Universal Glia to Neurone Lactate Transfer in the Nervous System: Physiological Functions and Pathological Consequences

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Techniques
2.1. Dialysis
2.2. Biosensors
2.3. FRET
2.4. Spectroscopy
3. Lactate in the Extracellular Fluid
3.1. Lactate Efflux from Astrocytes
3.2. Brain Glycogen
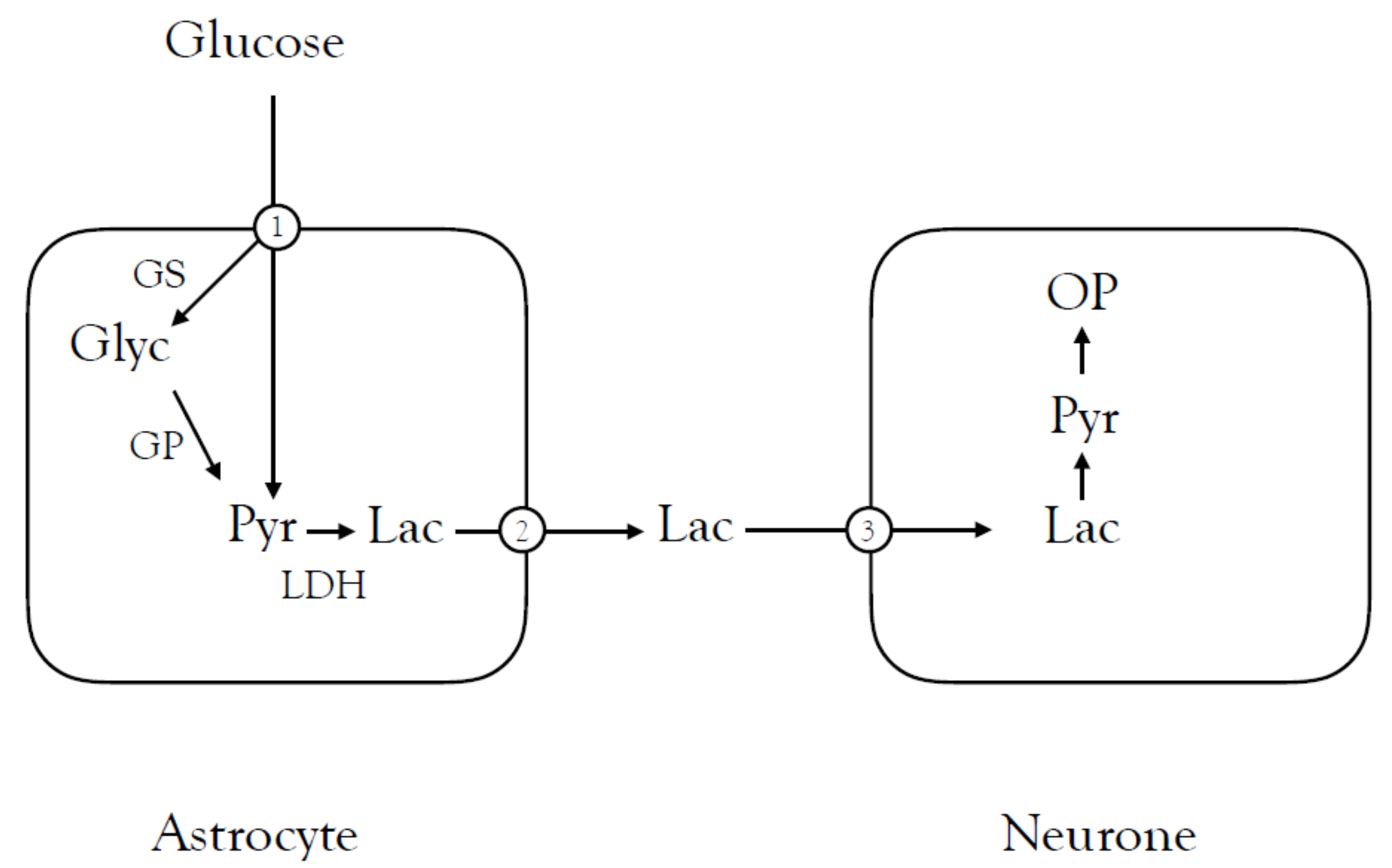
3.3. Lactate Shuttling between Astrocytes and Neurones
3.4. Lactate Shuttling in the Optic Nerve
4. Physiological Roles for Lactate Shutting
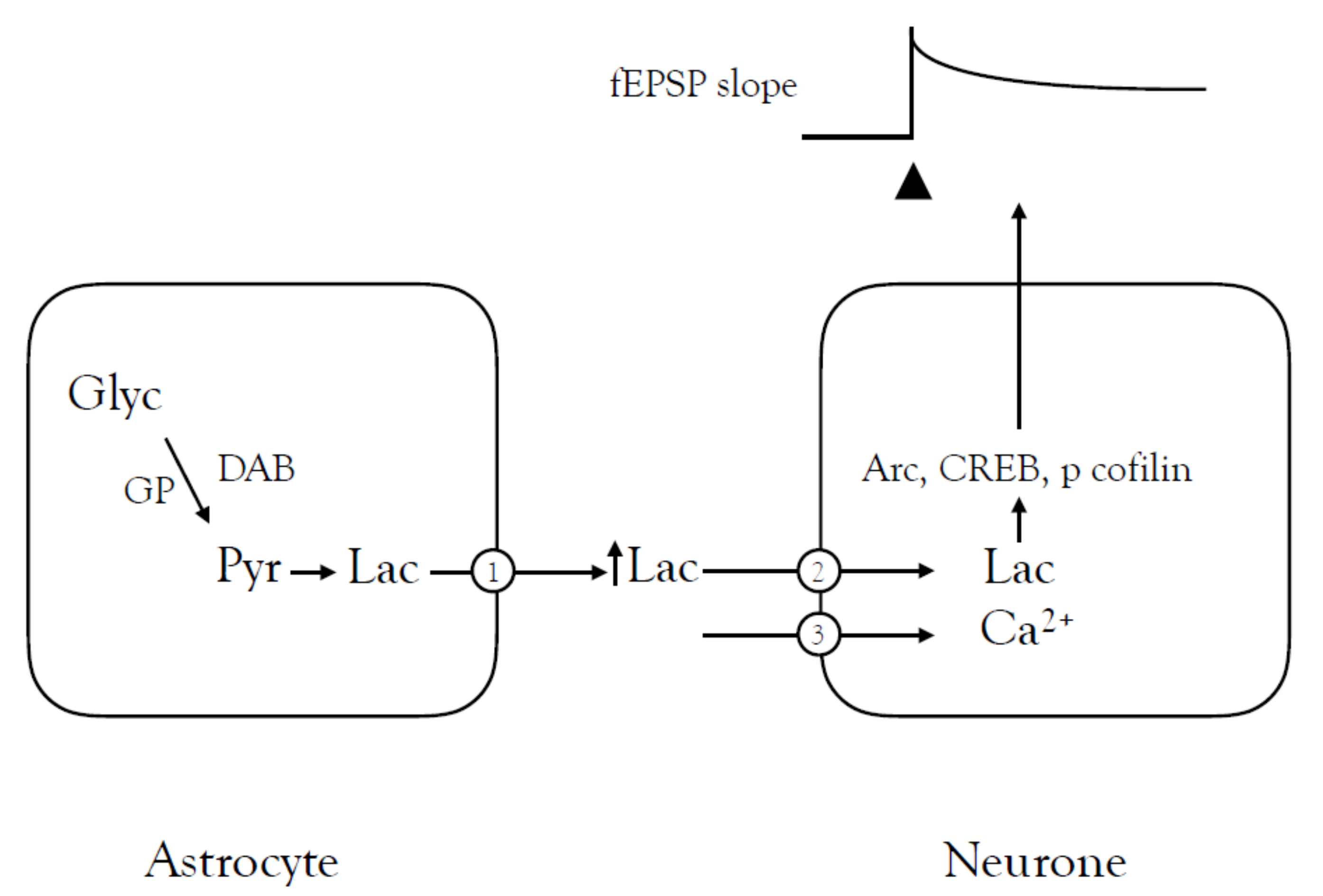
4.1. Learning and Memory
4.2. Sleep
4.3. Decision Making
5. Pathological Conditions
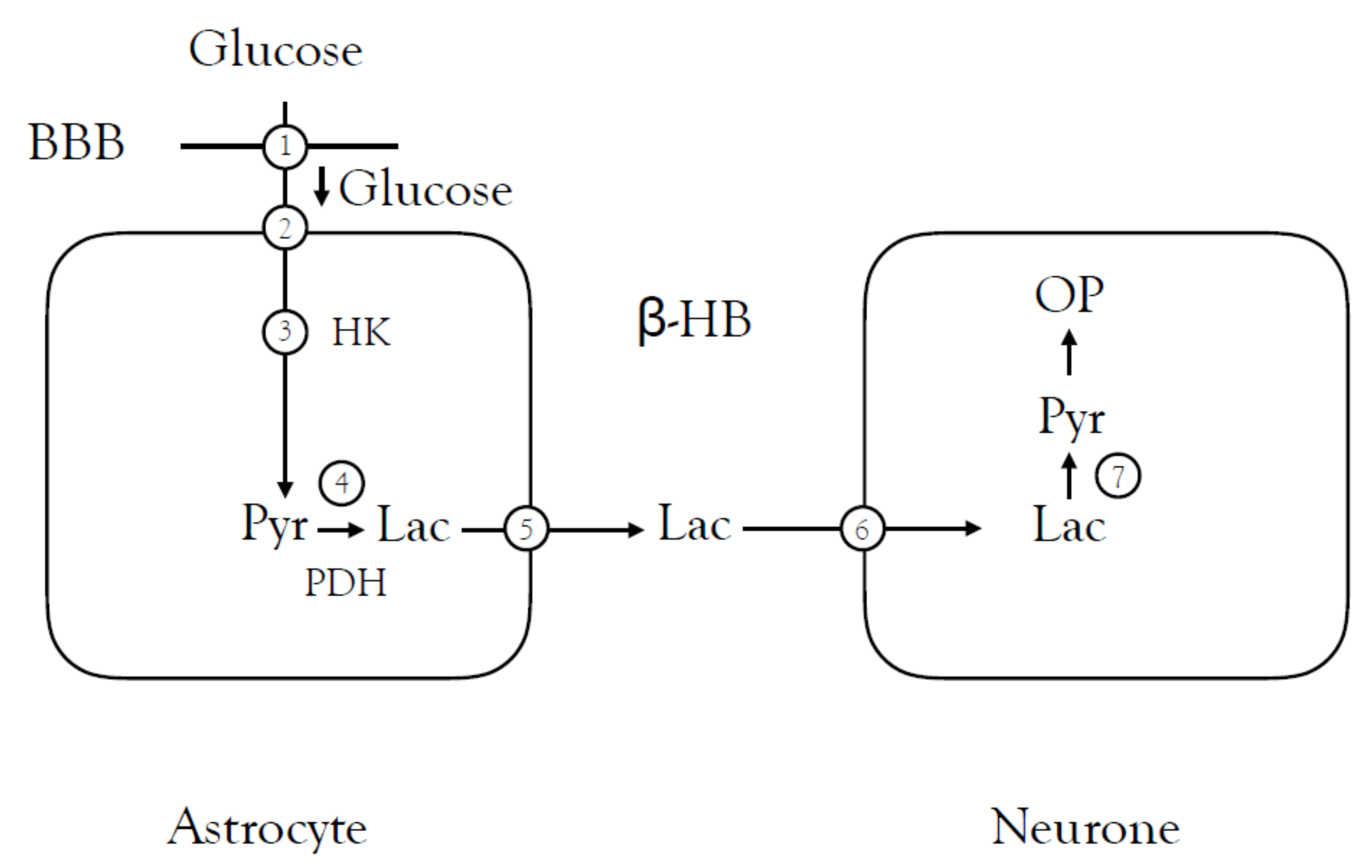
5.1. Alzheimer’s Disease (AD)
5.2. Amyotrophic Lateral Sclerosis (ALS)
5.3. Depression
5.4. Schizophrenia
5.5. Stress
6. Regeneration
6.1. CNS Regeneration
6.2. PNS Regeneration
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dienel, G.A. Energy Metabolism in the Brain, In From Molecules to Networks: An Introduction to Cellular and Molecular Neuroscience, 2nd ed.; Byrne, J.H., Roberts, J.L., Eds.; Academic Press: Amsterdam, The Netherlands, 2009. [Google Scholar]
- Stryer, L. Biochemistry, 4th ed.; W.H. Freeman & Co.: New York, NY, USA, 1995. [Google Scholar]
- Frier, B.M.; Heller, S.R.; McCrimmon, R.J. Hypoglycaemia in Clinical Diabetes, 3rd ed.; Wiley and Sons: Chichester, UK, 2014. [Google Scholar]
- Cryer, P.E. Hypoglycemia: The Limiting Factor in the Management of IDDM. Diabetes 1994, 43, 1378–1389. [Google Scholar] [CrossRef] [PubMed]
