
Mechanisms of Antibiotic Resistance in Important Gram-Positive and Gram-Negative Pathogens and Novel Antibiotic Solutions

Abstract
:1. Introduction
2. Mechanisms of Antimicrobial Resistance in S. aureus
3. Mechanisms of Antimicrobial Resistance in Enterococcus spp.
4. Mechanisms of Antimicrobial Resistance in P. aeruginosa
5. Mechanisms of Antimicrobial Resistance in E. coli
6. Mechanisms of Antimicrobial Resistance in K. pneumoniae
7. Mechanisms of Antimicrobial Resistance in A. baumannii
8. Overcoming Resistance through Novel Antibiotics
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- ECDC. Antimicrobial Resistance in the EU/EEA—AER for 2019; ECDC: Stockholm, Sweden, 2020. [Google Scholar]
- Cassini, A.; Diaz Högberg, L.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Skov Simonsen, G. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. Lancet Infect. Dis. 2019. [Google Scholar] [CrossRef] [Green Version]
- Ambler, R.P. The structure of beta-lactamases. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 1980, 289, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Coronell-Rodríguez, W.; Arteta-Acosta, C.; Dueñas-Castell, C. Interpretive reading of the antibiogram: A tool for clinical practice. In Sepsis, 3rd ed.; Springer: New York, NY, USA, 2017; pp. 95–115. ISBN 9781493973347. [Google Scholar]
- Foster, T.J. Antibiotic resistance in Staphylococcus aureus. Current status and future prospects. FEMS Microbiol. Rev. 2017, 007, 430–449. [Google Scholar] [CrossRef]
- Chambers, H.F.; DeLeo, F.R. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 2009, 7, 629–641. [Google Scholar] [CrossRef]
- Tsiodras, S.; Gold, H.S.; Sakoulas, G.; Eliopoulos, G.M.; Wennersten, C.; Venkataraman, L.; Moellering, R.C.; Ferraro, M.J. Linezolid resistance in a clinical isolate of Staphylococcus aureus. Lancet 2001, 358, 207–208. [Google Scholar] [CrossRef]
- Govardhan, C.P.; Pratt, R.F. Kinetics and Mechanism of the Serine β-Lactamase Catalyzed Hydrolysis of Depsipeptides. Biochemistry 1987, 26, 3385–3395. [Google Scholar] [CrossRef]
- Moisan, H.; Pruneau, M.; Malouin, F. Binding of ceftaroline to penicillin-binding proteins of Staphylococcus aureus and Streptococcus pneumoniae. J. Antimicrob. Chemother. 2010, 65, 713–716. [Google Scholar] [CrossRef]
- Pantosti, A.; Sanchini, A.; Monaco, M. Mechanisms of antibiotic resistance in Staphylococcus aureus. Future Microbiol. 2007, 2, 323–334. [Google Scholar] [CrossRef]
- Banerjee, T.; Anupurba, S. Colonization with vancomycin-intermediate Staphylococcus aureus strains containing the vanA resistance gene in a tertiary-care center in North India. J. Clin. Microbiol. 2012, 50, 1730–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardete, S.; Tomasz, A. Mechanisms of vancomycin resistance in Staphylococcus aureus. J. Clin. Investig. 2014, 124, 2836–2840. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Sievert, D.M.; Hageman, J.C.; Boulton, M.L.; Tenover, F.C.; Downes, F.P.; Shah, S.; Rudrik, J.T.; Pupp, G.R.; Brown, W.J.; et al. Infection with Vancomycin-Resistant Staphylococcus aureus Containing the vanA Resistance Gene. N. Engl. J. Med. 2003, 348, 1342–1347. [Google Scholar] [CrossRef]
- Hiramatsu, K.; Kayayama, Y.; Matsuo, M.; Aiba, Y.; Saito, M.; Hishinuma, T.; Iwamoto, A. Vancomycin-intermediate resistance in Staphylococcus aureus. J. Glob. Antimicrob. Resist. 2014, 2, 213–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, A.; Dick, J.D.; Perl, T.M. Vancomycin Resistance in Staphylococci. Clin. Microbiol. Rev. 2002, 15, 430–438. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.R.; Bayer, A.S.; Arias, C.A. Mechanism of action and resistance to daptomycin in Staphylococcus aureus and enterococci. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, J.A.; Mortin, L.I.; VanPraagh, A.D.G.; Li, T.; Alder, J. Inhibition of daptomycin by pulmonary surfactant: In vitro modeling and clinical impact. J. Infect. Dis. 2005, 191, 2149–2152. [Google Scholar] [CrossRef]
- Jiang, J.H.; Bhuiyan, M.S.; Shen, H.H.; Cameron, D.R.; Rupasinghe, T.W.T.; Wu, C.M.; Le Brun, A.P.; Kostoulias, X.; Domene, C.; Fulcher, A.J.; et al. Antibiotic resistance and host immune evasion in Staphylococcus aureus mediated by a metabolic adaptation. Proc. Natl. Acad. Sci. USA 2019, 116, 3722–3727. [Google Scholar] [CrossRef] [Green Version]
- Sedaghat, H.; Nasr Esfahani, B.; Mobasherizadeh, S.; Jazi, A.S.; Halaji, M.; Sadeghi, P.; Emaneini, M.; Havaei, S.A. Phenotypic and genotypic characterization of macrolide resistance among staphylococcus aureus isolates in Isfahan, Iran. Iran. J. Microbiol. 2017, 9, 264–270. [Google Scholar]
- Gardiner, B.J.; Grayson, M.L.; Wood, G.M. Inducible resistance to clindamycin in Staphylococcus aureus: Validation of Vitek-2 against CLSI D-test. Pathology 2013, 45, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Long, K.S.; Poehlsgaard, J.; Kehrenberg, C.; Schwarz, S.; Vester, B. The Cfr rRNA methyltransferase confers resistance to phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramin A antibiotics. Antimicrob. Agents Chemother. 2006, 50, 2500–2505. [Google Scholar] [CrossRef] [Green Version]
- Long, K.S.; Vester, B. Resistance to linezolid caused by modifications at its binding site on the ribosome. Antimicrob. Agents Chemother. 2012, 56, 603–612. [Google Scholar] [CrossRef] [Green Version]
- Garrido-Mesa, N.; Zarzuelo, A.; Gálvez, J. Minocycline: Far beyond an antibiotic. Br. J. Pharmacol. 2013, 169, 337–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nau, R.; Sörgel, F.; Eiffert, H. Penetration of drugs through the blood-cerebrospinal fluid/blood-brain barrier for treatment of central nervous system infections. Clin. Microbiol. Rev. 2010, 23, 858–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, N.D. Tigecycline (Tygacil): The First in the Glycylcycline Class of Antibiotics. Baylor Univ. Med. Cent. Proc. 2006, 19, 155–161. [Google Scholar] [CrossRef]
