
Bacterial Resistance to Antimicrobial Agents

 ,
,  , and
, and
Abstract
:1. Introduction
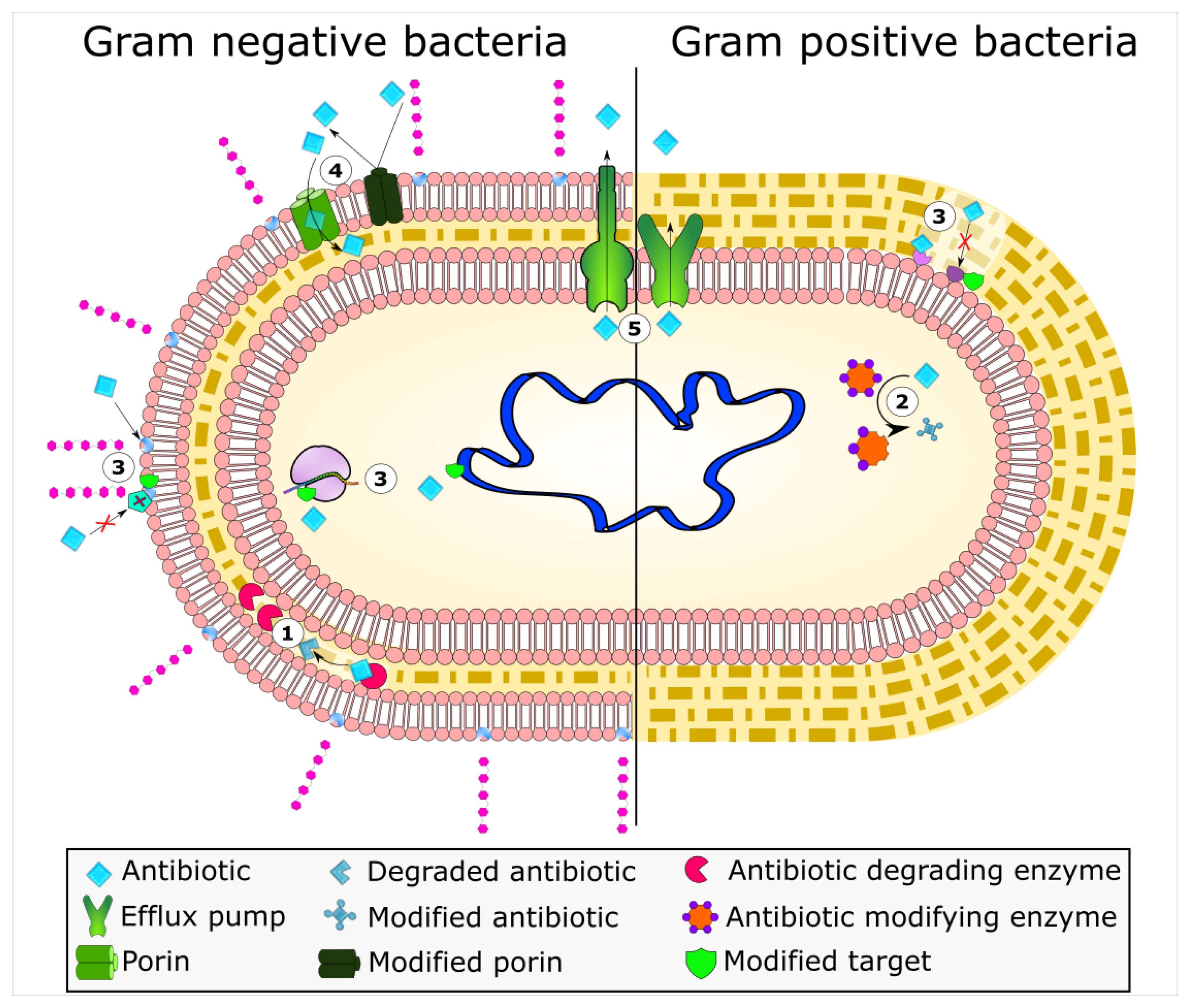
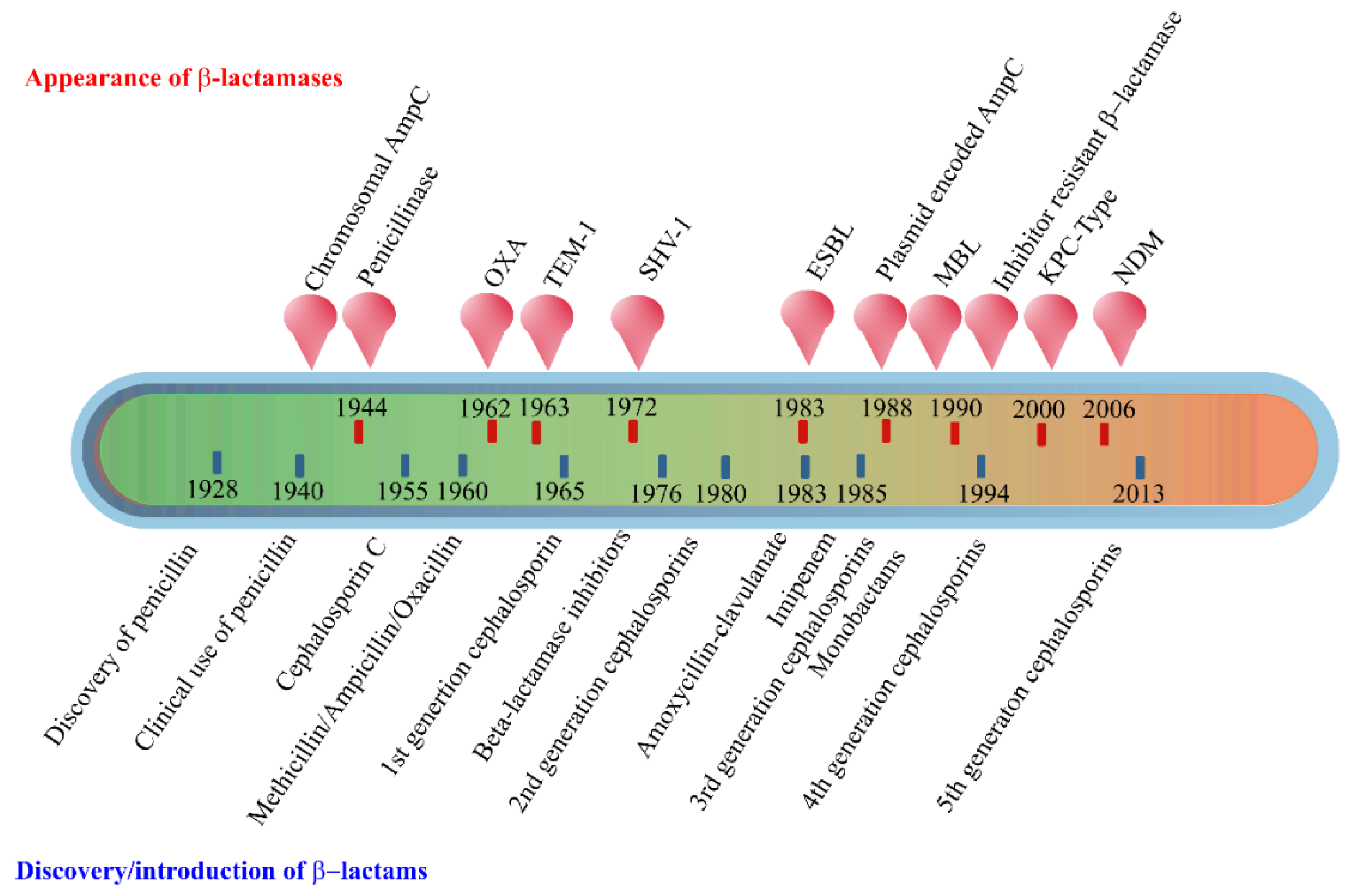
2. Enzyme-Based Antimicrobial-Inactivation Systems
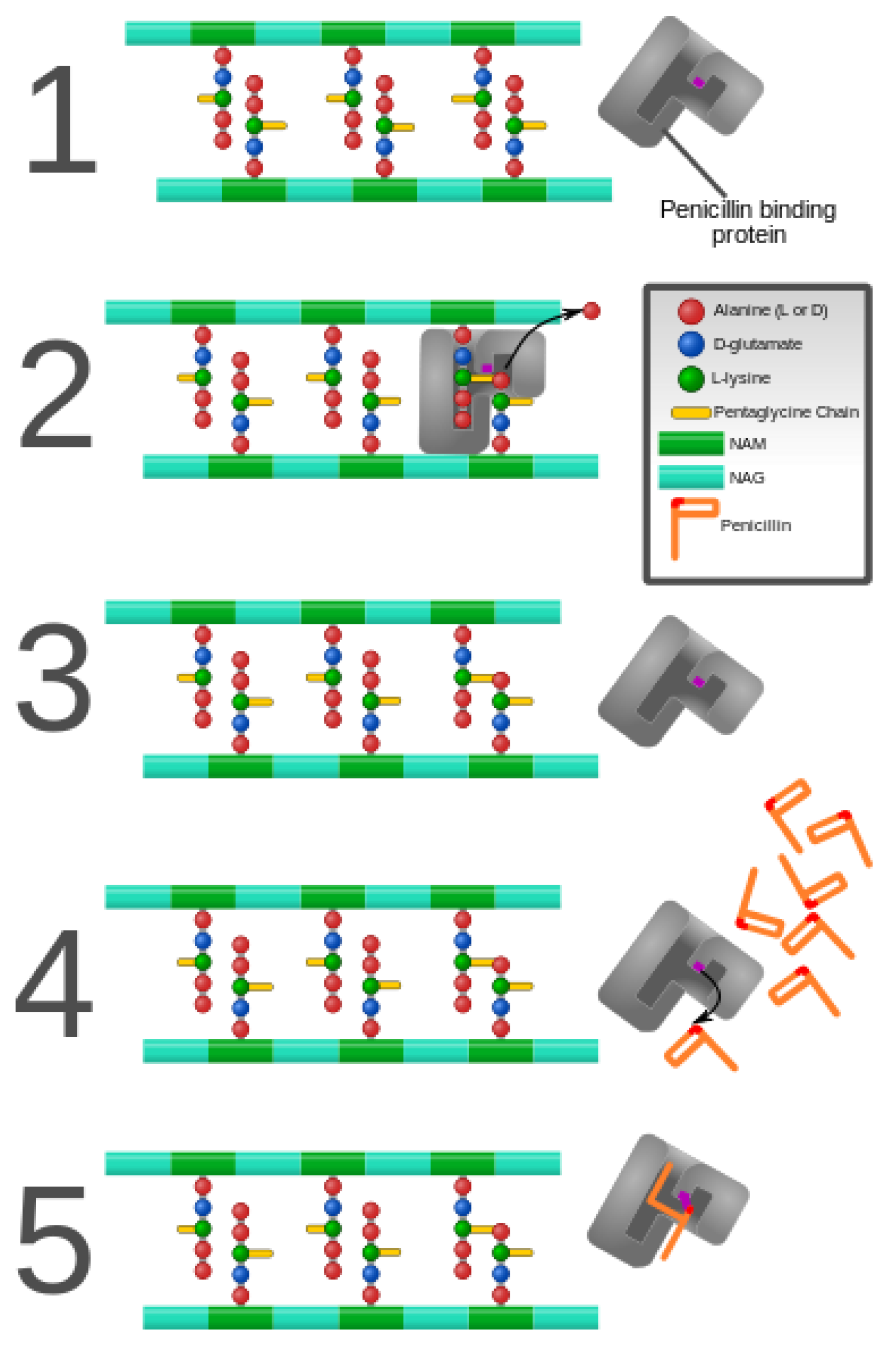
3. Alteration of Antimicrobial Targets
4. Protection of Antimicrobial Targets
4.1. Ribosomal Protection Protein (RPP)
4.2. Quinolone Resistance Proteins
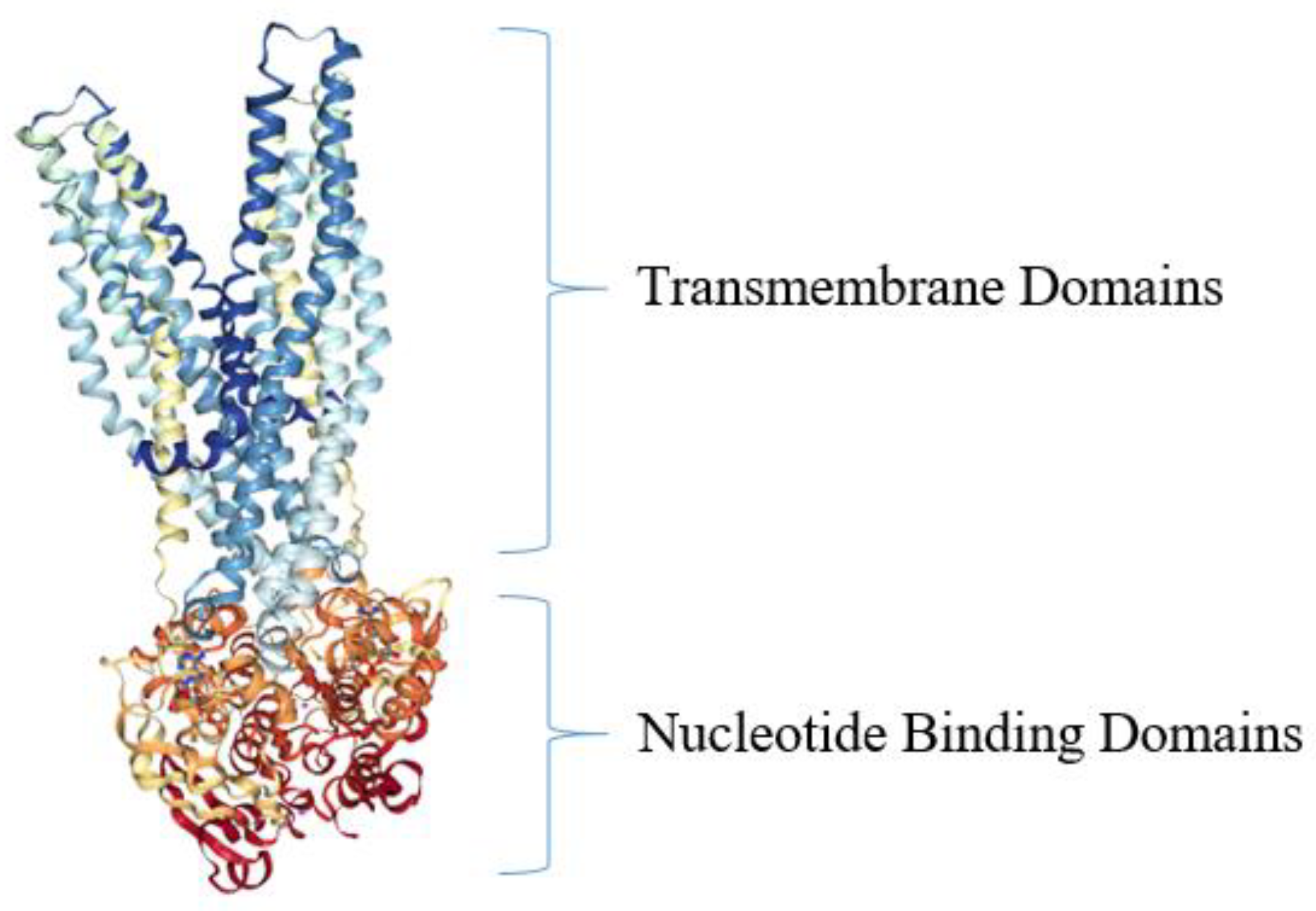
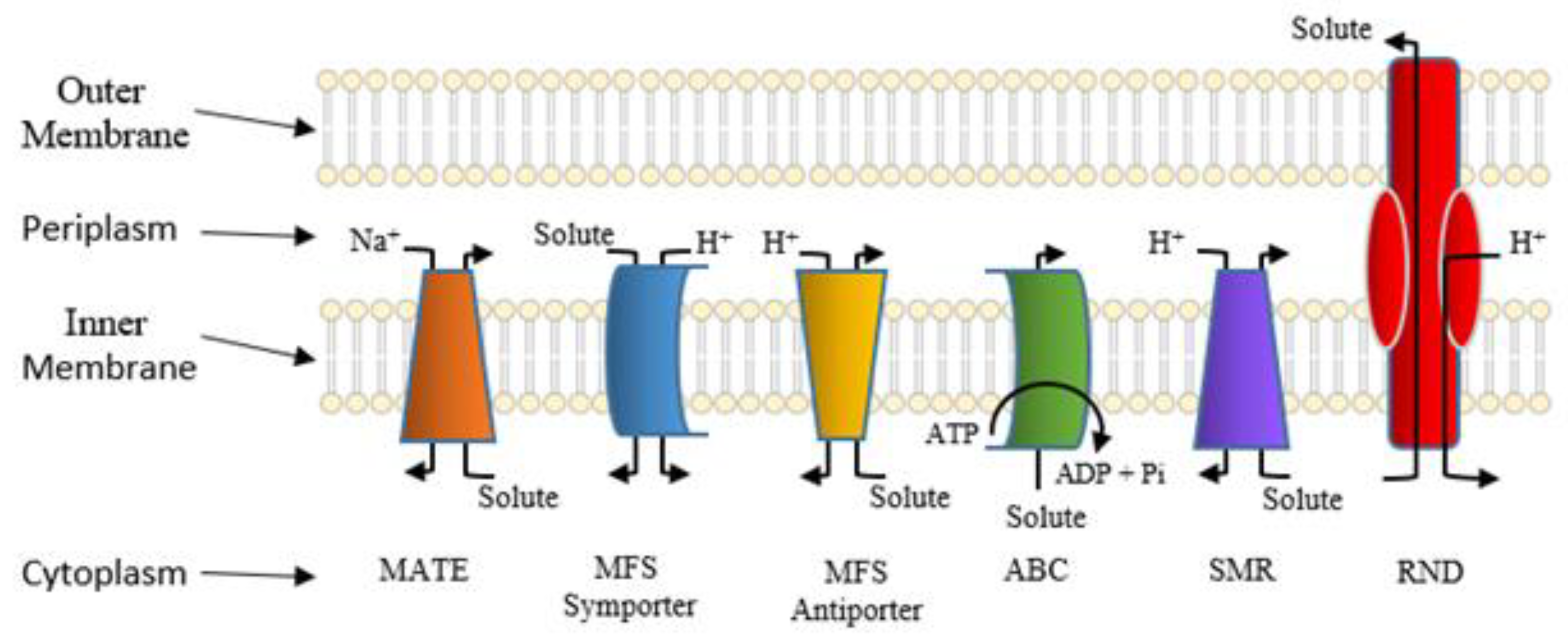
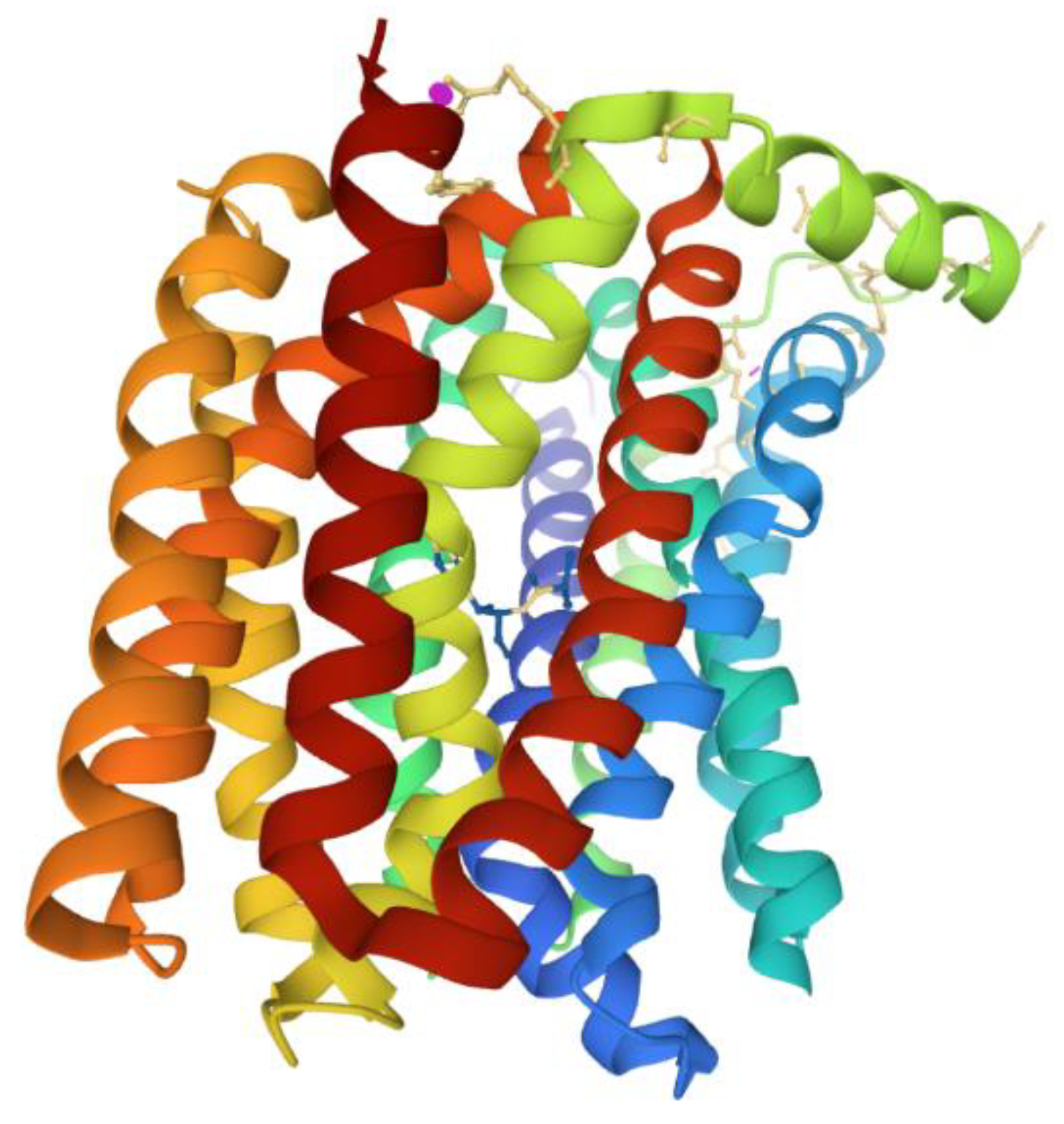
5. Active Efflux Pumps of Antimicrobial Agents
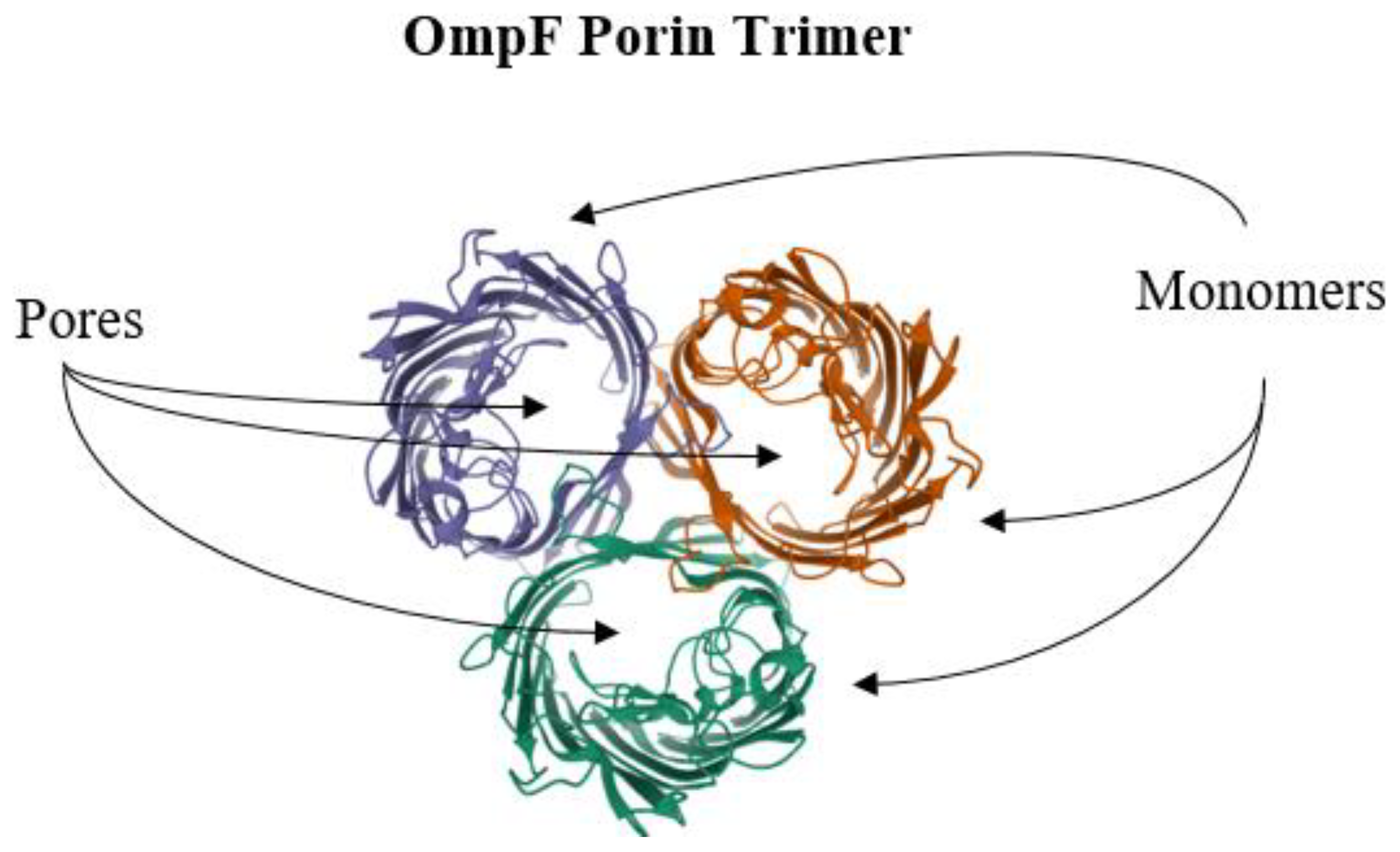
6. Reduction of Antimicrobial Permeability into Bacterial Cells
7. Future Directions
8. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| AAC | N-acetyltransferases |
| AME | aminoglycoside-modifying enzymes |
| ANT | O-adenyltransferases |
| APH | O-phosphotransferases |
| CAT | chloramphenicol acetyltransferase |
| CRE | carbapenem-resistant Enterobacteriaceae |
| CTX-M | cefotaximase |
| ESBLs | extended-spectrum β-lactamases |
| ESKAPE | Enterococcus, S. aureus, K. pneumoniae, A. baumannii, P. aeruginosa, and E. coli |
| MATE | multidrug and toxic compound extrusion |
| MFS | major facilitator superfamily |
| MOP | multidrug/oligosaccharidyl-lipid/polysaccharide |
| NAG | N-acetylglucosamine |
| NAM | N-acetylmuramic acid |
| NBDs | nucleotide-binding domains |
| NDM | New Delhi Metallo-β-lactamase |
| PACE | proteobacterial antimicrobial compound efflux |
| PBPs | penicillin-binding proteins |
| RND | resistance-nodulation-cell division |
| TEM | temoneira |
| TMDs | transmembrane domains |
| SHV | sulfhydryl variable |
References
- Ziebuhr, W.; Ohlsen, K.; Karch, H.; Korhonen, T.; Hacker, J. Evolution of bacterial pathogenesis. Cell. Mol. Life Sci. CMLS 1999, 56, 719–728. [Google Scholar] [CrossRef]
- Walsh, C. Molecular mechanisms that confer antibacterial drug resistance. Nature 2000, 406, 775–781. [Google Scholar] [CrossRef]
- Neu, H.C. The crisis in antibiotic resistance. Science 1992, 257, 1064–1073. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Varela, M.F. Molecular mechanisms of bacterial resistance to antimicrobial agents. Microbial Pathogens and Strategies for Combating Them: Science, Technology and Education. In Microbial Pathogens and Strategies for Combating Them: Science, Technology and Education; Méndez-Vilas, A., Ed.; Formatex Research Center: Badajoz, Spain, 2013; pp. 522–534. [Google Scholar]
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef]
- Wright, G.D. Molecular mechanisms of antibiotic resistance. Chem. Commun. 2011, 47, 4055–4061. [Google Scholar] [CrossRef]
- Hughes, D. Selection and Evolution of Resistance to Antimicrobial Drugs. Iubmb Life 2014, 66, 521–529. [Google Scholar] [CrossRef]
- Vivas, R.; Barbosa, A.A.T.; Dolabela, S.S.; Jain, S. Multidrug-Resistant Bacteria and Alternative Methods to Control Them: An Overview. Microb. Drug Resist. 2019, 25, 890–908. [Google Scholar] [CrossRef]
- Silbergeld, E.K.; Graham, J.; Price, L.B. Industrial Food Animal Production, Antimicrobial Resistance, and Human Health. Annu. Rev. Public Health 2008, 29, 151–169. [Google Scholar] [CrossRef]
- Varela, M.F.; Kumar, S. Strategies for Discovery of New Molecular Targets for Anti-Infective Drugs. Curr. Opin. Pharm. 2019, 48, 57–68. [Google Scholar] [CrossRef]
- Pollock, M.R. Origin and Function of Penicillinase: A Problem in Biochemical Evolution. Br. Med. J. 1967, 4, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Bush, K.; Jacoby, G.A. Updated Functional Classification of β-Lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [CrossRef] [Green Version]
- Abraham, E.P. A Retrospective View of β-Lactamases. J. Chemother. 1991, 3, 67–74. [Google Scholar] [CrossRef]
- Bush, K. Past and Present Perspectives on β-Lactamases. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naas, T.; Oxacelay, C.; Nordmann, P. Identification of CTX-M-Type Extended-Spectrum-β-Lactamase Genes Using Real-Time PCR and Pyrosequencing. Antimicrob. Agents Chemother. 2007, 51, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, E.P.; Chain, E. An Enzyme from Bacteria Able to Destroy Penicillin. 1940. Rev. Infect. Dis. 1988, 10, 677–678. [Google Scholar] [PubMed]
- Rammelkamp, C.H.; Maxon, T. Resistance of Staphylococcus aureus to the Action of Penicillin. Proc. Soc. Exp. Biol. Med. 1942, 51, 386–389. [Google Scholar] [CrossRef]
- Paterson, D.L.; Bonomo, R.A. Extended-Spectrum β-Lactamases: A Clinical Update. Clin. Microbiol. Rev. 2005, 18, 657–686. [Google Scholar] [CrossRef] [Green Version]
- Sirot, J.; Chanal, C.; Petit, A.; Sirot, D.; Labia, R.; Gerbaud, G. Klebsiella pneumoniae and Other Enterobacteriaceae Producing Novel Plasmid-Mediated β-Lactamases Markedly Active against Third-Generation Cephalosporins: Epidemiologic Studies. Rev. Infect. Dis. 1988, 10, 850–859. [Google Scholar] [CrossRef]
- Abraham, E.P.; Newton, G.G. The Structure of Cephalesporin C. Biochem. J. 1961, 79, 377–393. [Google Scholar] [CrossRef] [Green Version]
- González-Candelas, F.; Comas, I.; Martínez, J.; Luis; Galán, J.; Carlos; Baquero, F. 12—The Evolution of Antibiotic Resistance. In Genetics and Evolution of Infectious Disease; Tibayrenc, M., Ed.; Elsevier: London, UK, 2011; pp. 305–337. ISBN 978-0-12-384890-1. [Google Scholar]