- Sokoloff, L.; Reivich, M.; Kennedy, C.; Rosiers, M.H.D.; Patlak, C.S.; Pettigrew, K.D.; Sakurada, O.; Shinohara, M. The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: Theory, procedure, and normal values in the conscious and anesthetized albino rat. J. Neurochem. 1977, 28, 897–916. [Google Scholar] [CrossRef] [PubMed]
- Ames, A. CNS energy metabolism as related to function. Brain Res. Rev. 2000, 34, 42–68. [Google Scholar] [CrossRef]
- Aubert, A.; Costalat, R.; Magistretti, P.J.; Pellerin, L. Brain lactate kinetics: Modeling evidence for neuronal lactate uptake upon activation. Proc. Natl. Acad. Sci. USA 2005, 102, 16448–16453. [Google Scholar] [CrossRef] [Green Version]
- DiNuzzo, M.; Mangia, S.; Maraviglia, B.; Giove, F. Glycogenolysis in Astrocytes Supports Blood-Borne Glucose Channeling Not Glycogen-Derived Lactate Shuttling to Neurons: Evidence from Mathematical Modeling. Br. J. Pharmacol. 2010, 30, 1895–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robergs, R.A.; McNulty, C.R.; Minett, G.M.; Holland, J.; Trajano, G. Lactate, not Lactic Acid, is Produced by Cellular Cytosolic Energy Catabolism. Physiology 2018, 33, 10–12. [Google Scholar] [CrossRef]
- McNay, E.C.; Gold, P.E. Extracellular glucose concentrations in the rat hippocampus measured by zero-net-flux: Effects of microdialysis flow rate, strain, and age. J. Neurochem. 1999, 72, 785–790. [Google Scholar] [CrossRef]