- Nazarian, S.; Akhondi, H. Minocycline; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- McAleese, F.; Petersen, P.; Ruzin, A.; Dunman, P.M.; Murphy, E.; Projan, S.J.; Bradford, P.A. A novel MATE family efflux pump contributes to the reduced susceptibility of laboratory-derived Staphylococcus aureus mutants to tigecycline. Antimicrob. Agents Chemother. 2005, 49, 1865–1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, C. Antibiotics That Block Protein Synthesis. In Antibiotics: Challenges, Mechanisms, Opportunities; ASM Press: Washington, DC, USA, 2016; pp. 114–146. [Google Scholar]
- Kelmani Chandrakanth, R.; Raju, S.; Patil, S.A. Aminoglycoside-resistance mechanisms in multidrug-resistant Staphylococcus aureus clinical isolates. Curr. Microbiol. 2008, 56, 558–562. [Google Scholar] [CrossRef]
- Villar, M.; Marimon, J.M.; Garcia-Arenzana, J.M.; de la Campa, A.G.; Ferrandiz, M.J.; Perez-Trallero, E. Epidemiological and molecular aspects of rifampicin-resistant Staphylococcus aureus isolated from wounds, blood and respiratory samples. J. Antimicrob. Chemother. 2011, 66, 997–1000. [Google Scholar] [CrossRef]
- Walsh, C. Major Classes of Antibiotics and Their Modes of Action. In Antibiotics: Challenges, Mechanisms, Opportunities; ASM Press: Washington, DC, USA, 2016; pp. 16–32. [Google Scholar]
- Hollenbeck, B.L.; Rice, L.B. Intrinsic and acquired resistance mechanisms in enterococcus. Virulence 2012, 3, 421–569. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.R.; Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance in enterococci. Expert Rev. Anti-Infect. Ther. 2014, 12, 1221. [Google Scholar] [CrossRef]
- Kristich, C.J.; Rice, L.B.; Arias, C.A. Enterococcal Infection—Treatment and Antibiotic Resistance; Massachusetts Eye and Ear Infirmary: Boston, MA, USA, 2014. [Google Scholar]
- Portillo, A.; Ruiz-Larrea, F.; Zarazaga, M.; Alonso, A.; Martinez, J.L.; Torres, C. Macrolide resistance genes in Enterococcus spp. Antimicrob. Agents Chemother. 2000, 44, 967–971. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.V.; Weinstock, G.M.; Murray, B.E. An Enterococcus faecalis ABC Homologue (Lsa) Is Required for the Resistance of This Species to Clindamycin and Quinupristin-Dalfopristin. Antimicrob. Agents Chemother. 2002, 46, 1845–1850. [Google Scholar] [CrossRef] [Green Version]
- Bozdogan, B.; Berrezouga, L.; Kou, M.S.; Yurek, D.A.; Farley, K.A.; Stockman, B.J.; Leclercq, R. A new resistance gene, linB, conferring resistance to lincosamides by nucleotidylation in Enterococcus faecium HM1025. Antimicrob. Agents Chemother. 1999, 43, 925–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcks, A.; Andersen, S.R.; Licht, T.R. Characterization of transferable tetracycline resistance genes in Enterococcus faecalis isolated from raw food. FEMS Microbiol. Lett. 2005, 243, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, S.; Bender, J.K.; Klare, I.; Halbedel, S.; Grohmann, E.; Szewzyk, U.; Werner, G. Tigecycline resistance in clinical isolates of Enterococcus faecium is mediated by an upregulation of plasmid-encoded tetracycline determinants tet (L) and tet (M). J. Antimicrob. Chemother. 2016, 71, 871–881. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.O.; Baptiste, K.E. Vancomycin-Resistant Enterococci: A Review of Antimicrobial Resistance Mechanisms and Perspectives of Human and Animal Health. Microb. Drug Resist. 2018, 24, 590–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Hua, X.; Qu, T.; Jiang, Y.; Zhou, Z.; Yu, Y. Molecular characterization of Rifr mutations in Enterococcus faecalis and Enterococcus faecium. J. Chemother. 2014, 26, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Behzadi, P.; Baráth, Z.; Gajdács, M. It’s Not Easy Being Green: A Narrative Review on the Microbiology, Virulence and Therapeutic Prospects of Multidrug-Resistant Pseudomonas aeruginosa. Antibiotics 2021, 10, 42. [Google Scholar] [CrossRef]
- Pang, Z.; Raudonis, R.; Glick, B.R.; Lin, T.J.; Cheng, Z. Antibiotic resistance in Pseudomonas aeruginosa: Mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 2019, 37, 177–192. [Google Scholar] [CrossRef]
- Zhao, W.H.; Hu, Z.Q. β-Lactamases identified in clinical isolates of Pseudomonas aeruginosa. Crit. Rev. Microbiol. 2010, 36, 245–258. [Google Scholar] [CrossRef]
- Jacoby, G.A. AmpC Β-Lactamases. Clin. Microbiol. Rev. 2009, 22, 161–182. [Google Scholar] [CrossRef] [Green Version]
- Halat, D.H.; Moubareck, C.A. The current burden of carbapenemases: Review of significant properties and dissemination among gram-negative bacteria. Antibiotics 2020, 9, 186. [Google Scholar] [CrossRef]
- Arias-León, G. Resistance mechanisms: A problem and an approach to the solution. In Sepsis, 3rd ed.; Springer: New York, NY, USA, 2017; pp. 73–93. ISBN 9781493973347. [Google Scholar]
- Poole, K. Aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2005, 49, 479–487. [Google Scholar] [CrossRef] [Green Version]
- Eljaaly, K.; Alharbi, A.; Alshehri, S.; Ortwine, J.K.; Pogue, J.M. Plazomicin: A Novel Aminoglycoside for the Treatment of Resistant Gram-Negative Bacterial Infections. Drugs 2019, 79, 243–269. [Google Scholar] [CrossRef]
- Gad, G.F.; Mohamed, H.A.; Ashour, H.M. Aminoglycoside Resistance Rates, Phenotypes, and Mechanisms of Gram-Negative Bacteria from Infected Patients in Upper Egypt. PLoS ONE 2011, 6, e17224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalal, S.; Ciofu, O.; Høiby, N.; Gotoh, N.; Wretlind, B. Molecular mechanisms of fluoroquinolone resistance in Pseudomonas aeruginosa isolates from cystic fibrosis patients. Antimicrob. Agents Chemother. 2000, 44, 710–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, C.R.; Visalli, M.A.; Projan, S.J.; Sum, P.E.; Bradford, P.A. Efflux-mediated resistance to tigecycline (GAR-936) in Pseudomonas aeruginosa PAO1. Antimicrob. Agents Chemother. 2003, 47, 972–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustafa, M.H.; Khandekar, S.; Tunney, M.M.; Elborn, J.S.; Kahl, B.C.; Denis, O.; Plésiat, P.; Traore, H.; Tulkens, P.M.; Vanderbist, F.; et al. Acquired resistance to macrolides in Pseudomonas aeruginosa from cystic fibrosis patients. Eur. Respir. J. 2017, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huovinen, P. Resistance to trimethoprim-sulfamethoxazole. Clin. Infect. Dis. 2001, 32, 1608–1614. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.R.; Iles, J.C.; MacLean, R.C. The Fitness Cost of Rifampicin Resistance in Pseudomonas aeruginosa Depends on Demand for RNA Polymerase. Genetics 2011, 187, 817–822. [Google Scholar] [CrossRef] [Green Version]
