Label-Free Long-Term Methods for Live Cell Imaging of Neurons: New Opportunities

 ,
,  , , and
, , and 
Abstract
:
1. Introduction
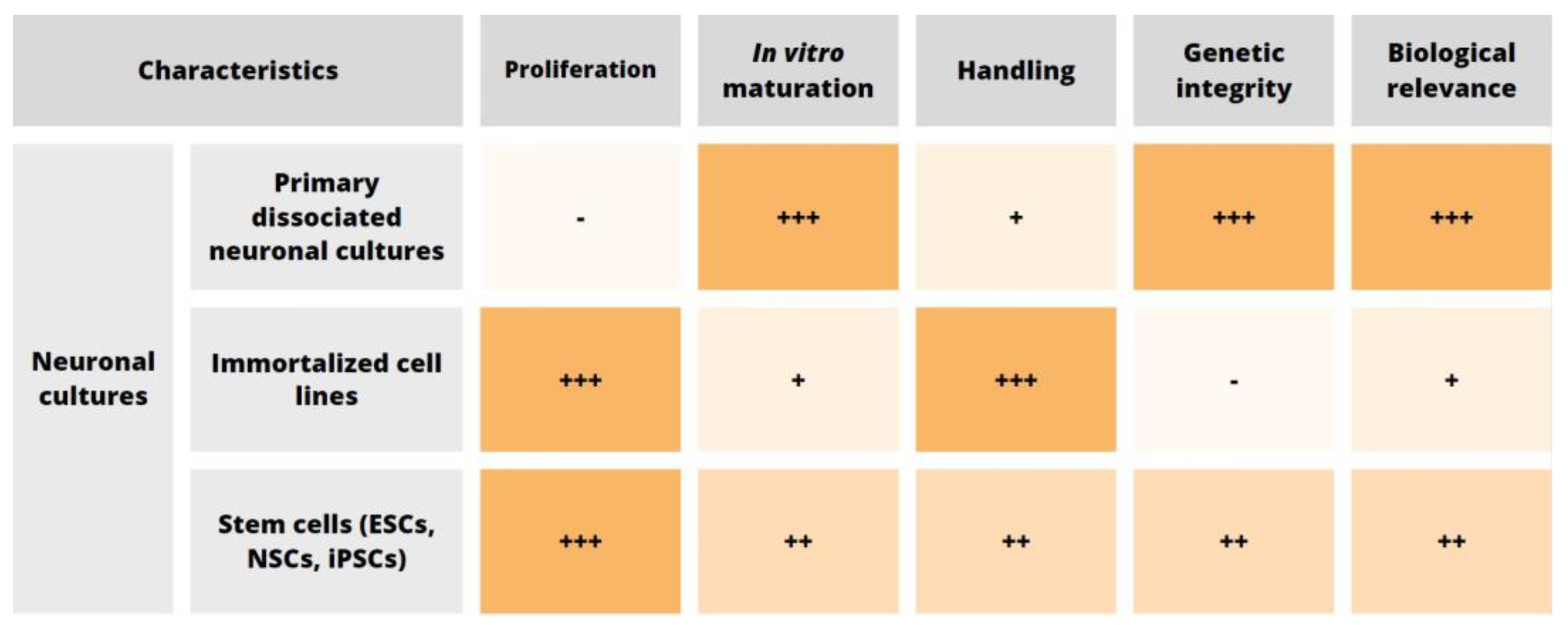
2. Cell Source
2.1. Primary Neuronal Cultures
2.2. Neuronal Cell Lines
2.3. In Vitro Stem Cell-Derived Neurons
2.4. Neuronal Cell Death In Vitro