- Humeniuk, C.; Arlet, G.; Gautier, V.; Grimont, P.; Labia, R.; Philippon, A. β-Lactamases of Kluyvera ascorbata, Probable Progenitors of Some Plasmid-Encoded CTX-M Types. Antimicrob. Agents Chemother. 2002, 46, 3045–3049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bush, K. Bench-to-Bedside Review: The Role of β-Lactamases in Antibiotic-Resistant Gram-Negative Infections. Crit. Care 2010, 14, 224. [Google Scholar] [CrossRef] [Green Version]
- Bush, K.; Jacoby, G.A.; Medeiros, A.A. A Functional Classification Scheme for β-Lactamases and Its Correlation with Molecular Structure. Antimicrob. Agents Chemother. 1995, 39, 1211–1233. [Google Scholar] [CrossRef] [Green Version]
- Davidson, A.L.; Dassa, E.; Orelle, C.; Chen, J. Structure, Function, and Evolution of Bacterial ATP-Binding Cassette Systems. Microbiol. Mol. Biol. Rev. 2008, 72, 317–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edlund, T.; Grundström, T.; Normark, S. Isolation and Characterization of DNA Repetitions Carrying the Chromosomal β-Lactamase Gene of Escherichia coli K-12. Mol. Gen. Genet. 1979, 173, 115–125. [Google Scholar] [CrossRef]
- Jaurin, B.; Grundström, T. AmpC Cephalosporinase of Escherichia coli K-12 Has a Different Evolutionary Origin from That of β-Lactamases of the Penicillinase Type. Proc. Natl. Acad. Sci. USA 1981, 78, 4897–4901. [Google Scholar] [CrossRef] [Green Version]
- Queenan, A.M.; Bush, K. Carbapenemases: The Versatile β-Lactamases. Clin. Microbiol. Rev. 2007, 20, 440–458. [Google Scholar] [CrossRef] [Green Version]
- Yong, D.; Toleman, M.A.; Giske, C.G.; Cho, H.S.; Sundman, K.; Lee, K.; Walsh, T.R. Characterization of a New Metallo-β-Lactamase Gene, Bla(NDM-1), and a Novel Erythromycin Esterase Gene Carried on a Unique Genetic Structure in Klebsiella pneumoniae Sequence Type 14 from India. Antimicrob. Agents Chemother. 2009, 53, 5046–5054. [Google Scholar] [CrossRef] [Green Version]
- Antunes, N.T.; Fisher, J.F. Acquired Class D β-Lactamases. Antibiotics 2014, 3, 398–434. [Google Scholar] [CrossRef]
- Evans, B.A.; Amyes, S.G.B. OXA β-Lactamases. Clin. Microbiol. Rev. 2014, 27, 241–263. [Google Scholar] [CrossRef] [Green Version]
- Afzal-Shah, M.; Woodford, N.; Livermore, D.M. Characterization of OXA-25, OXA-26, and OXA-27, Molecular Class D β-Lactamases Associated with Carbapenem Resistance in Clinical Isolates of Acinetobacter baumannii. Antimicrob. Agents Chemother. 2001, 45, 583–588. [Google Scholar] [CrossRef] [Green Version]
- Founou, R.C.; Founou, L.L.; Essack, S.Y. Clinical and Economic Impact of Antibiotic Resistance in Developing Countries: A Systematic Review and Meta-Analysis. PLoS ONE 2017, 12, 0189621. [Google Scholar] [CrossRef] [Green Version]
- Zhen, X.; Lundborg, C.S.; Sun, X.; Hu, X.; Dong, H. Economic burden of antibiotic resistance in ESKAPE organisms: A systematic review. Antimicrob. Resist. Infect. Control 2019, 8, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y. Discovery, Research, and Development of New Antibiotics: The Who Priority List of Antibiotic-Resistant Bacteria and Tuberculosis. Lancet. Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Barthelemy, P.; Autissier, D.; Gerbaud, G.; Courvalin, P. Enzymic Hydrolysis of Erythromycin by a Strain of Escherichia coli. J. Antibiot. 1984, 37, 1692–1696. [Google Scholar]
- Morar, M.; Pengelly, K.; Koteva, K.; Wright, G.D. Mechanism and Diversity of the Erythromycin Esterase Family of Enzymes. Biochemistry 2012, 51, 1740–1751. [Google Scholar] [CrossRef]
- Golkar, T.; Zieliński, M.; Berghuis, A.M. Look and Outlook on Enzyme-Mediated Macrolide Resistance. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Castaneda-Garcia, A.; Blazquez, J.; Rodriguez-Rojas, A. Molecular mechanisms and clinical impact of acquired and intrinsic fosfomycin resistance. Antibiotics 2013, 2, 217–236. [Google Scholar] [CrossRef] [Green Version]
- Silver, L.L. Fosfomycin: Mechanism and Resistance. Cold Spring Harb. Perspect. Med. 2017, 7, a025262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, C. Antibiotics: Actions, Origins, Resistance; American Society for Microbiology: Washington, DC, USA, 2003. [Google Scholar]
- Yang, W.; Moore, I.F.; Koteva, K.P.; Bareich, D.C.; Hughes, D.W.; Wright, G.D. TetX Is a Flavin-Dependent Monooxygenase Conferring Resistance to Tetracycline Antibiotics. J. Biol. Chem. 2004, 279, 52346–52352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koteva, K.; Cox, G.; Kelso, J.K.; Surette, M.D.; Zubyk, H.L.; Ejim, L.; Stogios, P.; Savchenko, A.; Sørensen, D.; Wright, G.D. Rox, a Rifamycin Resistance Enzyme with an Unprecedented Mechanism of Action. Cell Chem. Biol. 2018, 25, 403–412.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, G.D. Bacterial Resistance to Antibiotics: Enzymatic Degradation and Modification. Adv. Drug Deliv. Rev. 2005, 57, 1451–1470. [Google Scholar] [CrossRef] [PubMed]
- Garneau-Tsodikova, S.; Labby, K.J. Mechanisms of Resistance to Aminoglycoside Antibiotics: Overview and Perspectives. Medchemcomm 2016, 7, 11–27. [Google Scholar] [CrossRef] [Green Version]