- Aghapour, Z.; Gholizadeh, P.; Ganbarov, K.; Bialvaei, A.Z.; Mahmood, S.S.; Tanomand, A.; Yousefi, M.; Asgharzadeh, M.; Yousefi, B.; Kafil, H.S. Molecular mechanisms related to colistin resistance in enterobacteriaceae. Infect. Drug Resist. 2019, 12, 965–975. [Google Scholar] [CrossRef] [Green Version]
- Andrade, F.F.; Silva, D.; Rodrigues, A.; Pina-Vaz, C. Colistin update on its mechanism of action and resistance, present and future challenges. Microorganisms 2020, 8, 1716. [Google Scholar] [CrossRef]
- Bialvaei, A.Z.; Kafil, H.S. Colistin, mechanisms and prevalence of resistance. Curr. Med. Res. Opin. 2015, 31. [Google Scholar] [CrossRef]
- Poirel, L.; Madec, J.-Y.; Lupo, A.; Schink, A.-K.; Kieffer, N.; Nordmann, P.; Schwarz, S. Antimicrobial Resistance in Escherichia coli. In Antimicrobial Resistance in Bacteria from Livestock and Companion Animals; American Society of Microbiology: Washington, DC, USA, 2018; Volume 6, pp. 289–316. [Google Scholar]
- Bajaj, P.; Singh, N.S.; Virdi, J.S. Escherichia coli β-lactamases: What really matters. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Al-Agamy, M.H.; Aljallal, A.; Radwan, H.H.; Shibl, A.M. Characterization of carbapenemases, ESBLs, and plasmid-mediated quinolone determinants in carbapenem-insensitive Escherichia coli and Klebsiella pneumoniae in Riyadh hospitals. J. Infect. Public Health 2018, 11, 64–68. [Google Scholar] [CrossRef]
- Paitan, Y. Current trends in antimicrobial resistance of escherichia coli. In Current Topics in Microbiology and Immunology; Springer Verlag: Berlin/Heidelberg, Germany, 2018; Volume 416, pp. 181–211. [Google Scholar]
- Stewart, A.; Harris, P.; Henderson, A.; Paterson, D. Treatment of infections by OXA-48-producing Enterobacteriaceae. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Zhou, Y.N.; Goldstein, B.P.; Jin, D.J. Cross-resistance of Escherichia coli RNA polymerases conferring rifampin resistance to different antibiotics. J. Bacteriol. 2005, 187, 2783–2792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, M.C.P.; Woerther, P.L.; Bouvet, M.; Andremont, A.; Leclercq, R.; Canu, A. Escherichia coli as reservoir for macrolide resistance genes. Emerg. Infect. Dis. 2009, 15, 1648–1650. [Google Scholar] [CrossRef] [PubMed]
- Von Baum, H.; Marre, R. Antimicrobial resistance of Escherichia coli and therapeutic implications. Int. J. Med. Microbiol. 2005, 295, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Linkevicius, M.; Sandegren, L.; Andersson, D.I. Mechanisms and fitness costs of tigecycline resistance in Escherichia coli. J. Antimicrob. Chemother. 2013, 68, 2809–2819. [Google Scholar] [CrossRef] [Green Version]
- Bodendoerfer, E.; Marchesi, M.; Imkamp, F.; Courvalin, P.; Böttger, E.C.; Mancini, S. Co-occurrence of aminoglycoside and β-lactam resistance mechanisms in aminoglycoside- non-susceptible Escherichia coli isolated in the Zurich area, Switzerland. Int. J. Antimicrob. Agents 2020, 56, 106019. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Wang, Y.; Walsh, T.R.; Yi, L.X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet Infect. Dis. 2016, 16, 161–168. [Google Scholar] [CrossRef]
- Xavier, B.B.; Lammens, C.; Ruhal, R.; Kumar-Singh, S.; Butaye, P.; Goossens, H.; Malhotra-Kumar, S. Identification of a novel plasmid-mediated colistin-resistance gene, mcr-2, in Escherichia coli, Belgium, June 2016. Eurosurveillance 2016, 21, 30280. [Google Scholar] [CrossRef]
- Tzouvelekis, L.S.; Markogiannakis, A.; Psichogiou, M.; Tassios, P.T.; Daikos, G.L. Carbapenemases in Klebsiella pneumoniae and Other Enterobacteriaceae: An Evolving Crisis of Global Dimensions. Clin. Microbiol. Rev. 2012. [Google Scholar] [CrossRef] [Green Version]
- Arzanlou, M.; Chai, W.C.; Venter, H. Intrinsic, adaptive and acquired antimicrobial resistance in Gram-negative bacteria. Essays Biochem. 2017, 61, 49–59. [Google Scholar] [CrossRef]
- Cruz-Córdova, A.; Esteban-Kenel, V.; Espinosa-Mazariego, K.; Ochoa, S.A.; Moreno Espinosa, S.; de la Garza Elhain, A.; Fernández Rendón, E.; López Villegas, E.O.; Xicohtencatl-Cortes, J. Pathogenic determinants of clinical Klebsiella pneumoniae strains associated with their persistence in the hospital environment. Bol. Med. Hosp. Infant. Mex. 2014, 71, 15–24. [Google Scholar]
- Bouza, E.; Cercenado, E. Klebsiella and Enterobacter: Antibiotic resistance and treatment implications. Semin. Respir. Infect. 2002, 17, 215–230. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, G.; Chao, X.; Xie, L.; Wang, H. The characteristic of virulence, biofilm and antibiotic resistance of klebsiella pneumoniae. Int. J. Environ. Res. Public Health 2020, 17, 6278. [Google Scholar] [CrossRef]
- Lan, P.; Jiang, Y.; Zhou, J.; Yu, Y. A Global Perspective on the Convergence of Hypervirulence and Carbapenem-Resistance in Klebsiella pneumoniae. J. Glob. Antimicrob. Resist. 2021. [Google Scholar] [CrossRef] [PubMed]
- Girlich, D.; Poirel, L.; Nordmann, P. Value of the modified hodge test for detection of emerging carbapenemases in Enterobacteriaceae. J. Clin. Microbiol. 2012, 50, 477–479. [Google Scholar] [CrossRef] [Green Version]
- Doi, Y.; Arakawa, Y. 16S ribosomal RNA methylation: Emerging resistance mechanism against aminoglycosides. Clin. Infect. Dis. 2007, 45, 88–94. [Google Scholar] [CrossRef]
- Navon-Venezia, S.; Kondratyeva, K.; Carattoli, A. Klebsiella pneumoniae: A major worldwide source and shuttle for antibiotic resistance. FEMS Microbiol. Rev. 2017, 41, 252–275. [Google Scholar] [CrossRef]