3. Label-Free Live Cell Imaging—Key Experimental Settings
4. Label-Free Methods for Long-Term and High-Resolution Imaging of Neurons
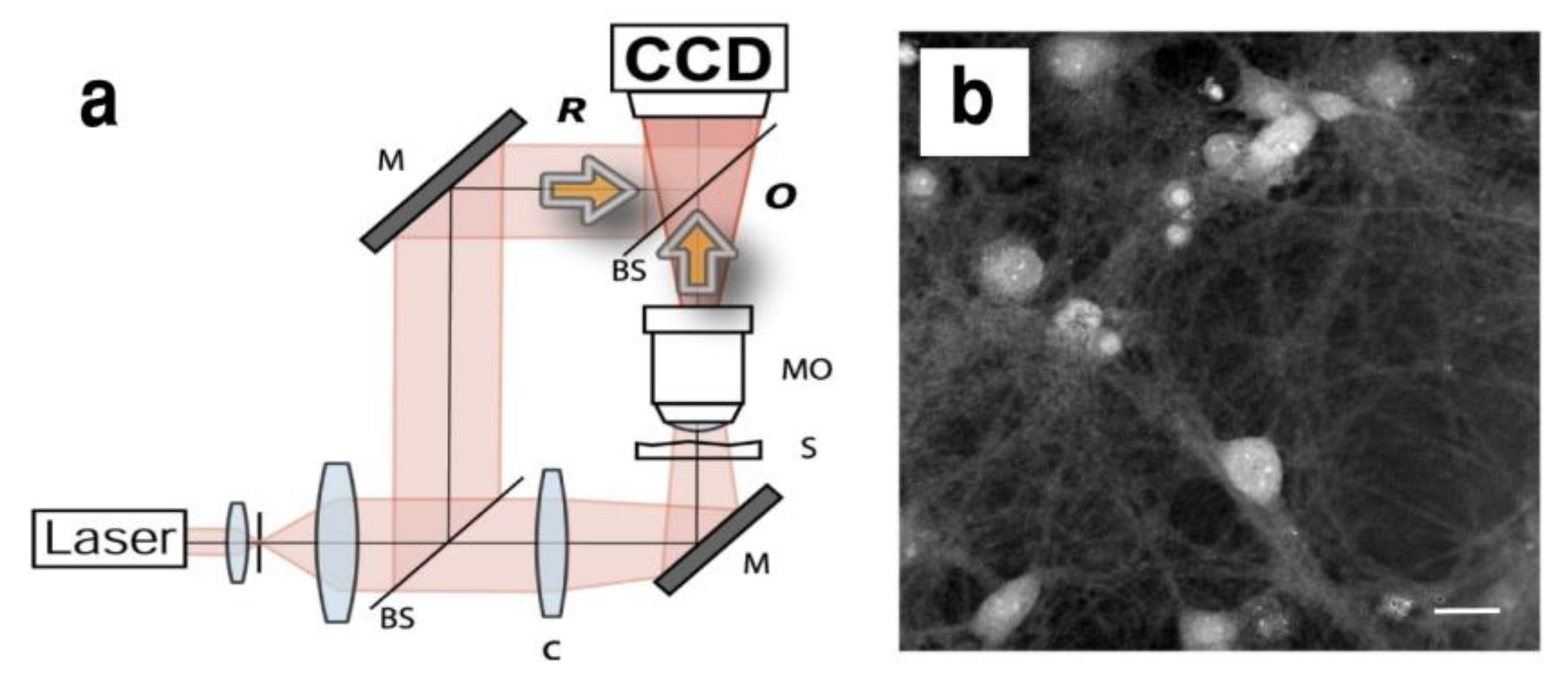
4.1. Digital Holography Microscopy
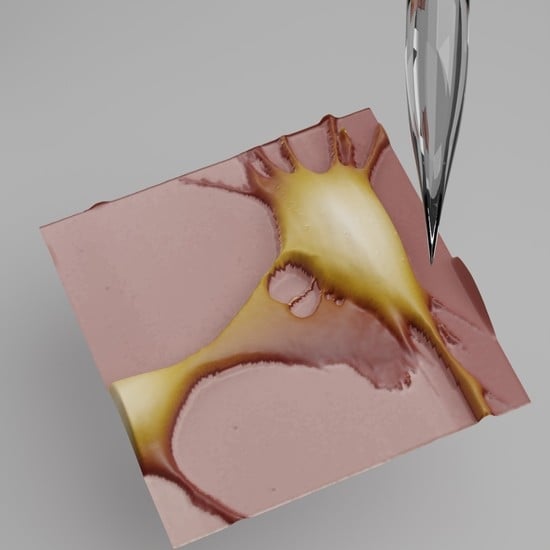
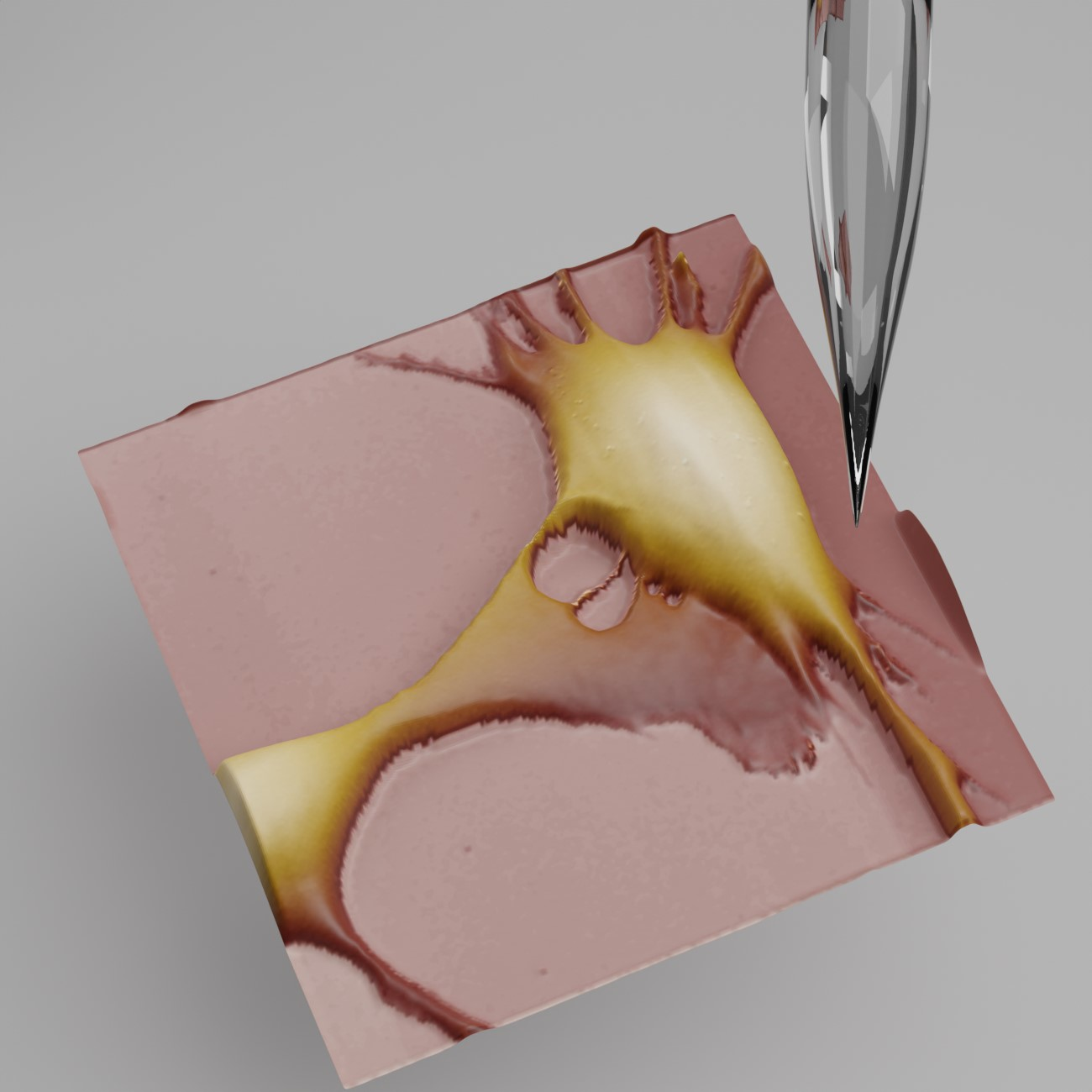
4.2. Scanning Probe Microscopy (SPM)
5. Conclusions
Author Contributions
Funding
 .
.Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Geschwind, D.H.; Rakic, P. Cortical Evolution: Judge the Brain by Its Cover. Neuron 2013, 80, 633–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merryweather, D.; Roach, P. The Need for Advanced Three-Dimensional Neural Models and Developing Enabling Technologies. MRS Commun. 2017, 7, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Bryant, D.M.; Mostov, K.E. From Cells to Organs: Building Polarized Tissue. Nat. Rev. Mol. Cell Biol. 2008, 9, 887–901. [Google Scholar] [CrossRef] [PubMed]
- Banker, G. The Development of Neuronal Polarity: A Retrospective View. J. Neurosci. 2018, 38, 1867–1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cáceres, A.; Ye, B.; Dotti, C.G. Neuronal Polarity: Demarcation, Growth and Commitment. Curr. Opin. Cell Biol. 2012, 24, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Barnes, A.P.; Polleux, F. Establishment of Axon-Dendrite Polarity in Developing Neurons. Annu. Rev. Neurosci. 2009, 32, 347–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, A.; Cavalli, V. Signaling Over Distances. Mol. Cell. Proteom. 2016, 15, 382–393. [Google Scholar] [CrossRef] [Green Version]
- Hirokawa, N.; Niwa, S.; Tanaka, Y. Molecular Motors in Neurons: Transport Mechanisms and Roles in Brain Function, Development, and Disease. Neuron 2010, 68, 610–638. [Google Scholar] [CrossRef] [Green Version]
- De Anda, F.C.; Madabhushi, R.; Rei, D.; Meng, J.; Gräff, J.; Durak, O.; Meletis, K.; Richter, M.; Schwanke, B.; Mungenast, A.; et al. Cortical Neurons Gradually Attain a Post-Mitotic State. Cell Res. 2016, 26, 1033–1047. [Google Scholar] [CrossRef] [Green Version]
- Herrup, K.; Yang, Y. Cell Cycle Regulation in the Postmitotic Neuron: Oxymoron or New Biology? Nat. Rev. Neurosci. 2007, 8, 368–378. [Google Scholar] [CrossRef]
- Fawcett, J.W. The Struggle to Make CNS Axons Regenerate: Why Has It Been so Difficult? Neurochem. Res. 2020, 45, 144–158. [Google Scholar] [CrossRef] [Green Version]
- Rodemer, W.; Gallo, G.; Selzer, M.E. Mechanisms of Axon Elongation Following CNS Injury: What Is Happening at the Axon Tip? Front. Cell. Neurosci. 2020, 14, 177. [Google Scholar] [CrossRef] [PubMed]
- Mladinic, M.; Muller, K.J.; Nicholls, J.G. Central Nervous System Regeneration: From Leech to Opossum. J. Physiol. 2009, 587, 2775–2782. [Google Scholar] [CrossRef]
- Rakic, P. Neuroscience. No More Cortical Neurons for You. Science 2006, 313, 928–929. [Google Scholar] [CrossRef]
- Schaefers, A.T.U.; Teuchert-Noodt, G. Developmental Neuroplasticity and the Origin of Neurodegenerative Diseases. World J. Biol. Psychiatry 2016, 17, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Glavač, D.; Mladinić, M.; Ban, J.; Mazzone, G.L.; Sámano, C.; Tomljanović, I.; Jezernik, G.; Ravnik-Glavač, M. The Potential Connection between Molecular Changes and Biomarkers Related to ALS and the Development and Regeneration of CNS. Int. J. Mol. Sci. 2022, 23, 11360. [Google Scholar] [CrossRef]
- Dotti, C.G.; Sullivan, C.A.; Banker, G.A. The Establishment of Polarity by Hippocampal Neurons in Culture. J. Neurosci. 1988, 8, 1454–1468. [Google Scholar] [CrossRef] [Green Version]
- Kaech, S.; Huang, C.-F.; Banker, G. General Considerations for Live Imaging of Developing Hippocampal Neurons in Culture. Cold Spring Harb. Protoc. 2012, 2012, pdb.ip068221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Ali, H.; Beckerman, S.R.; Bixby, J.L.; Lemmon, V.P. In Vitro Models of Axon Regeneration. Exp. Neurol. 2017, 287, 423–434. [Google Scholar] [CrossRef] [Green Version]
- The Principles of Humane Experimental Technique. Med. J. Aust. 1960, 1, 500. [CrossRef]
- Xu, K.; Zhong, G.; Zhuang, X. Actin, Spectrin, and Associated Proteins Form a Periodic Cytoskeletal Structure in Axons. Science 2013, 339, 452–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Este, E.; Kamin, D.; Göttfert, F.; El-Hady, A.; Hell, S.W. STED Nanoscopy Reveals the Ubiquity of Subcortical Cytoskeleton Periodicity in Living Neurons. Cell Rep. 2015, 10, 1246–1251. [Google Scholar] [CrossRef] [Green Version]
- Schnell, U.; Dijk, F.; Sjollema, K.A.; Giepmans, B.N.G. Immunolabeling Artifacts and the Need for Live-Cell Imaging. Nat. Methods 2012, 9, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Whelan, D.R.; Bell, T.D.M. Image Artifacts in Single Molecule Localization Microscopy: Why Optimization of Sample Preparation Protocols Matters. Sci. Rep. 2015, 5, 7924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaudoin, G.M.J.; Lee, S.-H.; Singh, D.; Yuan, Y.; Ng, Y.-G.; Reichardt, L.F.; Arikkath, J. Culturing Pyramidal Neurons from the Early Postnatal Mouse Hippocampus and Cortex. Nat. Protoc. 2012, 7, 1741–1754. [Google Scholar] [CrossRef]
- Kaech, S.; Banker, G. Culturing Hippocampal Neurons. Nat. Protoc. 2006, 1, 2406–2415. [Google Scholar] [CrossRef]
- Koseki, H.; Donegá, M.; Lam, B.Y.; Petrova, V.; van Erp, S.; Yeo, G.S.; Kwok, J.C.; Ffrench-Constant, C.; Eva, R.; Fawcett, J.W. Selective Rab11 Transport and the Intrinsic Regenerative Ability of CNS Axons. eLife 2017, 6, e26956. [Google Scholar] [CrossRef]