- Baysarowich, J.; Koteva, K.; Hughes, D.W.; Ejim, L.; Griffiths, E.; Zhang, K.; Junop, M.; Wright, G.D. Rifamycin Antibiotic Resistance by ADP-Ribosylation: Structure and Diversity of Arr. Proc. Natl. Acad. Sci. USA 2008, 105, 4886–4891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, W.V. Chloramphenicol Acetyltransferase: Enzymology and Molecular Biology. Crit. Rev. Biochem. 1983, 14, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Lambert, P.A. Bacterial Resistance to Antibiotics: Modified Target Sites. Adv. Drug Deliv. Rev. 2005, 57, 1471–1485. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.E. Structure, Biochemistry and Mechanism of Action of Glycopeptide Antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 1989, 8, 943–950. [Google Scholar] [CrossRef]
- Bugg, T.D.; Wright, G.D.; Dutka-Malen, S.; Arthur, M.; Courvalin, P.; Walsh, C.T. Molecular Basis for Vancomycin Resistance in Enterococcus faecium Bm4147: Biosynthesis of a Depsipeptide Peptidoglycan Precursor by Vancomycin Resistance Proteins VanH and VanA. Biochemistry 1991, 30, 10408–10415. [Google Scholar] [CrossRef]
- Billot-Klein, D.; Gutmann, L.; Sable, S.; Guittet, E.; Van Heijenoort, J. Modification of Peptidoglycan Precursors Is a Common Feature of the Low-Level Vancomycin-Resistant Vanb-Type Enterococcus D366 and of the Naturally Glycopeptide-Resistant Species Lactobacillus casei, Pediococcus pentosaceus, Leuconostoc mesenteroides, and Enterococcus gallinarum. J. Bacteriol. 1994, 176, 2398–2405. [Google Scholar]
- Cooper, M.A.; Fiorini, M.T.; Abell, C.; Williams, D.H. Binding of Vancomycin Group Antibiotics to D-Alanine and d-Lactate Presenting Self-Assembled Monolayers. Bioorganic Med. Chem. 2000, 8, 2609–2616. [Google Scholar] [CrossRef]
- Perichon, B.; Reynolds, P.; Courvalin, P. Vand-Type Glycopeptide-Resistant Enterococcus faecium Bm4339. Antimicrob. Agents Chemother. 1997, 41, 2016–2018. [Google Scholar] [CrossRef] [Green Version]
- Fines, M.; Perichon, B.; Reynolds, P.; Sahm, D.F.; Courvalin, P. Vane, a New Type of Acquired Glycopeptide Resistance in Enterococcus faecalis Bm4405. Antimicrob. Agents Chemother. 1999, 43, 2161–2164. [Google Scholar] [CrossRef] [Green Version]
- McKessar, S.J.; Berry, A.M.; Bell, J.M.; Turnidge, J.D.; Paton, J.C. Genetic Characterization of VanG, a Novel Vancomycin Resistance Locus of Enterococcus faecalis. Antimicrob. Agents Chemother. 2000, 44, 3224–3228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Champney, W.S. Bacterial Ribosomal Subunit Assembly Is an Antibiotic Target. Curr. Top. Med. Chem. 2003, 3, 929–947. [Google Scholar] [CrossRef] [PubMed]
- Weisblum, B. Erythromycin Resistance by Ribosome Modification. Antimicrob. Agents Chemother. 1995, 39, 577–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannuffel, P.; Di Giambattista, M.; Morgan, E.A.; Cocito, C. Identification of a Single Base Change in Ribosomal RNA Leading to Erythromycin Resistance. J. Biol. Chem. 1992, 267, 8377–8382. [Google Scholar] [CrossRef]
- Wang, G.; Taylor, D.E. Site-Specific Mutations in the 23s rRNA Gene of Helicobacter pylori Confer Two Types of Resistance to Macrolide-Lincosamide-Streptogramin B Antibiotics. Antimicrob. Agents Chemother. 1998, 42, 1952–1958. [Google Scholar] [CrossRef] [Green Version]
- Ross, J.I.; Eady, E.A.; Cove, J.H.; Jones, C.E.; Ratyal, A.H.; Miller, Y.W.; Vyakrnam, S.; Cunliffe, W.J. Clinical Resistance to Erythromycin and Clindamycin in Cutaneous Propionibacteria Isolated from Acne Patients is Associated with Mutations in 23s rRNA. Antimicrob. Agents Chemother. 1997, 41, 1162–1165. [Google Scholar] [CrossRef] [Green Version]
- Reinert, R.R.; Wild, A.; Appelbaum, P.; Lutticken, R.; Cil, M.Y.; Al-Lahham, A. Ribosomal Mutations Conferring Resistance to Macrolides in Streptococcus pneumoniae Clinical Strains Isolated in Germany. Antimicrob. Agents Chemother. 2003, 47, 2319–2322. [Google Scholar] [CrossRef] [Green Version]
- Canu, A.; Malbruny, B.; Coquemont, M.; Davies, T.A.; Appelbaum, P.C.; Leclercq, R. Diversity of Ribosomal Mutations Conferring Resistance to Macrolides, Clindamycin, Streptogramin, and Telithromycin in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2002, 46, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Swaney, S.M.; Aoki, H.; Ganoza, M.C.; Shinabarger, D.L. The Oxazolidinone Linezolid Inhibits Initiation of Protein Synthesis in Bacteria. Antimicrob. Agents Chemother. 1998, 42, 3251–3255. [Google Scholar] [CrossRef] [Green Version]
- Auckland, C.; Teare, L.; Cooke, F.; Kaufmann, M.E.; Warner, M.; Jones, G.; Bamford, K.; Ayles, H.; Johnson, A.P. Linezolid-Resistant Enterococci: Report of the First Isolates in the United Kingdom. J. Antimicrob. Chemother. 2002, 50, 743–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prystowsky, J.; Siddiqui, F.; Chosay, J.; Shinabarger, D.L.; Millichap, J.; Peterson, L.R.; Noskin, G.A. Resistance to Linezolid: Characterization of Mutations in rRNA and Comparison of Their Occurrences in Vancomycin-Resistant Enterococci. Antimicrob. Agents Chemother. 2001, 45, 2154–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliopoulos, G.M.; Meka, V.G.; Gold, H.S. Antimicrobial resistance to linezolid. Clin. Infect. Dis. 2004, 39, 1010–1015. [Google Scholar]
- Xiong, L.; Kloss, P.; Douthwaite, S.; Andersen, N.M.; Swaney, S.; Shinabarger, D.L.; Mankin, A.S. Oxazolidinone Resistance Mutations in 23s rRNA of Escherichia coli Reveal the Central Region of Domain V as the Primary Site of Drug Action. J. Bacteriol. 2000, 182, 5325–5331. [Google Scholar] [CrossRef] [Green Version]