- Dale, N.; Hatz, S.; Tian, F.; Llaudet, E. Listening to the brain: Microelectrode biosensors for neurochemicals. Trends Biotechnol. 2005, 23, 420–428. [Google Scholar] [CrossRef]
- Fillenz, M.; Lowry, J.P. Studies of the Source of Glucose in the Extracellular Compartment of the Rat Brain. Dev. Neurosci. 1998, 20, 365–368. [Google Scholar] [CrossRef]
- Netchiporouk, L.; Shram, N.; Salvert, D.; Cespuglio, R. Brain extracellular glucose assessed by voltammetry throughout the rat sleep-wake cycle. Eur. J. Neurosci. 2001, 13, 1429–1434. [Google Scholar] [CrossRef]
- Gramsbergen, J.B.; Leegsma-Vogt, G.; Venema, K.; Noraberg, J.; Korf, J. Quantitative on-line monitoring of hippocampus glucose and lactate metabolism in organotypic cultures using biosensor technology. J. Neurochem. 2003, 85, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frenguelli, B.G.; Wigmore, G.; Llaudet, E.; Dale, N. Temporal and mechanistic dissociation of ATP and adenosine release during ischaemia in the mammalian hippocampus. J. Neurochem. 2007, 101, 1400–1413. [Google Scholar] [CrossRef] [PubMed]
- Martín, A.S.; Ceballo, S.; Ruminot, I.; Lerchundi, R.; Frommer, W.B.; Barros, L.F. A Genetically Encoded FRET Lactate Sensor and Its Use To Detect the Warburg Effect in Single Cancer Cells. PLoS ONE 2013, 8, e57712. [Google Scholar] [CrossRef] [PubMed]