- Jo, Y.-H.; Stoeckel, M.-E.; Freund-Mercier, M.-J.; Schlichter, R. Oxytocin Modulates Glutamatergic Synaptic Transmission between Cultured Neonatal Spinal Cord Dorsal Horn Neurons. J. Neurosci. 1998, 18, 2377–2386. [Google Scholar] [CrossRef] [Green Version]
- Hugel, S.; Schlichter, R. Presynaptic P2X Receptors Facilitate Inhibitory GABAergic Transmission between Cultured Rat Spinal Cord Dorsal Horn Neurons. J. Neurosci. 2000, 20, 2121–2130. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.-J.; Glauner, K.S.; Gereau, R.W. ERK Integrates PKA and PKC Signaling in Superficial Dorsal Horn Neurons. I. Modulation of A-Type K+ Currents. J. Neurophysiol. 2003, 90, 1671–1679. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Nagao, M.; Sugimori, M.; Kosako, H.; Nakatomi, H.; Yamamoto, N.; Takebayashi, H.; Nabeshima, Y.; Kitamura, T.; Weinmaster, G.; et al. Transcription Factor Expression and Notch-Dependent Regulation of Neural Progenitors in the Adult Rat Spinal Cord. J. Neurosci. 2001, 21, 9814–9823. [Google Scholar] [CrossRef] [Green Version]
- Vargova, I.; Kriska, J.; Kwok, J.C.F.; Fawcett, J.W.; Jendelova, P. Long-Term Cultures of Spinal Cord Interneurons. Front. Cell. Neurosci. 2022, 16, 827628. [Google Scholar] [CrossRef]
- Cao, D.-L.; Peng, B.; Bao, C.; Gao, Y.-J. Primary Culture of Mouse Neurons from the Spinal Cord Dorsal Horn. Bio-Protocol 2017, 7, e2098. [Google Scholar] [CrossRef]
- Mikhailova, M.M.; Bolshakov, A.P.; Chaban, E.A.; Paltsev, M.A.; Panteleyev, A.A. Primary Culture of Mouse Embryonic Spinal Cord Neurons: Cell Composition and Suitability for Axonal Regeneration Studies. Int. J. Neurosci. 2019, 129, 762–769. [Google Scholar] [CrossRef]
- Eldeiry, M.; Yamanaka, K.; Reece, T.B.; Aftab, M. Spinal Cord Neurons Isolation and Culture from Neonatal Mice. J. Vis. Exp. 2017, 125, 55856. [Google Scholar] [CrossRef]
- Petrović, A.; Ban, J.; Tomljanović, I.; Pongrac, M.; Ivaničić, M.; Mikašinović, S.; Mladinic, M. Establishment of Long-Term Primary Cortical Neuronal Cultures from Neonatal Opossum Monodelphis Domestica. Front. Cell. Neurosci. 2021, 15, 661492. [Google Scholar] [CrossRef]
- Bartkowska, K.; Gajerska, M.; Turlejski, K.; Djavadian, R.L. Expression of TrkC Receptors in the Developing Brain of the Monodelphis Opossum and Its Effect on the Development of Cortical Cells. PLoS ONE 2013, 8, e74346. [Google Scholar] [CrossRef] [Green Version]
- Aubid, N.N.; Liu, Y.; Vidal, J.M.P.; Hall, V.J. Isolation and Culture of Porcine Primary Fetal Progenitors and Neurons from the Developing Dorsal Telencephalon. J. Vet Sci. 2019, 20, e3. [Google Scholar] [CrossRef]
- Reddy, R.C.; Amodei, R.; Estill, C.T.; Stormshak, F.; Meaker, M.; Roselli, C.E. Effect of Testosterone on Neuronal Morphology and Neuritic Growth of Fetal Lamb Hypothalamus-Preoptic Area and Cerebral Cortex in Primary Culture. PLoS ONE 2015, 10, e0129521. [Google Scholar] [CrossRef] [Green Version]
- Negishi, T.; Ishii, Y.; Kawamura, S.; Kuroda, Y.; Yoshikawa, Y. Cryopreservation and Primary Culture of Cerebral Neurons from Cynomolgus Monkeys (Macaca fascicularis). Neurosci. Lett. 2002, 328, 21–24. [Google Scholar] [CrossRef]
- Bonfanti, L.; Peretto, P. Adult Neurogenesis in Mammals—A Theme with Many Variations. Eur. J. Neurosci. 2011, 34, 930–950. [Google Scholar] [CrossRef]
- Todd, G.K.; Boosalis, C.A.; Burzycki, A.A.; Steinman, M.Q.; Hester, L.D.; Shuster, P.W.; Patterson, R.L. Towards Neuronal Organoids: A Method for Long-Term Culturing of High-Density Hippocampal Neurons. PLoS ONE 2013, 8, e58996. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, D.; Ban, J.; Iseppon, F.; Torre, V. An Improved Method for Growing Neurons: Comparison with Standard Protocols. J Neurosci. Methods 2017, 280, 1–10. [Google Scholar] [CrossRef]
- Kaneko, A.; Sankai, Y. Long-Term Culture of Rat Hippocampal Neurons at Low Density in Serum-Free Medium: Combination of the Sandwich Culture Technique with the Three-Dimensional Nanofibrous Hydrogel PuraMatrix. PLoS ONE 2014, 9, e102703. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Piechowicz, M.; Qiu, S. A Simplified Method for Ultra-Low Density, Long-Term Primary Hippocampal Neuron Culture. J. Vis. Exp. 2016, 109, e53797. [Google Scholar] [CrossRef] [Green Version]
- Calvo-Rodriguez, M.; Hernando-Pérez, E.; López-Vázquez, S.; Núñez, J.; Villalobos, C.; Núñez, L. Remodeling of Intracellular Ca2+ Homeostasis in Rat Hippocampal Neurons Aged In Vitro. Int. J. Mol. Sci. 2020, 21, 1549. [Google Scholar] [CrossRef] [Green Version]
- Biffi, E.; Regalia, G.; Menegon, A.; Ferrigno, G.; Pedrocchi, A. The Influence of Neuronal Density and Maturation on Network Activity of Hippocampal Cell Cultures: A Methodological Study. PLoS ONE 2013, 8, e83899. [Google Scholar] [CrossRef]
- Lei, W.-L.; Xing, S.-G.; Deng, C.-Y.; Ju, X.-C.; Jiang, X.-Y.; Luo, Z.-G. Laminin/Β1 Integrin Signal Triggers Axon Formation by Promoting Microtubule Assembly and Stabilization. Cell Res. 2012, 22, 954–972. [Google Scholar] [CrossRef] [Green Version]