- Recht, M.I.; Douthwaite, S.; Dahlquist, K.D.; Puglisi, J.D. Effect of Mutations in the A Site of 16 s rRNA on Aminoglycoside Antibiotic-Ribosome Interaction. J. Mol. Biol. 1999, 286, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Recht, M.I.; Puglisi, J.D. Aminoglycoside Resistance with Homogeneous and Heterogeneous Populations of Antibiotic-Resistant Ribosomes. Antimicrob. Agents Chemother. 2001, 45, 2414–2419. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Katsukawa, C.; Tamaru, A.; Abe, C.; Makino, M.; Mizuguchi, Y.; Taniguchi, H. Detection of Kanamycin-Resistant Mycobacterium tuberculosis by Identifying Mutations in the 16S rRNA Gene. J. Clin. Microbiol. 1998, 36, 1220–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prammananan, T.; Sander, P.; Brown, B.A.; Frischkorn, K.; Onyi, G.O.; Zhang, Y.; Böttger, E.C.; Wallace, J.R., Jr. A Single 16S Ribosomal RNA Substitution Is Responsible for Resistance to Amikacin and Other 2-Deoxystreptamine Aminoglycosides in Mycobacterium abscessus and Mycobacterium chelonae. J. Infect. Dis. 1998, 177, 1573–1581. [Google Scholar] [CrossRef] [Green Version]
- Koch, A.L. Bacterial Wall as Target for Attack: Past, Present, and Future Research. Clin. Microbiol. Rev. 2003, 16, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Weidel, W.; Pelzer, H. Bagshaped Macromolecules–a New Outlook on Bacterial Cell Walls. Adv. Enzymol. Relat. Subj. Biochem. 1964, 26, 193–232. [Google Scholar]
- Vollmer, W.; Bertsche, U. Murein (peptidoglycan) structure, architecture and biosynthesis in Escherichia coli. Biochim. Biophys. Acta 2008, 1778, 1714–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heijenoort, J. Formation of the Glycan Chains in the Synthesis of Bacterial Peptidoglycan. Glycobiology 2001, 11, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Demchick, P.; Koch, A.L. The Permeability of the Wall Fabric of Escherichia coli and Bacillus subtilis. J. Bacteriol. 1996, 178, 768–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseleau-Petit, D.; Liebart, J.C.; Ayala, J.A.; D’Ari, R. Unstable Escherichia coli l Forms Revisited: Growth Requires Peptidoglycan Synthesis. J. Bacteriol. 2007, 189, 6512–6520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salton, M.R.J.; Kim, K.S. Structure. In Medical Microbiology; Baron, B., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996; Chapter 2. [Google Scholar]
- Pandey, N.; Cascella, M. β lactam antibiotics In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Miyachiro, M.M.; Contreras-Martel, C.; Dessen, A. Penicillin-Binding Proteins (PBPs) and Bacterial Cell Wall Elongation Complexes. Macromol. Protein Complexes II Struct. Funct. 2019, 93, 273–289. [Google Scholar]
- Cooksey, R.; Swenson, J.; Clark, N.; Gay, E.; Thornsberry, C. Patterns and Mechanisms of β-Lactam Resistance among Isolates of Escherichia coli from Hospitals in the United States. Antimicrob. Agents Chemother. 1990, 34, 739–745. [Google Scholar] [CrossRef] [Green Version]
- Toth, M.; Antunes, N.T.; Stewart, N.K.; Frase, H.; Bhattacharya, M.; Smith, C.A.; Vakulenko, S.B. Class D β-lactamases do exist in gram-positive bacteria. Nat. Chem. Biol. 2016, 12, 9–14. [Google Scholar] [CrossRef]
- Bera, A.; Herbert, S.; Jakob, A.; Vollmer, W.; Gotz, F. Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan o-acetyltransferase oatA is the major determinant for lysozyme resistance of Staphylococcus aureus. Mol. Microbiol. 2005, 55, 778–787. [Google Scholar] [CrossRef]
- Wilson, D.N.; Hauryliuk, V.; Atkinson, G.C.; O’Neill, A.J. Target protection as a key antibiotic resistance mechanism. Nat. Rev. Microbiol. 2020, 18, 637–648. [Google Scholar] [CrossRef]
- Connell, S.R.; Tracz, D.M.; Nierhaus, K.H.; Taylor, D.E. Ribosomal Protection Proteins and Their Mechanism of Tetracycline Resistance. Antimicrob. Agents Chemother. 2003, 47, 3675–3681. [Google Scholar] [CrossRef] [Green Version]
- Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, M.C.; Kenny, G.E. Dissemination of the TetM tetracycline resistance determinant to Ureaplasma urealyticum. Antimicrob. Agents Chemother. 1986, 29, 350–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donhofer, A.; Franckenberg, S.; Wickles, S.; Berninghausen, O.; Beckmann, R.; Wilson, D.N. Structural Basis for TetM-Mediated Tetracycline Resistance. Proc. Natl. Acad. Sci. USA 2012, 109, 16900–16905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, F.; Starosta, A.L.; Arenz, S.; Sohmen, D.; Donhofer, A.; Wilson, D.N. Tetracycline antibiotics and resistance mechanisms. Biol. Chem. 2014, 395, 559–575. [Google Scholar] [CrossRef]
- Morse, S.A.; Johnson, S.R.; Biddle, J.W.; Roberts, M.C. High-Level Tetracycline Resistance in Neisseria gonorrhoeae is Result of Acquisition of Streptococcal TetM Determinant. Antimicrob. Agents Chemother. 1986, 30, 664–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdett, V. Tet(M)-Promoted Release of Tetracycline from Ribosomes Is GTP Dependent. J. Bacteriol. 1996, 178, 3246–3251. [Google Scholar] [CrossRef] [Green Version]
- Vetting, M.W.; Hegde, S.S.; Wang, M.; Jacoby, G.A.; Hooper, D.C.; Blanchard, J.S. Structure of QnrB1, a Plasmid-Mediated Fluoroquinolone Resistance Factor. J. Biol. Chem. 2011, 286, 25265–25273. [Google Scholar] [CrossRef] [Green Version]
- Trieber, C.A.; Burkhardt, N.; Nierhaus, K.H.; Taylor, D.E. Ribosomal Protection from Tetracycline Mediated by Tet(O): Tet(O) Interaction with Ribosomes Is GTP-Dependent. Biol. Chem. 1998, 379, 847–855. [Google Scholar] [CrossRef]