- Vetting, M.W.; Chi, H.P.; Hegde, S.S.; Jacoby, G.A.; Hooper, D.C.; Blanchard, J.S. Mechanistic and structural analysis of aminoglycoside N-acetyltransferase AAC(6′)-Ib and its bifunctional, fluoroquinolone-active AAC(6′)-Ib-cr variant. Biochemistry 2008, 47, 9825–9835. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, R.L.; Da Silva, B.C.M.; Rezende, G.S.; Nakamura-Silva, R.; Pitondo-Silva, A.; Campanini, E.B.; Brito, M.C.A.; Da Silva, E.M.L.; De Melo Freire, C.C.; Da Cunha, A.F.; et al. High prevalence of multidrug-resistant klebsiella pneumoniae harboring several virulence and β-lactamase encoding genes in a brazilian intensive care unit. Front. Microbiol. 2019, 10, 3198. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Z.K.; Hu, F.; Wang, W.; Guo, Q.; Chen, Z.; Xu, X.; Zhu, D.; Wang, M. Mechanisms of tigecycline resistance among klebsiella pneumoniae clinical isolates. Antimicrob. Agents Chemother. 2014, 58, 6982–6985. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.; Smith, K.P.; Whitfield, B.A.; Zucchi, P.C.; Lasco, T.M.; Bias, T.E.; Kirby, J.E.; Hirsch, E.B. Activity of minocycline against Klebsiella pneumoniae carbapenemase (KPC)-producing Enterobacteriaceae clinical isolates, with comparison to doxycycline and tigecycline. Diagn. Microbiol. Infect. Dis. 2017, 88, 365–367. [Google Scholar] [CrossRef]
- Warrier, A.R.; Babu, R. Susceptibility of Minocycline against Carbapenem Resistant Gram-Negative Bacilli. J. Microbiol. Infect. Dis. 2018, 8, 140–146. [Google Scholar] [CrossRef]
- Tascini, C.; Tagliaferri, E.; Giani, T.; Leonildi, A.; Flammini, S.; Casini, B.; Lewis, R.; Ferranti, S.; Rossolini, G.M.; Menichetti, F. Synergistic activity of colistin plus rifampin against colistin-resistant kpc-producing klebsiella pneumoniae. Antimicrob. Agents Chemother. 2013, 57, 3990–3993. [Google Scholar] [CrossRef] [Green Version]
- Arlet, G.; Nadjar, D.; Herrmann, J.; Donay, J.; Rouveau, M.; Lagrange, P.; Philippon, A. Plasmid-mediated rifampin resistance encoded by an arr-2-like gene cassette in Klebsiella pneumoniae producing an aCC-1 class c β-lactamase. Antimicrob. Agents Chemother. 2001, 45, 2971–2972. [Google Scholar] [CrossRef] [Green Version]
- Chung, P.Y. The emerging problems of Klebsiella pneumoniae infections: Carbapenem resistance and biofilm formation. FEMS Microbiol. Lett. 2016, 363. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial resistance in ESKAPE pathogens. Clin. Microbiol. Rev. 2020, 33. [Google Scholar] [CrossRef]
- Howard, A.; O’Donoghue, M.; Feeney, A.; Sleator, R.D. Acinetobacter baumannii An emerging opportunistic pathogen. Virulence 2012, 3, 5. [Google Scholar] [CrossRef]
- Rocha, I.V.; Xavier, D.E.; de Almeida, K.R.H.; de Oliveira, S.R.; Leal, N.C. Multidrug-resistant Acinetobacter baumannii clones persist on hospital inanimate surfaces. Brazilian J. Infect. Dis. 2018, 22, 438–441. [Google Scholar] [CrossRef]
- Bonomo, R.A.; Szabo, D. Mechanisms of Multidrug Resistance in Acinetobacter Species and Pseudomonas aeruginosa. Clin. Infect. Dis. 2006, 43, S49–S56. [Google Scholar] [CrossRef] [Green Version]
- Abdar, M.H.; Taheri-Kalani, M.; Taheri, K.; Emadi, B.; Hasanzadeh, A.; Sedighi, A.; Pirouzi, S.; Sedighi, M. Prevalence of extended-spectrum beta-lactamase genes in Acinetobacter baumannii strains isolated from nosocomial infections in Tehran, Iran. GMS Hyg. Infect. Control 2019, 14, Doc02. [Google Scholar] [CrossRef]
- Penwell, W.F.; Shapiro, A.B.; Giacobbe, R.A.; Gu, R.F.; Gao, N.; Thresher, J.; McLaughlin, R.E.; Huband, M.D.; DeJonge, B.L.M.; Ehmann, D.E.; et al. Molecular mechanisms of sulbactam antibacterial activity and resistance determinants in Acinetobacter baumannii. Antimicrob. Agents Chemother. 2015, 59, 1680–1689. [Google Scholar] [CrossRef] [Green Version]
- Betrosian, A.P.; Frantzeskaki, F.; Xanthaki, A.; Douzinas, E.E. Efficacy and safety of high-dose ampicillin/sulbactam vs. colistin as monotherapy for the treatment of multidrug resistant Acinetobacter baumannii ventilator-associated pneumonia. J. Infect. 2008, 56, 432–436. [Google Scholar] [CrossRef]
- Liu, J.; Shu, Y.; Zhu, F.; Feng, B.; Zhang, Z.; Liu, L.; Wang, G. Comparative efficacy and safety of combination therapy with high-dose sulbactam or colistin with additional antibacterial agents for multiple drug-resistant and extensively drug-resistant Acinetobacter baumannii infections: A systematic review and network meta-analysis. J. Glob. Antimicrob. Resist. 2021, 24, 136–147. [Google Scholar] [CrossRef]
- Poirel, L.; Nordmann, P. Carbapenem resistance in Acinetobacter baumannii: Mechanisms and epidemiology. Clin. Microbiol. Infect. 2006, 12, 826–836. [Google Scholar] [CrossRef] [Green Version]
- Jamal, S.; Al Atrouni, A.; Rafei, R.; Dabboussi, F.; Hamze, M.; Osman, M. Molecular mechanisms of antimicrobial resistance in Acinetobacter baumannii, with a special focus on its epidemiology in Lebanon. J. Glob. Antimicrob. Resist. 2018, 15, 154–163. [Google Scholar] [CrossRef]
- Chen, Q.; Li, X.; Zhou, H.; Jiang, Y.; Chen, Y.; Hua, X.; Yu, Y. Decreased susceptibility to tigecycline in Acinetobacter baumannii mediated by a mutation in trm encoding SAM-dependent methyltransferase. J. Antimicrob. Chemother. 2014, 69, 72–76. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, L.; Ji, J.; Chen, Q.; Hua, X.; Jiang, Y.; Feng, Y.; Yu, Y. Tigecycline resistance in Acinetobacter baumannii mediated by frameshift mutation in plsC, encoding 1-acyl-sn-glycerol-3-phosphate acyltransferase. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 625–631. [Google Scholar] [CrossRef]
- Lashinsky, J.N.; Henig, O.; Pogue, J.M.; Kaye, K.S. Minocycline for the Treatment of Multidrug and Extensively Drug-Resistant A. baumannii: A Review. Infect. Dis. Ther. 2017, 6, 199–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vázquez-López, R.; Solano-Gálvez, S.G.; Juárez Vignon-Whaley, J.J.; Abello Vaamonde, J.A.; Padró Alonzo, L.A.; Rivera Reséndiz, A.; Muleiro Álvarez, M.; Vega López, E.N.; Franyuti-Kelly, G.; Álvarez-Hernández, D.A.; et al. Acinetobacter baumannii Resistance: A Real Challenge for Clinicians. Antibiotics 2020, 9, 205. [Google Scholar] [CrossRef]
- Mohammadi, M.; Khayat, H.; Sayehmiri, K.; Soroush, S.; Sayehmiri, F.; Delfani, S.; Bogdanovic, L.; Taherikalani, M. Synergistic Effect of Colistin and Rifampin Against Multidrug Resistant Acinetobacter baumannii: A Systematic Review and Meta-Analysis. Open Microbiol. J. 2017, 11, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Pachón-Ibáñez, M.E.; Docobo-Pérez, F.; Jiménez-Mejias, M.E.; Ibáñez-Martínez, J.; García-Curiel, A.; Pichardo, C.; Pachón, J. Efficacy of rifampin, in monotherapy and in combinations, in an experimental murine pneumonia model caused by panresistant Acinetobacter baumannii strains. Eur. J. Clin. Microbiol. Infect. Dis. 2011, 30, 895–901. [Google Scholar] [CrossRef] [Green Version]