- Ban, J.; Bonifazi, P.; Pinato, G.; Broccard, F.D.; Studer, L.; Torre, V.; Ruaro, M.E. Embryonic Stem Cell-Derived Neurons Form Functional Networks in Vitro. Stem Cells 2007, 25, 738–749. [Google Scholar] [CrossRef]
- Baj, G.; Patrizio, A.; Montalbano, A.; Sciancalepore, M.; Tongiorgi, E. Developmental and Maintenance Defects in Rett Syndrome Neurons Identified by a New Mouse Staging System in Vitro. Front. Cell. Neurosci. 2014, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, A.M.; DeSimone, E.; Chwalek, K.; Kaplan, D.L. 3D in Vitro Modeling of the Central Nervous System. Prog. Neurobiol. 2015, 125, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brewer, G.J.; Torricelli, J.R.; Evege, E.K.; Price, P.J. Optimized Survival of Hippocampal Neurons in B27-Supplemented Neurobasal, a New Serum-Free Medium Combination. J. Neurosci. Res. 1993, 35, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Boehler, M.D.; Jones, T.T.; Wheeler, B.C. NbActiv4 Medium Improvement to Neurobasal/B27 Increases Neuron Synapse Densities and Network Spike Rates on Multielectrode Arrays. J. Neurosci. Methods 2008, 170, 181–187. [Google Scholar] [CrossRef] [Green Version]
- Cullen, D.K.; Gilroy, M.E.; Irons, H.R.; Laplaca, M.C. Synapse-to-Neuron Ratio Is Inversely Related to Neuronal Density in Mature Neuronal Cultures. Brain Res. 2010, 1359, 44–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivenshitz, M.; Segal, M. Neuronal Density Determines Network Connectivity and Spontaneous Activity in Cultured Hippocampus. J. Neurophysiol. 2010, 104, 1052–1060. [Google Scholar] [CrossRef] [Green Version]
- Dowell, J.A.; Johnson, J.A.; Li, L. Identification of Astrocyte Secreted Proteins with a Combination of Shotgun Proteomics and Bioinformatics. J. Proteome Res. 2009, 8, 4135–4143. [Google Scholar] [CrossRef] [Green Version]
- Etienne-Manneville, S. In Vitro Assay of Primary Astrocyte Migration as a Tool to Study Rho GTPase Function in Cell Polarization. Methods Enzymol. 2006, 406, 565–578. [Google Scholar] [CrossRef]
- Lange, S.C.; Bak, L.K.; Waagepetersen, H.S.; Schousboe, A.; Norenberg, M.D. Primary Cultures of Astrocytes: Their Value in Understanding Astrocytes in Health and Disease. Neurochem. Res. 2012, 37, 2569–2588. [Google Scholar] [CrossRef] [Green Version]
- Saura, J. Microglial Cells in Astroglial Cultures: A Cautionary Note. J. Neuroinflamm. 2007, 4, 26. [Google Scholar] [CrossRef] [Green Version]
- Wolfes, A.C.; Ahmed, S.; Awasthi, A.; Stahlberg, M.A.; Rajput, A.; Magruder, D.S.; Bonn, S.; Dean, C. A Novel Method for Culturing Stellate Astrocytes Reveals Spatially Distinct Ca2+ Signaling and Vesicle Recycling in Astrocytic Processes. J. Gen. Physiol. 2017, 149, 149–170. [Google Scholar] [CrossRef] [Green Version]
- Roqué, P.J.; Costa, L.G. Co-Culture of Neurons and Microglia. Curr. Protoc. Toxicol. 2017, 74, 11.24.1–11.24.17. [Google Scholar] [CrossRef]
- Crocker, S.J.; Frausto, R.F.; Whitton, J.L.; Milner, R. A Novel Method to Establish Microglia-Free Astrocyte Cultures: Comparison of Matrix Metalloproteinase Expression Profiles in Pure Cultures of Astrocytes and Microglia. Glia 2008, 56, 1187–1198. [Google Scholar] [CrossRef] [Green Version]
- Goshi, N.; Morgan, R.K.; Lein, P.J.; Seker, E. A Primary Neural Cell Culture Model to Study Neuron, Astrocyte, and Microglia Interactions in Neuroinflammation. J. Neuroinflamm. 2020, 17, 155. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.A.; Tischler, A.S. Establishment of a Noradrenergic Clonal Line of Rat Adrenal Pheochromocytoma Cells Which Respond to Nerve Growth Factor. Proc. Natl. Acad. Sci. USA 1976, 73, 2424–2428. [Google Scholar] [CrossRef] [Green Version]
- Lee, V.M.; Andrews, P.W. Differentiation of NTERA-2 Clonal Human Embryonal Carcinoma Cells into Neurons Involves the Induction of All Three Neurofilament Proteins. J. Neurosci. 1986, 6, 514–521. [Google Scholar] [CrossRef] [Green Version]
- Biedler, J.L.; Roffler-Tarlov, S.; Schachner, M.; Freedman, L.S. Multiple Neurotransmitter Synthesis by Human Neuroblastoma Cell Lines and Clones. Cancer Res. 1978, 38, 3751–3757. [Google Scholar] [PubMed]
- Gordon, J.; Amini, S.; White, M.K. General Overview of Neuronal Cell Culture. In Neuronal Cell Culture; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 1078, pp. 1–8. [Google Scholar] [CrossRef] [Green Version]
- David, A.; Raos, B.; Svirskis, D.; O’Carroll, S.J. Optimised Techniques for High-Throughput Screening of Differentiated SH-SY5Y Cells and Application for Neurite Outgrowth Assays. Sci. Rep. 2021, 11, 23935. [Google Scholar] [CrossRef] [PubMed]
- Campos Cogo, S.; da Costa do Nascimento, T.G.F.; de Almeida Brehm Pinhatti, F.; de França Junior, N.; Santos Rodrigues, B.; Cavalli, L.R.; Elifio-Esposito, S. An Overview of Neuroblastoma Cell Lineage Phenotypes and in Vitro Models. Exp. Biol. Med. 2020, 245, 1637–1647. [Google Scholar] [CrossRef]