- Li, W.; Atkinson, G.C.; Thakor, N.S.; Allas, U.; Lu, C.C.; Chan, K.Y.; Tenson, T.; Schulten, K.; Wilson, K.S.; Hauryliuk, V. Mechanism of tetracycline resistance by ribosomal protection protein Tet(O). Nat. Commun. 2013, 4, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Martinez, J.M.; Cano, M.E.; Velasco, C.; Martinez-Martinez, L.; Pascual, A. Plasmid-Mediated Quinolone Resistance: An Update. J. Infect. Chemother. Off. J. Jpn. Soc. Chemother. 2011, 17, 149–182. [Google Scholar] [CrossRef]
- Levy, S.B. Active efflux, a common mechanism for biocide and antibiotic resistance. J. Appl. Microbiol. 2002, 92, 65S–71S. [Google Scholar] [CrossRef]
- Kumar, S.; Varela, M.F. Biochemistry of Bacterial Multidrug Efflux Pumps. Int. J. Mol. Sci. 2012, 13, 4484–4495. [Google Scholar] [CrossRef]
- Davidson, A.L.; Maloney, P.C. ABC transporters: How small machines do a big job. Trends Microbiol. 2007, 15, 448–455. [Google Scholar] [CrossRef]
- Shi, Y. Common Folds and Transport Mechanisms of Secondary Active Transporters. Annu. Rev. Biophys. 2013, 42, 51–72. [Google Scholar] [CrossRef]
- West, I.C. Energy Coupling in Secondary Active Transport. Biochim. Biophys. Acta 1980, 604, 91–126. [Google Scholar] [CrossRef]
- Stieger, B.; Higgins, C.F. Twenty Years of ATP-Binding Cassette (ABC) Transporters. Pflug. Arch. Eur. J. Physiol. 2007, 453, 543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, I.B. Rise and Rise of the ABC Transporter Families. Res. Microbiol. 2019, 170, 304–320. [Google Scholar] [CrossRef]
- Lage, H. ABC-transporters: Implications on drug resistance from microorganisms to human cancers. Int. J. Antimicrob. Agents 2003, 22, 188–199. [Google Scholar] [CrossRef]
- Dawson, R.J.; Locher, K.P. Structure of a bacterial multidrug ABC transporter. Nature 2006, 443, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F. ABC transporters: Physiology, structure and mechanism–an overview. Res. Microbiol. 2001, 152, 205–210. [Google Scholar] [CrossRef]
- Rose, A.S.; Bradley, A.R.; Valasatava, Y.; Duarte, J.M.; Prlić, A.; Rose, P.W. Ngl Viewer: Web-Based Molecular Graphics for Large Complexes. Bioinformatics 2018, 34, 3755–3758. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Dawson, R.J.; Locher, K.P. Structure of the Multidrug ABC Transporter Sav1866 from Staphylococcus aureus in Complex with Amp-Pnp. FEBS Lett. 2007, 581, 935–938. [Google Scholar] [CrossRef] [Green Version]
- Poole, K. Bacterial Stress Responses as Determinants of Antimicrobial Resistance. J. Antimicrob. Chemother. 2012, 67, 2069–2089. [Google Scholar] [CrossRef] [Green Version]
- Ambrose, K.D.; Nisbet, R.; Stephens, D.S. Macrolide Efflux in Streptococcus pneumoniae is Mediated by a Dual Efflux Pump (Mel and Mef) and is Erythromycin Inducible. Antimicrob. Agents Chemother. 2005, 49, 4203–4209. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Pei, H.; Huang, B.; Zhu, X.; Zhang, J.; Zhou, B.; Zhu, L.; Zhang, Y.; Zhou, F.F. The Expression of ABC Efflux Pump, Rv1217c–Rv1218c, and Its Association with Multidrug Resistance of Mycobacterium tuberculosis in China. Curr. Microbiol. 2013, 66, 222–226. [Google Scholar] [CrossRef]
- Garvey, M.I.; Piddock, L.J. The Efflux Pump Inhibitor Reserpine Selects Multidrug-Resistant Streptococcus pneumoniae Strains That Overexpress the ABC Transporters PatA and PatB. Antimicrob. Agents Chemother. 2008, 52, 1677–1685. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, N.; Nishino, K.; Yamaguchi, A. Novel Macrolide-Specific ABC-Type Efflux Transporter in Escherichia coli. J. Bacteriol. 2001, 183, 5639–5644. [Google Scholar] [CrossRef] [Green Version]
- Bogomolnaya, L.M.; Andrews, K.D.; Talamantes, M.; Maple, A.; Ragoza, Y.; Vazquez-Torres, A.; Andrews-Polymenis, H. The ABC-type efflux pump MacAB protects Salmonella enterica serovar Typhimurium from oxidative stress. mBio 2013, 4, e00630-13. [Google Scholar] [CrossRef] [Green Version]
- Krämer, R. Functional principles of solute transport systems: Concepts and perspectives. Biochim. Biophys. Acta 1994, 1185, 1–34. [Google Scholar] [CrossRef]
- Poolman, B.; Konings, W.N. Secondary solute transport in bacteria. Biochim. Biophys. Acta 1993, 1183, 5–39. [Google Scholar] [CrossRef] [Green Version]
- Saier, M.H., Jr. Molecular Phylogeny as a Basis for the Classification of Transport Proteins from Bacteria, Archaea and Eukarya. Adv. Microb. Physiol. 1998, 40, 81–136. [Google Scholar]
- Varela, M.F. Antimicrobial efflux pumps. In Antibiotic Drug Resistance; Capelo-Martínez, J.-L., Igrejas, G., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2020; pp. 167–179. [Google Scholar]
- Pao, S.S.; Paulsen, I.T.; Saier, M.H. Major Facilitator Superfamily. Microbiol. Mol. Biol. Rev. 1998, 62, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, I.T.; Skurray, R.A.; Tam, R.; Saier, M.H., Jr.; Turner, R.J.; Weiner, J.H.; Goldberg, E.B.; Grinius, L.L. The SMR Family: A Novel Family of Multidrug Efflux Proteins Involved with the Efflux of Lipophilic Drugs. Mol. Microbiol. 1996, 19, 1167–1175. [Google Scholar] [CrossRef]
- Jack, D.L.; Yang, N.M.; Saier, M.H., Jr. The Drug/Metabolite Transporter Superfamily. Eur. J. Biochem. 2001, 268, 3620–3639. [Google Scholar] [CrossRef] [PubMed]
- Hvorup, R.N.; Winnen, B.; Chang, A.B.; Jiang, Y.; Zhou, X.F.; Saier, M.H., Jr. The Multidrug/Oligosaccharidyl-Lipid/Polysaccharide (MOP) Exporter Superfamily. Eur. J. Biochem. 2003, 270, 799–813. [Google Scholar] [CrossRef]