- Asif, M.; Alvi, I.A.; Ur Rehman, S. Insight into acinetobacter baumannii: Pathogenesis, global resistance, mechanisms of resistance, treatment options, and alternative modalities. Infect. Drug Resist. 2018, 11, 1249–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lertsrisatit, Y.; Santimaleeworagun, W.; Thunyaharn, S.; Traipattanakul, J. In vitro activity of colistin mono- and combination therapy against colistin-resistant Acinetobacter baumannii, mechanism of resistance, and clinical outcomes of patients infected with colistinresistant A. baumannii at a Thai university hospital. Infect. Drug Resist. 2017, 10, 437–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, R. Antibiotic development pipeline runs dry. Lancet 2003, 362, 1726–1727. [Google Scholar] [CrossRef]
- Renwick, M.J.; Brogan, D.M.; Mossialos, E. A systematic review and critical assessment of incentive strategies for discovery and development of novel antibiotics. J. Antibiot. 2016, 69, 73–88. [Google Scholar] [CrossRef] [Green Version]
- Evans, B.A.; Amyes, S.G.B. OXA β-lactamases. Clin. Microbiol. Rev. 2014, 27, 241–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupia, T.; Pallotto, C.; Corcione, S.; Boglione, L.; De Rosa, F.G. Ceftobiprole Perspective: Current and Potential Future Indications. Antibiotics 2021, 10, 170. [Google Scholar] [CrossRef] [PubMed]
- Kisgen, J.; Whitney, D. Ceftobiprole, a broad-spectrum cephalosporin with activity against methicillin-resistant Staphylococcus aureus (MRSA). Pharm. Ther. 2008, 33, 631–641. [Google Scholar]
- Shirley, D.A.T.; Heil, E.L.; Johnson, J.K. Ceftaroline Fosamil: A Brief Clinical Review. Infect. Dis. Ther. 2013, 2, 95–110. [Google Scholar] [CrossRef] [Green Version]
- Lupia, T.; Corcione, S.; Pinna, S.M.; de Rosa, F.G. New cephalosporins for the treatment of pneumonia in internal medicine wards. J. Thorac. Dis. 2020, 12, 3747–3763. [Google Scholar] [CrossRef]
- Abdul-Mutakabbir, J.C.; Alosaimy, S.; Morrisette, T.; Kebriaei, R.; Rybak, M.J. Cefiderocol: A Novel Siderophore Cephalosporin against Multidrug-Resistant Gram-Negative Pathogens. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2020, 40, 1228–1247. [Google Scholar] [CrossRef]
- Sato, T.; Yamawaki, K. Cefiderocol: Discovery, Chemistry, and In Vivo Profiles of a Novel Siderophore Cephalosporin. Clin. Infect. Dis. 2019, 69, S538–S543. [Google Scholar] [CrossRef] [Green Version]
- FDA. Highlights of Prescribing Information for Cefiderocol; FDA: White Oak, MD, USA, 2019.
- Zhanel, G.G.; Chung, P.; Adam, H.; Zelenitsky, S.; Denisuik, A.; Schweizer, F.; Lagacé-Wiens, P.R.S.; Rubinstein, E.; Gin, A.S.; Walkty, A.; et al. Ceftolozane/tazobactam: A novel cephalosporin/β-lactamase inhibitor combination with activity against multidrug-resistant gram-negative bacilli. Drugs 2014, 74, 31–51. [Google Scholar] [CrossRef]
- Sorbera, M.; Chung, E.; Ho, C.W.; Marzella, N. Ceftolozane/tazobactam: A new option in the treatment of complicated gram-negative infections. Pharm. Ther. 2014, 39, 825–832. [Google Scholar]
- FDA. Highlights of Prescribing Information for Ceftolozane/Tazobactam; FDA: White Oak, MD, USA, 2014.
- Kollef, M.H.; Nováček, M.; Kivistik, Ü.; Réa-Neto, Á.; Shime, N.; Martin-Loeches, I.; Timsit, J.F.; Wunderink, R.G.; Bruno, C.J.; Huntington, J.A.; et al. Ceftolozane–tazobactam versus meropenem for treatment of nosocomial pneumonia (ASPECT-NP): A randomised, controlled, double-blind, phase 3, non-inferiority trial. Lancet Infect. Dis. 2019, 19, 1299–1311. [Google Scholar] [CrossRef]
- Bassetti, M.; Peghin, M.; Vena, A.; Giacobbe, D.R. Treatment of Infections Due to MDR Gram-Negative Bacteria. Front. Med. 2019, 6, 74. [Google Scholar] [CrossRef]
- Karaiskos, I.; Lagou, S.; Pontikis, K.; Rapti, V.; Poulakou, G. The “Old” and the “New” antibiotics for MDR Gram-negative pathogens: For whom, when, and how. Front. Public Health 2019, 7, 151. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Lawrence, J.; Liu, C.; Yin, N. Advances in inhibitors of penicillin-binding proteins and β-lactamases as antibacterial agents. In Annual Reports in Medicinal Chemistry; Academic Press Inc.: Cambridge, MA, USA, 2014; Volume 49, pp. 249–266. [Google Scholar]
- Biedenbach, D.J.; Kazmierczak, K.; Bouchillon, S.K.; Sahm, D.F.; Bradford, P.A. In vitro activity of aztreonam-avibactam against a global collection of Gram-negative pathogens from 2012 and 2013. Antimicrob. Agents Chemother. 2015, 59, 4239–4248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zasowski, E.J.; Rybak, J.M.; Rybak, M.J. The β-Lactams Strike Back: Ceftazidime-Avibactam. Pharmacotherapy 2015, 35, 755–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransom, E.; Bhatnagar, A.; Patel, J.B.; Machado, M.J.; Boyd, S.; Reese, N.; Lutgring, J.D.; Lonsway, D.; Anderson, K.; Brown, A.C.; et al. Validation of aztreonam-avibactam susceptibility testing using digitally dispensed custom panels. J. Clin. Microbiol. 2020, 58. [Google Scholar] [CrossRef] [PubMed]
- Koulenti, D.; Xu, E.; Mok, I.; Song, A.; Karageorgopoulos, D.; Armaganidis, A.; Lipman, J.; Tsiodras, S. Novel Antibiotics for Multidrug-Resistant Gram-Positive Microorganisms. Microorganisms 2019, 7, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shortridge, D.; Pfaller, M.A.; Streit, J.M.; Flamm, R.K. Update on the activity of delafloxacin against acute bacterial skin and skin-structure infection isolates from European hospitals (2014–2019). J. Glob. Antimicrob. Resist. 2020, 23, 278–283. [Google Scholar] [CrossRef]
- Eljaaly, K.; Ortwine, J.K.; Shaikhomer, M.; Almangour, T.A.; Bassetti, M. Efficacy and safety of eravacycline: A meta-analysis. J. Glob. Antimicrob. Resist. 2021, 24, 424–428. [Google Scholar] [CrossRef]
- Sutcliffe, J.A.; O’Brien, W.; Fyfe, C.; Grossman, T.H. Antibacterial activity of eravacycline (TP-434), a novel fluorocycline, against hospital and community pathogens. Antimicrob. Agents Chemother. 2013, 57, 5548–5558. [Google Scholar] [CrossRef] [Green Version]
- FDA. Highlights of Prescribing Information for Plazomicin; FDA: White Oak, MD, USA, 2018.