- Yoshihara, M.; Hayashizaki, Y.; Murakawa, Y. Genomic Instability of IPSCs: Challenges Towards Their Clinical Applications. Stem Cell Rev. Rep. 2017, 13, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Edwards, M.A.; Loxley, R.A.; Williams, A.J.; Connor, M.; Phillips, J.K. Lack of Functional Expression of NMDA Receptors in PC12 Cells. Neurotoxicology 2007, 28, 876–885. [Google Scholar] [CrossRef]
- LePage, K.T.; Dickey, R.W.; Gerwick, W.H.; Jester, E.L.; Murray, T.F. On the Use of Neuro-2a Neuroblastoma Cells versus Intact Neurons in Primary Culture for Neurotoxicity Studies. Crit. Rev. Neurobiol. 2005, 17, 27–50. [Google Scholar] [CrossRef]
- Martin, G.R. Isolation of a Pluripotent Cell Line from Early Mouse Embryos Cultured in Medium Conditioned by Teratocarcinoma Stem Cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.J.; Kaufman, M.H. Establishment in Culture of Pluripotential Cells from Mouse Embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, B.A.; Weiss, S. Generation of Neurons and Astrocytes from Isolated Cells of the Adult Mammalian Central Nervous System. Science 1992, 255, 1707–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastrana, E.; Silva-Vargas, V.; Doetsch, F. Eyes Wide Open: A Critical Review of Sphere-Formation as an Assay For Stem Cells. Cell Stem Cell 2011, 8, 486–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriegstein, A.; Alvarez-Buylla, A. The Glial Nature of Embryonic and Adult Neural Stem Cells. Annu. Rev. Neurosci. 2009, 32, 149–184. [Google Scholar] [CrossRef] [Green Version]
- Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Südhof, T.C.; Wernig, M. Direct Conversion of Fibroblasts to Functional Neurons by Defined Factors. Nature 2010, 463, 1035–1041. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. A Decade of Transcription Factor-Mediated Reprogramming to Pluripotency. Nat. Rev. Mol. Cell Biol. 2016, 17, 183–193. [Google Scholar] [CrossRef]
- Stockley, J.H.; Evans, K.; Matthey, M.; Volbracht, K.; Agathou, S.; Mukanowa, J.; Burrone, J.; Káradóttir, R.T. Surpassing Light-Induced Cell Damage in Vitro with Novel Cell Culture Media. Sci. Rep. 2017, 7, 849. [Google Scholar] [CrossRef] [Green Version]
- Carvalho-de-Souza, J.L.; Treger, J.S.; Dang, B.; Kent, S.B.H.; Pepperberg, D.R.; Bezanilla, F. Photosensitivity of Neurons Enabled by Cell-Targeted Gold Nanoparticles. Neuron 2015, 86, 207–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamkim, A.; Kiseleva, I. Mechanosensitivity of the Nervous System. Springer: Dordrecht, The Netherlands, 2009; pp. 9–11. [Google Scholar]
- Cho, H.; Shin, J.; Shin, C.Y.; Lee, S.-Y.; Oh, U. Mechanosensitive Ion Channels in Cultured Sensory Neurons of Neonatal Rats. J. Neurosci. 2002, 22, 1238–1247. [Google Scholar] [CrossRef]
- Marcus, M.; Baranes, K.; Park, M.; Choi, I.S.; Kang, K.; Shefi, O. Interactions of Neurons with Physical Environments. Adv. Healthc. Mater. 2017, 6, 1700267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J. Neuronal Cell Death in Nervous System Development, Disease, and Injury (Review). Int. J. Mol. Med. 2001, 7, 455–478. [Google Scholar] [CrossRef]
- Pavillon, N.; Kühn, J.; Moratal, C.; Jourdain, P.; Depeursinge, C.; Magistretti, P.J.; Marquet, P. Early Cell Death Detection with Digital Holographic Microscopy. PLoS ONE 2012, 7, e30912. [Google Scholar] [CrossRef]
- Pattison, D.I.; Davies, M.J. Actions of Ultraviolet Light on Cellular Structures. In Cancer: Cell Structures, Carcinogens and Genomic Instability; Experientia Supplementum; Birkhäuser: Basel, Switzerland, 2006; pp. 131–157. ISBN 978-3-7643-7378-8. [Google Scholar]
- Wäldchen, S.; Lehmann, J.; Klein, T.; van de Linde, S.; Sauer, M. Light-Induced Cell Damage in Live-Cell Super-Resolution Microscopy. Sci. Rep. 2015, 5, 15348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frigault, M.M.; Lacoste, J.; Swift, J.L.; Brown, C.M. Live-Cell Microscopy—Tips and Tools. J. Cell Sci. 2009, 122, 753–767. [Google Scholar] [CrossRef] [Green Version]
- Marquet, P.; Depeursinge, C.; Magistretti, P.J. Review of Quantitative Phase-Digital Holographic Microscopy: Promising Novel Imaging Technique to Resolve Neuronal Network Activity and Identify Cellular Biomarkers of Psychiatric Disorders. Neurophotonics 2014, 1, 020901. [Google Scholar] [CrossRef] [Green Version]
- Hell, S.W. Microscopy and Its Focal Switch. Nat. Methods 2009, 6, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Honig, M.G.; Hume, R.I. Dil and DiO: Versatile Fluorescent Dyes for Neuronal Labelling and Pathway Tracing. Trends Neurosci. 1989, 12, 333–341. [Google Scholar] [CrossRef] [Green Version]
- Levy, S.L.; White, J.J.; Lackey, E.P.; Schwartz, L.; Sillitoe, R.V. WGA-Alexa Conjugates for Axonal Tracing. Curr. Protoc. Neurosci. 2017, 79, 1.28.1–1.28.24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsien, R.Y. The Green Fluorescent Protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar] [CrossRef] [PubMed]