- Kuroda, T.; Tsuchiya, T. Multidrug efflux transporters in the MATE family. Biochim. Biophys. Acta 2009, 1794, 763–768. [Google Scholar] [CrossRef]
- Hassan, K.A.; Liu, Q.; Elbourne, L.D.H.; Ahmad, I.; Sharples, D.; Naidu, V.; Chan, C.L.; Li, L.; Harborne, S.P.D.; Pokhrel, A. Pacing across the Membrane: The Novel PACE Family of Efflux Pumps Is Widespread in Gram-Negative Pathogens. Res. Microbiol. 2018, 169, 450–454. [Google Scholar] [CrossRef]
- Tseng, T.T.; Gratwick, K.S.; Kollman, J.; Park, D.; Nies, D.H.; Goffeau, A.; Saier, M.H., Jr. The RND permease superfamily: An ancient, ubiquitous and diverse family that includes human disease and development proteins. J. Mol. Microbiol. Biotechnol. 1999, 1, 107–125. [Google Scholar]
- Kumar, S.; He, G.; Kakarla, P.; Shrestha, U.; Ranjana, K.C.; Ranaweera, I.; Willmon, T.M.; Barr, S.R.; Hernandez, A.J.; Varela, M.F. Bacterial Multidrug Efflux Pumps of the Major Facilitator Superfamily as Targets for Modulation. Infect Disord. Drug Targets 2016, 16, 28–43. [Google Scholar] [CrossRef]
- Lekshmi, M.; Ammini, P.; Adjei, J.; Sanford, L.M.; Shrestha, U.; Kumar, S.; Varela, M.F. Modulation of Antimicrobial Efflux Pumps of the Major Facilitator Superfamily in Staphylococcus aureus. AIMS Microbiol. 2018, 4, 1–18. [Google Scholar] [CrossRef]
- Ranaweera, I.; Shrestha, U.; Ranjana, K.C.; Kakarla, P.; Willmon, T.M.; Hernandez, A.J.; Mukherjee, M.M.; Barr, S.R.; Varela, M.F. Structural Comparison of Bacterial Multidrug Efflux Pumps of the Major Facilitator Superfamily. Trends Cell Mol. Biol. 2015, 10, 131–140. [Google Scholar]
- Kakarla, P.; Ranjana, K.; Shrestha, U.; Ranaweera, I.; Mukherjee, M.M.; Willmon, T.M.; Hernandez, A.J.; Barr, S.R.; Varela, M.F. Functional roles of highly conserved amino acid sequence motifs A and C in solute transporters of the major facilitator superfamily. In Drug Resistance in Bacteria, Fungi, Malaria, and Cancer; Springer: Berlin/Heidelberg, Germany, 2017; pp. 111–140. [Google Scholar]
- Heng, J.; Zhao, Y.; Liu, M.; Liu, Y.; Fan, J.; Wang, X.; Zhang, X.C. Substrate-Bound Structure of the E. coli Multidrug Resistance Transporter MdfA. Cell Res. 2015, 25, 1060–1073. [Google Scholar] [CrossRef]
- Liu, M.; Heng, J.; Gao, Y.; Wang, X. Crystal Structures of MdfA Complexed with Acetylcholine and Inhibitor Reserpine. Biophys. Rep. 2016, 2, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Nagarathinam, K.; Nakada-Nakura, Y.; Parthier, C.; Terada, T.; Juge, N.; Jaenecke, F.; Liu, K.; Hotta, Y.; Miyaji, T.; Omote, H. Outward Open Conformation of a Major Facilitator Superfamily Multidrug/H(+) Antiporter Provides Insights into Switching Mechanism. Nat. Commun. 2018, 9, 4005. [Google Scholar] [CrossRef] [PubMed]
- Varela, M.F.; Sansom, C.E.; Griffith, J.K. Mutational analysis and molecular modelling of an amino acid sequence motif conserved in antiporters but not symporters in a transporter superfamily. Mol. Membr. Biol. 1995, 12, 313–319. [Google Scholar] [CrossRef]
- Varela, M.F.; Griffith, J.K. Nucleotide and deduced protein sequences of the class D tetracycline resistance determinant: Relationship to other antimicrobial transport proteins. Antimicrob. Agents Chemother. 1993, 37, 1253–1258. [Google Scholar] [CrossRef] [Green Version]
- Lowrence, R.C.; Subramaniapillai, S.G.; Ulaganathan, V.; Nagarajan, S. Tackling Drug Resistance with Efflux Pump Inhibitors: From Bacteria to Cancerous Cells. Crit. Rev. Microbiol. 2019, 45, 334–353. [Google Scholar] [CrossRef]
- Andersen, J.L.; He, G.-X.; Kakarla, P.; KC, R.; Kumar, S.; Lakra, W.S.; Mukherjee, M.M.; Ranaweera, I.; Shrestha, U.; Tran, T.; et al. Multidrug Efflux Pumps from Enterobacteriaceae, Vibrio cholerae and Staphylococcus aureus Bacterial Food Pathogens. Int. J. Environ. Res. Public Health 2015, 12, 1487–1547. [Google Scholar] [CrossRef] [Green Version]
- Papkou, A.; Hedge, J.; Kapel, N.; Young, B.; MacLean, R.C. Efflux pump activity potentiates the evolution of antibiotic resistance across S. aureus isolates. Nat. Commun. 2020, 11, 3970. [Google Scholar] [CrossRef]
- Zimmermann, S.; Klinger-Strobel, M.; Bohnert, J.A.; Wendler, S.; Rodel, J.; Pletz, M.W.; Loffler, B.; Tuchscherr, L. Clinically Approved Drugs Inhibit the Staphylococcus aureus Multidrug NorA Efflux Pump and Reduce Biofilm Formation. Front. Microbiol. 2019, 10, 2762. [Google Scholar] [CrossRef] [Green Version]
- Uddin, G.M.N.; Larsen, M.H.; Barco, L.; Phu, T.M.; Dalsgaard, A. Clonal Occurrence of Salmonella Weltevreden in Cultured Shrimp in the Mekong Delta, Vietnam. PLoS ONE 2015, 10, e0134252. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.J.; Saier, M.H., Jr. SMR-type multidrug resistance pumps. Curr. Opin. Drug Discov. Dev. 2001, 4, 237–245. [Google Scholar]
- Chen, Y.J.; Pornillos, O.; Lieu, S.; Ma, C.; Chen, A.P.; Chang, G. X-Ray Structure of EmrE Supports Dual Topology Model. Proc. Natl. Acad. Sci. USA 2007, 104, 18999–19004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuldiner, S. Undecided Membrane Proteins Insert in Random Topologies. Up, down and Sideways: It Does Not Really Matter. Trends Biochem. Sci. 2012, 37, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Leninger, M.; Sae Her, A.; Traaseth, N.J. Inducing Conformational Preference of the Membrane Protein Transporter EmrE through Conservative Mutations. eLife 2019, 8, e48909. [Google Scholar] [CrossRef]