- Zhanel, G.G.; Calic, D.; Schweizer, F.; Zelenitsky, S.; Adam, H.; Lagac-Wiens, P.R.S.; Rubinstein, E.; Gin, A.S.; Hoban, D.J.; Karlowsky, J.A. New lipoglycopeptides: A comparative review of dalbavancin, oritavancin and telavancin. Drugs 2010, 70, 859–886. [Google Scholar] [CrossRef] [PubMed]
- FDA. Highlights of Prescribing Information for Oritavancin; FDA: White Oak, MD, USA, 2014.
- FDA. Highlights of Prescribing Information for Telavancin; FDA: White Oak, MD, USA, 2009.
- FDA. Highlights of Prescribing Information for Dalbavancin; FDA: White Oak, MD, USA, 2014.
- Zhanel, G.G.; Love, R.; Adam, H.; Golden, A.; Zelenitsky, S.; Schweizer, F.; Gorityala, B.; Lagacé-Wiens, P.R.S.; Rubinstein, E.; Walkty, A.; et al. Tedizolid: A novel oxazolidinone with potent activity against multidrug-resistant gram-positive pathogens. Drugs 2015, 75, 253–270. [Google Scholar] [CrossRef]
- FDA. Highlights of Prescribing Information for Tedizolid; FDA: White Oak, MD, USA, 2014.



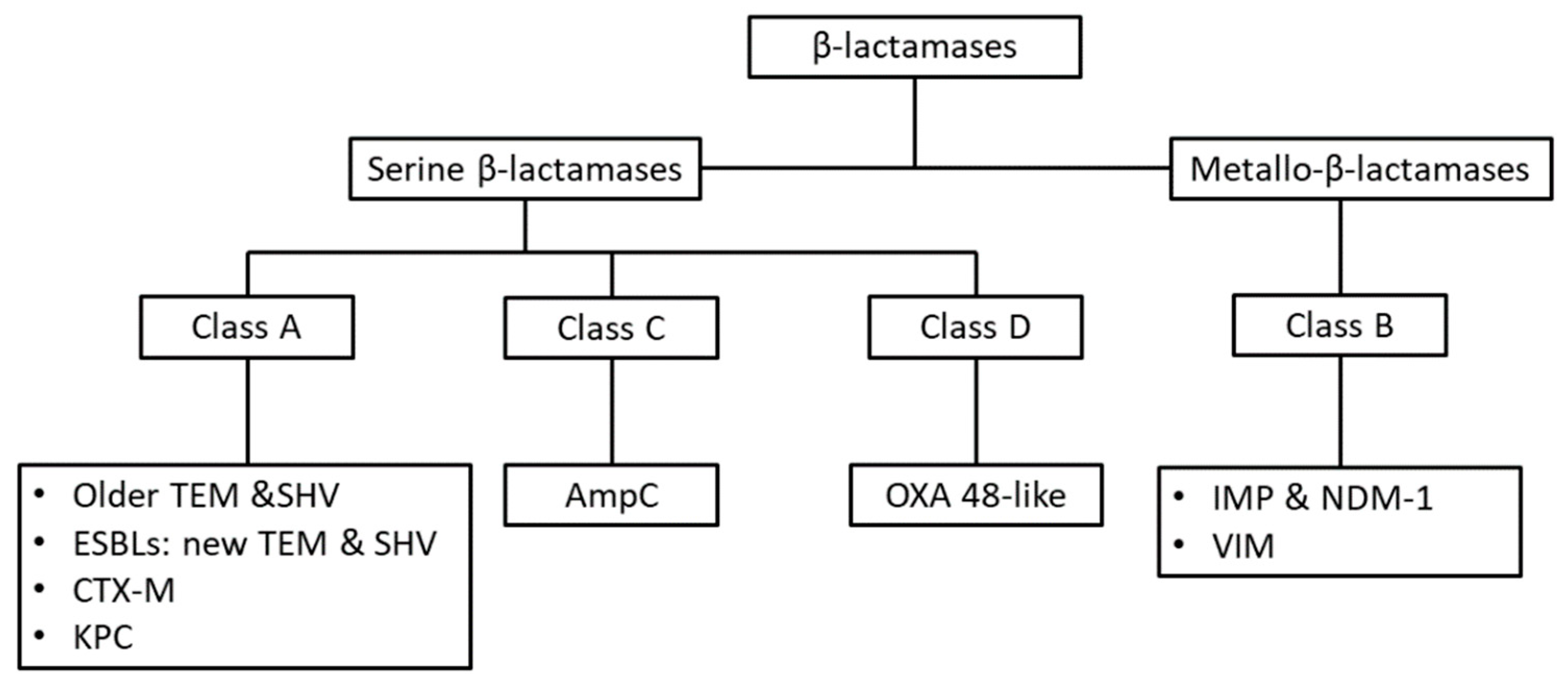{kind=link}
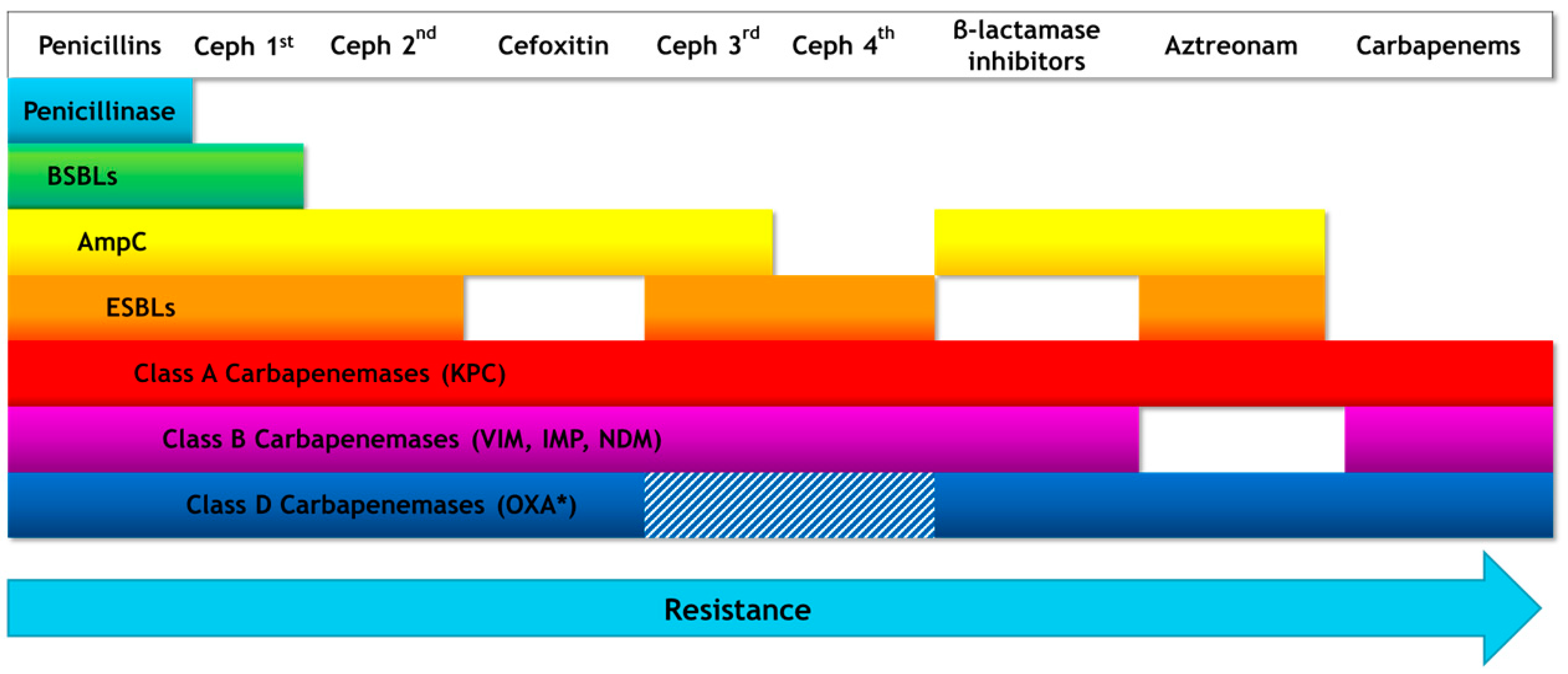{kind=link}
| Pathogen | Antibiotic Resistance | Median Number of Infections | Median Number of Attributable Deaths |
|---|---|---|---|
| E. coli | Third-Generation Cephalosporin * | 297,416 | 9066 |
| Carbapenem # | 2619 | 141 | |
| Colistin | 7156 | 621 | |
| Overall | 307,191 | 9828 | |
| S. aureus | Methicillin-resistant (MRSA) | 148,727 | 7049 |
| P. aeruginosa | > = 3 antibiotic groups * | 9028 | 572 |
| Carbapenem # | 61,892 | 4155 | |
| Colistin | 1262 | 84.5 | |
| Overall | 72,182 | 4811.5 | |
| K. pneumoniae | Third-Generation Cephalosporin * | 68,588 | 3687 |
| Carbapenem# | 15,947 | 2118 | |
| Colistin | 7450 | 1635 | |
| Overall | 91,985 | 7440 | |
| Enterococcus spp. | Vancomycin | 16,146 | 1081 |
| Acinetobacter spp. | Aminoglycoside and Fluoroquinolone | 2182 | 100 |
| Carbapenem # | 27,343 | 2363 | |
| Colistin | 1084 | 94.5 | |
| Overall | 30,609 | 2557.5 | |
| Overall | 666,840 | 32,767 |
| Antibiotic Class | S. aureus | Enterococcus spp. |
|---|---|---|
| Penicillins | Penicillinase, production of PBP2a | Low affinity PBPs |
| Cephalosporins 1st gen. | PBP2a | Low affinity PBPs |
| Cephalosporins 2nd gen. | PBP2a | Low affinity PBPs |
| Cephalosporins 3rd gen. | PBP2a | Low affinity PBPs |
| Cephalosporins 4th gen. | PBP2a | Low affinity PBPs |
| b-lactamase inhibitors | PBP2a | |
| Carbapenems | Development of PBP2a | Low affinity PBPs |
| Tetracyclines | Ribosomal methylation of binding sites, efflux pumps | Ribosomal methylation of binding sites, efflux pumps |
| Tigecyclines | Efflux pumps | Ribosomal methylation of binding sites, efflux pumps |
| Macrolides and clindamycin | Ribosomal methylation of binding sites, efflux pumps | Efflux pumps, clindamycin inactivating enzymes |
| Fluoroquinolones | Mutations in topoisomerase IV and DNA gyrase, efflux pumps | Mutations in topoisomerase IV and DNA gyrase, production of protection proteins |
| Rifampicin | Mutations in RNA polymerase gene | Mutations in RNA polymerase gene |
| TMP/SMX | Mutations in DHPS and DHFR | Folate absorption from environment |
| Aminoglycosides | Aminoglycoside degradation enzymes | Aminoglycoside degradation enzymes, ribosomal mutations |
| Daptomycin | Electrostatic repulsion through increase to the cell-surface charge | E faeccium: electrostatic repulsion through increase to the cell-surface charge. E. faecalis: redistribution of cardiolipin away from septum plane |