- Wiedenmann, J.; Oswald, F.; Nienhaus, G.U. Fluorescent Proteins for Live Cell Imaging: Opportunities, Limitations, and Challenges. IUBMB Life 2009, 61, 1029–1042. [Google Scholar] [CrossRef]
- Costantini, L.M.; Baloban, M.; Markwardt, M.L.; Rizzo, M.A.; Guo, F.; Verkhusha, V.V.; Snapp, E.L. A Palette of Fluorescent Proteins Optimized for Diverse Cellular Environments. Nat. Commun. 2015, 6, 7670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishin, A.S.; Subach, F.V.; Yampolsky, I.V.; King, W.; Lukyanov, K.A.; Verkhusha, V.V. The First Mutant of the Aequorea Victoria Green Fluorescent Protein That Forms a Red Chromophore. Biochemistry 2008, 47, 4666–4673. [Google Scholar] [CrossRef] [Green Version]
- Karra, D.; Dahm, R. Transfection Techniques for Neuronal Cells. J. Neurosci. 2010, 30, 6171–6177. [Google Scholar] [CrossRef] [Green Version]
- Deibler, M.; Spatz, J.P.; Kemkemer, R. Actin Fusion Proteins Alter the Dynamics of Mechanically Induced Cytoskeleton Rearrangement. PLoS ONE 2011, 6, e22941. [Google Scholar] [CrossRef]
- Riedl, J.; Crevenna, A.H.; Kessenbrock, K.; Yu, J.H.; Neukirchen, D.; Bista, M.; Bradke, F.; Jenne, D.; Holak, T.A.; Werb, Z.; et al. Lifeact: A Versatile Marker to Visualize F-Actin. Nat. Methods 2008, 5, 605–607. [Google Scholar] [CrossRef]
- Melak, M.; Plessner, M.; Grosse, R. Actin Visualization at a Glance. J. Cell Sci. 2017, 130, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukinavičius, G.; Reymond, L.; D’Este, E.; Masharina, A.; Göttfert, F.; Ta, H.; Güther, A.; Fournier, M.; Rizzo, S.; Waldmann, H.; et al. Fluorogenic Probes for Live-Cell Imaging of the Cytoskeleton. Nat. Methods 2014, 11, 731–733. [Google Scholar] [CrossRef]
- Park, Y.; Depeursinge, C.; Popescu, G. Quantitative Phase Imaging in Biomedicine. Nat. Photon. 2018, 12, 578–589. [Google Scholar] [CrossRef]
- Marquet, P.; Rappaz, B.; Magistretti, P.J.; Cuche, E.; Emery, Y.; Colomb, T.; Depeursinge, C. Digital Holographic Microscopy: A Noninvasive Contrast Imaging Technique Allowing Quantitative Visualization of Living Cells with Subwavelength Axial Accuracy. Opt. Lett. 2005, 30, 468–470. [Google Scholar] [CrossRef] [Green Version]
- Bon, P.; Lécart, S.; Fort, E.; Lévêque-Fort, S. Fast Label-Free Cytoskeletal Network Imaging in Living Mammalian Cells. Biophys. J. 2014, 106, 1588–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernecker, C.; Lima, M.A.R.B.F.; Ciubotaru, C.D.; Schlenke, P.; Dorn, I.; Cojoc, D. Biomechanics of Ex Vivo-Generated Red Blood Cells Investigated by Optical Tweezers and Digital Holographic Microscopy. Cells 2021, 10, 552. [Google Scholar] [CrossRef]
- Do RBF Lima, M.A.; Cojoc, D. Monitoring Human Neutrophil Differentiation by Digital Holographic Microscopy. Front. Phys. 2021, 9, 653353. [Google Scholar]
- Picazo-Bueno, J.Á.; Cojoc, D.; Iseppon, F.; Torre, V.; Micó, V. Single-Shot, Dual-Mode, Water-Immersion Microscopy Platform for Biological Applications. Appl. Opt. AO 2018, 57, A242–A249. [Google Scholar] [CrossRef] [PubMed]
- Lévesque, S.A.; Mugnes, J.-M.; Bélanger, E.; Marquet, P. Sample and Substrate Preparation for Exploring Living Neurons in Culture with Quantitative-Phase Imaging. Methods 2018, 136, 90–107. [Google Scholar] [CrossRef]
- Yang, S.; Emiliani, V.; Tang, C.-M. The Kinetics of Multibranch Integration on the Dendritic Arbor of CA1 Pyramidal Neurons. Front. Cell. Neurosci. 2014, 8, 127. [Google Scholar] [CrossRef] [Green Version]
- Cairns, N.J.; Lee, V.M.-Y.; Trojanowski, J.Q. The Cytoskeleton in Neurodegenerative Diseases. J. Pathol. 2004, 204, 438–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotte, Y.; Toy, F.; Jourdain, P.; Pavillon, N.; Boss, D.; Magistretti, P.; Marquet, P.; Depeursinge, C. Marker-Free Phase Nanoscopy. Nat. Photon. 2013, 7, 113–117. [Google Scholar] [CrossRef]
- Binnig, G.; Quate, C.F.; Gerber, C. Atomic Force Microscope. Phys. Rev. Lett. 1986, 56, 930–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, A.; Sakai, N.; Uekusa, Y.; Imaoka, Y.; Itagaki, Y.; Suzuki, Y.; Yoshimura, S.H. Morphological Changes of Plasma Membrane and Protein Assembly during Clathrin-Mediated Endocytosis. PLoS Biol. 2018, 16, e2004786. [Google Scholar] [CrossRef] [Green Version]
- Seifert, J.; Rheinlaender, J.; Novak, P.; Korchev, Y.E.; Schäffer, T.E. Comparison of Atomic Force Microscopy and Scanning Ion Conductance Microscopy for Live Cell Imaging. Langmuir 2015, 31, 6807–6813. [Google Scholar] [CrossRef]
- Hansma, P.K.; Drake, B.; Marti, O.; Gould, S.A.; Prater, C.B. The Scanning Ion-Conductance Microscope. Science 1989, 243, 641–643. [Google Scholar] [CrossRef]
- Shevchuk, A.I.; Frolenkov, G.I.; Sánchez, D.; James, P.S.; Freedman, N.; Lab, M.J.; Jones, R.; Klenerman, D.; Korchev, Y.E. Imaging Proteins in Membranes of Living Cells by High-Resolution Scanning Ion Conductance Microscopy. Angew. Chem. Int. Ed. 2006, 45, 2212–2216. [Google Scholar] [CrossRef]
- Korchev, Y.E.; Bashford, C.L.; Milovanovic, M.; Vodyanoy, I.; Lab, M.J. Scanning Ion Conductance Microscopy of Living Cells. Biophys. J. 1997, 73, 653–658. [Google Scholar] [CrossRef] [Green Version]