- Ovchinnikov, V.; Stone, T.A.; Deber, C.M.; Karplus, M. Structure of the Emre Multidrug Transporter and Its Use for Inhibitor Peptide Design. Proc. Natl. Acad. Sci. USA 2018, 115, 7932–7941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermaas, J.V.; Rempe, S.B.; Tajkhorshid, E. Electrostatic Lock in the Transport Cycle of the Multidrug Resistance Transporter EmrE. Proc. Natl. Acad. Sci. USA 2018, 115, 7502–7511. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, C.J.; Stone, T.A.; Deber, C.M. Peptide-Based Efflux Pump Inhibitors of the Small Multidrug Resistance Protein from Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Wynne, S.A.; Thomas, N.E.; Uhlemann, E.M.; Tate, C.G.; Henzler-Wildman, K.A. Identification of an Alternating-Access Dynamics Mutant of EmrE with Impaired Transport. J. Mol. Biol. 2019, 431, 2777–2789. [Google Scholar] [CrossRef]
- Murakami, S.; Nakashima, R.; Yamashita, E.; Yamaguchi, A. Crystal Structure of Bacterial Multidrug Efflux Transporter AcrB. Nature 2002, 419, 587–593. [Google Scholar] [CrossRef]
- Jones, P.M.; O’Mara, M.L.; George, A.M. Abc transporters: A riddle wrapped in a mystery inside an enigma. Trends Biochem. Sci. 2009, 34, 520–531. [Google Scholar] [CrossRef] [Green Version]
- Zwama, M.; Hayashi, K.; Sakurai, K.; Nakashima, R.; Kitagawa, K.; Nishino, K.; Yamaguchi, A. Hoisting-Loop in Bacterial Multidrug Exporter AcrB is a Highly Flexible Hinge That Enables the Large Motion of the Subdomains. Front. Microbiol. 2017, 8, 2095. [Google Scholar] [CrossRef]
- Kobylka, J.; Kuth, M.S.; Muller, R.T.; Geertsma, E.R.; Pos, K.M. AcrB: A mean, keen, drug efflux machine. Ann. N. Y. Acad. Sci. 2020, 1459, 38–68. [Google Scholar] [CrossRef] [Green Version]
- Murakami, S. Multidrug Efflux Transporter, AcrB–the Pumping Mechanism. Curr. Opin. Struct. Biol. 2008, 18, 459–465. [Google Scholar] [CrossRef]
- Seeger, M.A.; Diederichs, K.; Eicher, T.; Brandstatter, L.; Schiefner, A.; Verrey, F.; Pos, K.M. The AcrB Efflux Pump: Conformational Cycling and Peristalsis Lead to Multidrug Resistance. Curr. Drug Targets 2008, 9, 729–749. [Google Scholar] [CrossRef]
- Misra, R.; Bavro, V.N. Assembly and transport mechanism of tripartite drug efflux systems. Biochim. Biophys. Acta 2009, 1794, 817–825. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, A.; Nakashima, R.; Sakurai, K. Structural Basis of RND-Type Multidrug Exporters. Front. Microbiol. 2015, 6, 327. [Google Scholar] [CrossRef] [Green Version]
- Piddock, L.J. Multidrug-Resistance Efflux Pumps—Not Just for Resistance. Nature reviews. Microbiology 2006, 4, 629–636. [Google Scholar]
- Pumbwe, L.; Chang, A.; Smith, R.L.; Wexler, H.M. Clinical Significance of Overexpression of Multiple RND-Family Efflux Pumps in Bacteroides fragilis Isolates. J. Antimicrob. Chemother. 2006, 58, 543–548. [Google Scholar] [CrossRef]
- El Meouche, I.; Dunlop, M.J. Heterogeneity in efflux pump expression predisposes antibiotic-resistant cells to mutation. Science 2018, 362, 686–690. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, U.; Lekshmi, M.; Kumar, S.; Adjei, J.; Jones, K.; Hernandez, A.; Leslie, S.; Varela, M.F. Bioactive Compounds as Modulators of Multidrug Resistant Efflux Pumps of Major Facilitator Superfamily in Key Bacterial Pathogens. Curr. Trends Microbiol. 2019, 12, 15–37. [Google Scholar]
- Sharma, A.; Gupta, V.K.; Pathania, R. Efflux Pump Inhibitors for Bacterial Pathogens: From Bench to Bedside. Indian J. Med. Res. 2019, 149, 129–145. [Google Scholar] [PubMed]
- Kumar, S.; Lekshmi, M.; Parvathi, A.; Ojha, M.; Wenzel, N.; Varela, M.F. Functional and Structural Roles of the Major Facilitator Superfamily Bacterial Multidrug Efflux Pumps. Microorganisms 2020, 8, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikaido, H. Molecular Basis of Bacterial Outer Membrane Permeability Revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delcour, A.H. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta 2009, 1794, 808–816. [Google Scholar] [CrossRef] [Green Version]
- Ghai, I.; Ghai, S. Understanding Antibiotic Resistance via Outer Membrane Permeability. Infect. Drug Resist. 2018, 11, 523–530. [Google Scholar] [CrossRef] [Green Version]
- Pages, J.M.; James, C.E.; Winterhalter, M. The Porin and the Permeating Antibiotic: A Selective Diffusion Barrier in Gram-Negative Bacteria. Nat. Rev. Microbiol. 2008, 6, 893–903. [Google Scholar] [CrossRef] [Green Version]
- Koebnik, R.; Locher, K.P.; Van Gelder, P. Structure and Function of Bacterial Outer Membrane Proteins: Barrels in a Nutshell. Mol. Microbiol. 2000, 37, 239–253. [Google Scholar] [CrossRef]
- Bauer, W.R.; Nadler, W. Molecular Transport through Channels and Pores: Effects of in-Channel Interactions and Blocking. Proc. Natl. Acad. Sci. USA 2006, 103, 11446–11451. [Google Scholar] [CrossRef] [Green Version]
- Guillier, M.; Gottesman, S.; Storz, G. Modulating the Outer Membrane with Small rRNAs. Genes Dev. 2006, 20, 2338–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vila, J.; Marti, S.; Sanchez-Cespedes, J. Porins, Efflux Pumps and Multidrug Resistance in Acinetobacter Baumannii. J. Antimicrob. Chemother. 2007, 59, 1210–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevalier, S.; Bouffartigues, E.; Bodilis, J.; Maillot, O.; Lesouhaitier, O.; Feuilloley, M.G.J.; Orange, N.; Dufour, A.; Cornelis, P. Structure, Function and Regulation of Pseudomonas aeruginosa Porins. FEMS Microbiol. Rev. 2017, 41, 698–722. [Google Scholar] [CrossRef] [PubMed]