- Barros, L.F.; Martín, A.S.; Sotelo-Hitschfeld, T.; Lerchundi, R.; Fernández-Moncada, I.; Ruminot, I.; Gutiérrez, R.; Valdebenito, R.; Ceballo, S.; Alegria, K.; et al. Small is fast: Astrocytic glucose and lactate metabolism at cellular resolution. Front. Cell. Neurosci. 2013, 7, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotelo-Hitschfeld, T.; Niemeyer, M.I.; Mächler, P.; Ruminot, I.; Lerchundi, R.; Wyss, M.T.; Stobart, J.; Fernández-Moncada, I.; Valdebenito, R.; Garrido-Gerter, P.; et al. Channel-Mediated Lactate Release by K+-Stimulated Astrocytes. J. Neurosci. 2015, 35, 4168–4178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruminot, I.; Schmälzle, J.; Leyton, B.; Barros, L.F.; Deitmer, J.W. Tight coupling of astrocyte energy metabolism to synaptic activity revealed by genetically encoded FRET nanosensors in hippocampal tissue. Br. J. Pharmacol. 2017, 39, 513–523. [Google Scholar] [CrossRef]
- Taylor, A.; McLean, M.; Morris, P.; Bachelard, H. Approaches to Studies on Neuronal/Glial Relationships by 13C-MRS Analysis. Dev. Neurosci. 1996, 18, 434–442. [Google Scholar] [CrossRef]
- Gruetter, R.; Adriany, G.; Merkle, H.; Andersen, P.M. Broadband decoupled, 1H-localized 13C MRS of the human brain at 4 tesla. Magn. Reson. Med. 1996, 36, 659–664. [Google Scholar] [CrossRef] [Green Version]
- Bachelard, H. Landmarks in the Application of 13C-Magnetic Resonance Spectroscopy to Studies of Neuronal/Glial Relationships. Dev. Neurosci. 1998, 20, 277–288. [Google Scholar] [CrossRef]
- Morris, P.; Bachelard, H. Reflections on the application of 13C-MRS to research on brain metabolism. NMR Biomed. 2003, 16, 303–312. [Google Scholar] [CrossRef]