| Vancomycin | VRSA: altered structure of peptidoglycan precursors from D-Ala-D-Ala to D-Ala-D-Lac; VISA: increased production of peptidoglycan, thicker cell wall, decoy D-Ala-D-Ala dipeptides on cell surface | Altered structure of peptidoglycan precursors from D-Ala-D-Ala to D-Ala-D-Lac |
| Linezolid | Mutations to the 23S rRNA, altering required modifications to the 23S rRNA, mutations to the 50S ribosomal L3 protein | Mutations to the 23S rRNA |
| Antibiotic Class | P. auruginosa | E. coli | K. pneumoniae | A. baumanii |
|---|---|---|---|---|
| Penicillins | AmpC, ESBLs, other b-lactamases | AmpC, ESBLs, other b-lactamases | AmpC, ESBLs, other b-lactamases | AmpC, ESBLs, other b-lactamases |
| Cephalosporins 1st gen. | AmpC, ESBLs | AmpC, ESBLs | AmpC, ESBLs | AmpC, ESBLs |
| Cephalosporins 2nd gen. | AmpC, ESBLs | AmpC, ESBLs | AmpC, ESBLs | AmpC, ESBLs |
| Cephalosporins 3rd gen. | AmpC, ESBLs | AmpC, ESBLs | AmpC, ESBLs | AmpC, ESBLs |
| Cephalosporins 4th gen. | ESBLs | ESBLs | ESBLs | ESBLs |
| b-lactamase inhibitors | AmpC | AmpC | AmpC | AmpC |
| Aztreonam | ESBLs | ESBLs | ESBLs | ESBLs |
| Carbapenems | Class B & D carbapenemases | Class A, B & D carbapenemases | Class A, B & D carbapenemases | Class B & D carbapenemases |
| Tetracyclines | Efflux pumps | Efflux pumps | Efflux pumps | Efflux pumps |
| Tigecycline | Efflux pumps | Efflux pumps, porin downregulation | acrAB efflux pump | AdeABC efflux pump, reduced membrane permeability |
| Macrolides and clindamycin | Efflux pumps | Efflux pumps, macrolide inactivating enzymes, target site modification | ||
| Fluoroquinolones | Mutations in topoisomerase IV and DNA gyrase genes, efflux pumps | Mutations in DNA gyrase gene | Mutations in DNA gyrase gene, efflux pumps, enzyme protection proteins, fluoroquinolone degradation enzymes | Mutations in genes for DNA gyrase and topoisomerase IV, efflux pumps, enzyme protection proteins, fluoroquinolone degradation enzymes |
| Rifampicin | Mutations in RNA polymerase gene | Mutations in RNA polymerase gene | Enzymatic degradation | Mutations in RNA polymerase gene, efflux pumps, enzymatic degradation |
| TMP/SMX | Efflux pumps | Overproduction of DHFR, mutation of DHPS | ||
| Aminoglycosides | Aminoglycoside degradation enzymes, efflux pumps | Aminoglycoside degradation enzymes | Aminoglycoside degradation enzymes, production of 16SrRNA | Aminoglycoside degradation enzymes |
| Colistin | Reduction of membrane negative charge through addition of N4-aminoarabinose to lipid A | Reduction of membrane negative charge through addition of phosphoethanolamine to lipid A | Reduction of membrane negative charge through addition of phosphoethanolamine to lipid A | efflux pumps, loss of LPS production, alterations in the structure of lipid A |
| Antibiotic | Resistance Mechanisms Designed to Overcome | Active Against | Inactive Against | Indications |
|---|---|---|---|---|
| Cephalosporins | ||||
| Ceftobiprole | Active against altered PBPs, such as PBP2a and PBP2x | MRSA, VRSA, PRSP, Gram-negative bacteria | ESBLs, AmpC, Class A, B and D carbapenemases | CAP, SSTI |
| Ceftaroline | Active against altered PBPs, such as PBP2a | MRSA | ESBLs, AmpC, Class A, B and D carbapenemases | CAP, SSTI |
| Cefiderocol | Utilizes iron to bypass porins, accumulates in periplasmic space. Resistant to hydrolysis to all β-lactamases: ESBLs, AmpC, Class A, B and D carbapenemases | MDR Gram-negative bacteria | - | HAP, VAP, cUTI |