- Schulz, G.E. Bacterial Porins: Structure and Function. Curr. Opin. Cell Biol. 1993, 5, 701–707. [Google Scholar] [CrossRef]
- Cowan, S.W.; Schirmer, T.; Rummel, G.; Steiert, M.; Ghosh, R.; Pauptit, R.A.; Jansonius, J.N.; Rosenbusch, J.P. Crystal Structures Explain Functional Properties of Two E. coli Porins. Nature 1992, 358, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Modi, N.; Ganguly, S.; Barcena-Uribarri, I.; Benz, R.; Berg, B.; Kleinekathofer, U. Structure, Dynamics, and Substrate Specificity of the OprO Porin from Pseudomonas aeruginosa. Biophys. J. 2015, 109, 1429–1438. [Google Scholar] [CrossRef] [Green Version]
- Cowan, S.W.; Garavito, R.M.; Jansonius, J.N.; Jenkins, J.A.; Karlsson, R.; Konig, N.; Pai, E.F.; Pauptit, R.A.; Rizkallah, P.J.; Rosenbusch, J.P. The Structure of OmpF Porin in a Tetragonal Crystal Form. Structure 1995, 3, 1041–1050. [Google Scholar] [CrossRef]
- Masi, M.; Pages, J.M. Structure, Function and Regulation of Outer Membrane Proteins Involved in Drug Transport in Enterobactericeae: The OmpF/C—TolC Case. Open Microbiol. J. 2013, 7, 22–33. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeld, Y.; Shai, Y. Lipopolysaccharide (Endotoxin)-host defense antibacterial peptides interactions: Role in bacterial resistance and prevention of sepsis. Biochim. Biophys. Acta 2006, 1758, 1513–1522. [Google Scholar] [CrossRef] [Green Version]
- Ebbensgaard, A.; Mordhorst, H.; Aarestrup, F.M.; Hansen, E.B. The role of outer membrane proteins and lipopolysaccharides for the sensitivity of Escherichia coli to antimicrobial peptides. Front. Microbiol. 2018, 9, 2153. [Google Scholar] [CrossRef] [Green Version]
- Dixon, R.A.; Chopra, I. Leakage of periplasmic proteins from Escherichia coli mediated by polymyxin B nonapeptide. Antimicrob. Agents Chemother. 1986, 29, 781–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallardo, A.; Ugarte-Ruiz, M.; Hernández, M.; Miguela-Villoldo, P.; Rodríguez-Lázaro, D.; Domínguez, L.; Quesada, A. Involvement of hpap2 and dgkA genes in colistin resistance mediated by mcr determinants. Antibiotics 2020, 9, 531. [Google Scholar] [CrossRef] [PubMed]
- Rubin, E.J.; Herrera, C.M.; Crofts, A.A.; Trent, M.S. PmrD is required for modifications to Escherichia coli endotoxin that promote antimicrobial resistance. Antimicrob. Agents Chemother. 2015, 59, 2051–2061. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Zheng, L.; Mu, Y.; Ng, W.J.; Bhattacharjya, S. Structure and interactions of a host defense antimicrobial peptide thanatin in lipopolysaccharide micelles reveal mechanism of bacterial cell agglutination. Sci. Rep. 2017, 7, 17795. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Fang, C.; Lu, L.; Wang, M.; Xue, X.; Zhou, Y.; Li, M.; Hu, Y.; Luo, X.; Hou, Z. The antimicrobial peptide thanatin disrupts the bacterial outer membrane and inactivates the NDM-1 metallo-β-lactamase. Nat. Commun. 2019, 10, 3517. [Google Scholar] [CrossRef] [Green Version]
- Dash, R.; Bhattacharjya, S. Thanatin: An fmerging host defense antimicrobial peptide with multiple modes of action. Int. J. Mol. Sci. 2021, 22, 1522. [Google Scholar] [CrossRef]
- Peterson, E.; Kaur, P. Antibiotic Resistance Mechanisms in Bacteria: Relationships between Resistance Determinants of Antibiotic Producers, Environmental Bacteria, and Clinical Pathogens. Front. Microbiol. 2018, 9, 2928. [Google Scholar] [CrossRef]
- Marshall, B.M.; Levy, S.B. Food Animals and Antimicrobials: Impacts on Human Health. Clin. Microbiol. Rev. 2011, 24, 718–733. [Google Scholar] [CrossRef] [Green Version]
- Levy, S.B. The Future of Antibiotics: Facing Antibiotic Resistance. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2000, 6 (Suppl. 3), 101–106. [Google Scholar] [CrossRef] [Green Version]
- Doron, S.; Davidson, L.E. Antimicrobial Stewardship. Mayo Clin. Proc. 2011, 86, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- Cunha, C.B. Antimicrobial stewardship programs: Principles and practice. Med. Clin. N. Am. 2018, 102, 797–803. [Google Scholar] [CrossRef]
- Rice, L.B. Antimicrobial stewardship and antimicrobial resistance. Med. Clin. N. Am. 2018, 102, 805–818. [Google Scholar] [CrossRef]
- Lewis, K. The Science of Antibiotic Discovery. Cell 2020, 181, 29–45. [Google Scholar] [CrossRef]
- Lagadinou, M.; Onisor, M.O.; Rigas, A.; Musetescu, D.V.; Gkentzi, D.; Assimakopoulos, S.F.; Panos, G.; Marangos, M. Antimicrobial properties on non-antibiotic drugs in the era of increased bacterial resistance. Antibiotics 2020, 9, 107. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varela, M.F.; Stephen, J.; Lekshmi, M.; Ojha, M.; Wenzel, N.; Sanford, L.M.; Hernandez, A.J.; Parvathi, A.; Kumar, S.H. Bacterial Resistance to Antimicrobial Agents. Antibiotics 2021, 10, 593. https://doi.org/10.3390/antibiotics10050593
Varela MF, Stephen J, Lekshmi M, Ojha M, Wenzel N, Sanford LM, Hernandez AJ, Parvathi A, Kumar SH. Bacterial Resistance to Antimicrobial Agents. Antibiotics. 2021; 10(5):593. https://doi.org/10.3390/antibiotics10050593
Chicago/Turabian StyleVarela, Manuel F., Jerusha Stephen, Manjusha Lekshmi, Manisha Ojha, Nicholas Wenzel, Leslie M. Sanford, Alberto J. Hernandez, Ammini Parvathi, and Sanath H. Kumar. 2021. "Bacterial Resistance to Antimicrobial Agents" Antibiotics 10, no. 5: 593. https://doi.org/10.3390/antibiotics10050593