are required. | [51] |
| Ebselen | Anti-inflammatory, antioxidative and antiatherosclerotic (safety proven, but not clinically used) | MRSA, VRSA, MSSA, VISA, E. coli, E. faecium, E. fecalis | Inhibits the TrxR system. | [47,52] |
| Ibuprofen | Anti-inflammatory | P. aeruginosa, Burkholderia species, E. faecalis | Suggested to uncouple oxidative phosphorylation in bacteria, to alter bacterial hydrophobicity, hemolysin production and inhibit fimbriae. Reduces biofilm biomass. | [53,54] |
| Diclofenac | Anti-inflammatory | E. faecalis | Suggested inhibition of bacterial DNA synthesis or impairment of membrane mechanisms. However, further studies are required. | [54] |
| Ticagrelor | Antiplatelet | MSSA, GISA, MRSA, VRE | Inhibits MRSA and VRE biofilm formation. | [55] |
| Auranofin | Antirheumatic | MRSA, E. faecium, E. faecalis | MRSA: Downregulates the proteins of 5 major biosynthetic pathways (DNA, RNA, protein, cell-wall, and lipid synthesis). Reduces MRSA toxins. Eradicates intracellular MRSA in infected macrophages. Enterococcus: Antibiofilm activity. Inhibits selenium metabolism and selenoenzymes. | [56,57] |
| Glatiramer acetate | Multiple sclerosis | E. coli, A. baumannii, P. aeruginosa (CF patients isolates) | Forms intracellular condensates. | [58] |
| Ciclopirox | Antifungal | P. aeruginosa, Proteus mirabilis, E. coli, K. pneumoniae, S. aureus, Corynebacterium spp., A. baumannii | Affects LPS composition and galactose metabolism in E. coli. | [59,60] |
| Drug | Primary Medical Indication | Synergy with | Antimicrobial Activity | Reference |
|---|---|---|---|---|
| Gallium | Anticancer | Colistin | A. baumannii | [30] |
| Curcumin | Anticancer | Ciprofloxacin | P. aeruginosa; S. aureus | [48] |
| Penfluridol | Antipsychotic | Amikacin, GentamycinAdditive effect: Vancomycin, Teicoplanin | E. faecalis | [49] |
| Thioridazine | Antipsychotic | Vancomycin, Ampicillin | Enterococcus species | [50] |
| Sertraline | Antidepressant | Ciprofloxacin, Norfloxacin, Moxifloxacin, Gentamicin, Levofloxacin. Resistance of E. coli to cefexime was reversed. | S. aureus, E. coli, P. aeruginosa | [51] |
| Salicylanilides (Oxyclozanide, Rafoxanide, Closantel) | Internal antiparasitic for veterinary use | Colistin | E. cloacae, A. baumannii, K. pneumoniae, E. coli, P. aeruginosa, | [61] |
| Disulfiram | Alcoholism treatment | Minocycline | S. aureus | [62] |
| Loperamide | Diarrhea treatment | Minocycline | P. aeruginosa; E. coli | [62] |
| Ticagrelor | Antiplatelet | Rifampicin, Ciprofloxacin and Vancomycin | MRSA | [55] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sousa, S.A.; Feliciano, J.R.; Pita, T.; Soeiro, C.F.; Mendes, B.L.; Alves, L.G.; Leitão, J.H. Bacterial Nosocomial Infections: Multidrug Resistance as a Trigger for the Development of Novel Antimicrobials. Antibiotics 2021, 10, 942. https://doi.org/10.3390/antibiotics10080942
Sousa SA, Feliciano JR, Pita T, Soeiro CF, Mendes BL, Alves LG, Leitão JH. Bacterial Nosocomial Infections: Multidrug Resistance as a Trigger for the Development of Novel Antimicrobials. Antibiotics. 2021; 10(8):942. https://doi.org/10.3390/antibiotics10080942
Chicago/Turabian StyleSousa, Sílvia A., Joana R. Feliciano, Tiago Pita, Catarina F. Soeiro, Beatriz L. Mendes, Luis G. Alves, and Jorge H. Leitão. 2021. "Bacterial Nosocomial Infections: Multidrug Resistance as a Trigger for the Development of Novel Antimicrobials" Antibiotics 10, no. 8: 942. https://doi.org/10.3390/antibiotics10080942