| Ceftolozane-tazobactam | Ceftolozane overcomes P. aeuruginosa resistance mechanisms: efflux pumps, altered PBPs and porins. Tazobactam confers resistance to ESBLs | MDR P. aeuruginosa | cUTI, cIAI | |
| Novel β-lactam inhibitor combination | ||||
| Meropenem-vaborbactam | Vaborbactam inhibits ESBLs, Class C cephalosporinases and Class A carbapenemases | MDR Gram-negative bacteria | Class B carbapenemases | HAP, VAP, cUTI, cIAI |
| Imipenem-relebactam | Relebactam inhibits ESBLs, Class C cephalosporinases and Class A carbapenemases | MDR Gram-negative bacteria | Class B and D carbapenemases | |
| Aztreonam-avibactam | Avibactam inhibits all Class A and Class C, and some Class D β-lactamases. Aztreonam inhibits Class B β-lactamases. | MDR Gram-negative bacteria | - | Approval pending |
| Ceftazidime-avibactam | Avibactam inhibits all Class A and Class C, and some Class D β-lactamases. | MDR Gram-negative bacteria | Class B carbapenemases | HAP, VAP, cUTI, cIAI |
| Fluoroquinolones | ||||
| Delafloxacin | Balanced inhibition of topoisomerase IV and DNA gyrase, decreasing resistance potential. Enhanced penetration and activity in acidic environments, such as infection sites | Fluoroquinolone-resistant S. aureus, Gram-negative bacteria | CAP, SSTI | |
| Tetracyclines | ||||
| Omadacycline | Active against tetracycline efflux pumps or ribosomal protection proteins | MRSA, VRE, PRSP, Gram-negative bacteria | P. aeruginosa | CAP, SSTI |
| Eravacycline | Active against bacteria that have efflux pumps or ribosomal protection proteins | MDR Acinetobacter spp., ESBL-producing Enterobacteriaceae, VRE, MRSA and S. pneumoniae | cIAI | |
| Aminoglycoside | ||||
| Plazomicin | Resistant to degradation by aminoglycoside nucleotidyltransferases, phosphotransferases and acetyltransferases | Gram-negative bacteria that produce aminoglycoside degradation enzymes | 16SrRNA methylase producing bacteria | cUTI |
| Lipoglycopeptides | ||||
| Dalbavancin | Increased membrane anchoring | MRSA, VISA and VRE exhibiting the VanB phenotype | VRSA and VRE exhibiting the VanA phenotype | SSTI |
| Telavancin | Increased membrane anchoring, disruption of membrane integrity, permeability and potential | MRSA, VISA and VRE exhibiting the VanB phenotype | VRSA and VRE exhibiting the VanA phenotype | SSTI, HAP & VAP by S. aureus |
| Oritavancin | Increased membrane anchoring, disruption of membrane integrity, permeability and potential, RNA synthesis inhibition, binding to both D-Ala-D-Ala & D-Ala-D-Lac dipeptides | MRSA, VISA & VRE with VanB phenotype, VRSA & VRE with VanA phenotype | - | SSTI |
| Oxazolidinone | ||||
| Tedizolid | More potent binding to the 23S rRNA binding site than linezolid | VRE, MRSA and linezolid-resistant isolates | - | SSTI |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kakoullis, L.; Papachristodoulou, E.; Chra, P.; Panos, G. Mechanisms of Antibiotic Resistance in Important Gram-Positive and Gram-Negative Pathogens and Novel Antibiotic Solutions. Antibiotics 2021, 10, 415. https://doi.org/10.3390/antibiotics10040415
Kakoullis L, Papachristodoulou E, Chra P, Panos G. Mechanisms of Antibiotic Resistance in Important Gram-Positive and Gram-Negative Pathogens and Novel Antibiotic Solutions. Antibiotics. 2021; 10(4):415. https://doi.org/10.3390/antibiotics10040415
Chicago/Turabian StyleKakoullis, Loukas, Eleni Papachristodoulou, Paraskevi Chra, and George Panos. 2021. "Mechanisms of Antibiotic Resistance in Important Gram-Positive and Gram-Negative Pathogens and Novel Antibiotic Solutions" Antibiotics 10, no. 4: 415. https://doi.org/10.3390/antibiotics10040415