- Simeonov, S.; Schäffer, T.E. High-Speed Scanning Ion Conductance Microscopy for Sub-Second Topography Imaging of Live Cells. Nanoscale 2019, 11, 8579–8587. [Google Scholar] [CrossRef]
- Gorelik, J.; Shevchuk, A.I.; Frolenkov, G.I.; Diakonov, I.A.; Lab, M.J.; Kros, C.J.; Richardson, G.P.; Vodyanoy, I.; Edwards, C.R.W.; Klenerman, D.; et al. Dynamic Assembly of Surface Structures in Living Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 5819–5822. [Google Scholar] [CrossRef] [Green Version]
- Böcker, M.; Muschter, S.; Schmitt, E.K.; Steinem, C.; Schäffer, T.E. Imaging and Patterning of Pore-Suspending Membranes with Scanning Ion Conductance Microscopy. Langmuir 2009, 25, 3022–3028. [Google Scholar] [CrossRef]
- Novak, P.; Li, C.; Shevchuk, A.I.; Stepanyan, R.; Caldwell, M.; Hughes, S.; Smart, T.G.; Gorelik, J.; Ostanin, V.P.; Lab, M.J.; et al. Nanoscale Live-Cell Imaging Using Hopping Probe Ion Conductance Microscopy. Nat. Methods 2009, 6, 279–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y.; Zhou, Y.; Miyamoto, T.; Higashi, H.; Nakamichi, N.; Takeda, Y.; Kato, Y.; Korchev, Y.; Fukuma, T. High-Speed SICM for the Visualization of Nanoscale Dynamic Structural Changes in Hippocampal Neurons. Anal. Chem. 2020, 92, 2159–2167. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, M.; Orsini, P.; Pellegrini, M.; Baschieri, P.; Dinelli, F.; Petracchi, D.; Tognoni, E.; Ascoli, C. Weak Hydrostatic Forces in Far-Scanning Ion Conductance Microscopy Used to Guide Neuronal Growth Cones. Neurosci. Res. 2011, 69, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Leitao, S.M.; Drake, B.; Pinjusic, K.; Pierrat, X.; Navikas, V.; Nievergelt, A.P.; Brillard, C.; Djekic, D.; Radenovic, A.; Persat, A.; et al. Time-Resolved Scanning Ion Conductance Microscopy for Three-Dimensional Tracking of Nanoscale Cell Surface Dynamics. ACS Nano 2021, 15, 17613–17622. [Google Scholar] [CrossRef] [PubMed]
- Navikas, V.; Leitao, S.M.; Grussmayer, K.S.; Descloux, A.; Drake, B.; Yserentant, K.; Werther, P.; Herten, D.-P.; Wombacher, R.; Radenovic, A.; et al. Correlative 3D Microscopy of Single Cells Using Super-Resolution and Scanning Ion-Conductance Microscopy. Nat. Commun. 2021, 12, 4565. [Google Scholar] [CrossRef]
- Mahecic, D.; Stepp, W.L.; Zhang, C.; Griffié, J.; Weigert, M.; Manley, S. Event-Driven Acquisition for Content-Enriched Microscopy. Nat. Methods 2022, 19, 1262–1267. [Google Scholar] [CrossRef]
- Mahecic, D.; Carlini, L.; Kleele, T.; Colom, A.; Goujon, A.; Matile, S.; Roux, A.; Manley, S. Mitochondrial Membrane Tension Governs Fission. Cell Rep. 2021, 35, 108947. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
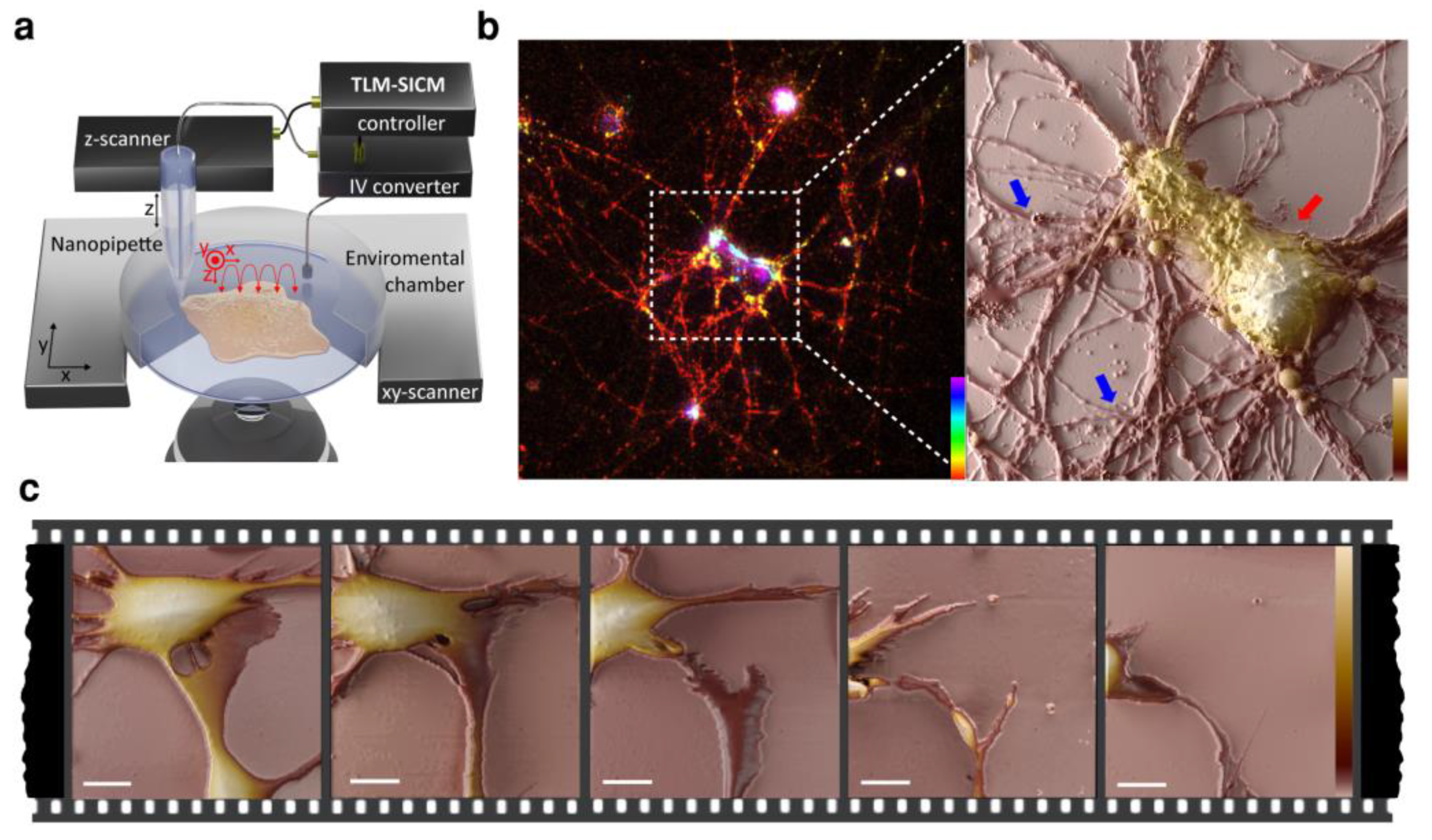{kind=link}
| Imaging Technique | Resolution * (Lateral; Axial) | Acquisition Frequency * (fps) | Advantages | Limitations | References |
|---|---|---|---|---|---|
| Label-free | |||||
| Transmitted light microscopy (BF, PhC, DIC) | ~200 nm; 400–700 | 1- over 200 | Ease of use Non-invasive Long-term recordings (hours, days) | Low contrast Low resolution Phototoxicity (lower than fluorescence-base methods) | [92,93,94] |
| DHM | ~260 nm; ~160–320 nm (90 nm; 150 nm with 2п-DHM) | 50–160 | Fast Non-invasive (low-light level of illumination intensity, ~200 μW/cm2) High axial sensitivity allows visualization of <10 nm structures Volumetric cell analysis | Sensitive to various sources of experimental noise | [107,108,109,112,115] |
| AFM | <10 nm | 0.1–10 | High resolution surface topography imaging Photobleaching- and phototoxicity-free | Resolution is dependent on the AFM tip Mechanical force induction | [116,118] |
| SICM | 180 nm; <5 nm | 2–4 | High resolution surface topography imaging Live cell imaging in physiological conditions Photobleaching- and phototoxicity-free | Resolution is dependent on pipette Lower imaging speed compared to other methods | [118,128] |
| Label-based | |||||
| Fluorescence Microscopy | 180 nm; 400 nm (widefield) ,~30 nm (superresolution imaging of live cells) | 1- over 200 | High signal-to-background ratio Molecular tracking In-cell imaging | Phototoxicity (several minutes at ~1 kW/cm2 light intensity) Photobleaching (depending on the fluorophore used) Interference of tags with cell physiology | [21,22,91,92,94] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baričević, Z.; Ayar, Z.; Leitao, S.M.; Mladinic, M.; Fantner, G.E.; Ban, J. Label-Free Long-Term Methods for Live Cell Imaging of Neurons: New Opportunities. Biosensors 2023, 13, 404. https://doi.org/10.3390/bios13030404
Baričević Z, Ayar Z, Leitao SM, Mladinic M, Fantner GE, Ban J. Label-Free Long-Term Methods for Live Cell Imaging of Neurons: New Opportunities. Biosensors. 2023; 13(3):404. https://doi.org/10.3390/bios13030404
Chicago/Turabian StyleBaričević, Zrinko, Zahra Ayar, Samuel M. Leitao, Miranda Mladinic, Georg E. Fantner, and Jelena Ban. 2023. "Label-Free Long-Term Methods for Live Cell Imaging of Neurons: New Opportunities" Biosensors 13, no. 3: 404. https://doi.org/10.3390/bios13030404