- Hu, Y.; Wilson, G.S. A Temporary Local Energy Pool Coupled to Neuronal Activity: Fluctuations of Extracellular Lactate Levels in Rat Brain Monitored with Rapid-Response Enzyme-Based Sensor. J. Neurochem. 1997, 69, 1484–1490. [Google Scholar] [CrossRef] [PubMed]
- Maggs, D.G.; Borg, W.P.; Sherwin, R.S. Microdialysis techniques in the study of brain and skeletal muscle. Diabetologia 1997, 40, S75–S82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalsgaard, M.K. Fuelling Cerebral Activity in Exercising Man. Br. J. Pharmacol. 2005, 26, 731–750. [Google Scholar] [CrossRef] [PubMed]
- Dalsgaard, M.K.; Quistorff, B.; Danielsen, E.R.; Selmer, C.; Vogelsang, T.; Secher, N.H. A reduced cerebral metabolic ratio in exercise reflects metabolism and not accumulation of lactate within the human brain. J. Physiol. 2004, 554, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A. Lactate muscles its way into consciousness: Fueling brain activation. Am. J. Physiol. Integr. Comp. Physiol. 2004, 287, R519–R521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienel, G.A.; Cruz, N.F. Nutrition during brain activation: Does cell-to-cell lactate shuttling contribute significantly to sweet and sour food for thought? Neurochem. Int. 2004, 45, 321–351. [Google Scholar] [CrossRef]
- Dienel, G.A.; Hertz, L. Glucose and lactate metabolism during brain activation. J. Neurosci. Res. 2001, 66, 824–838. [Google Scholar] [CrossRef]
- Chih, C.-P.; Roberts, E.L., Jr. Energy Substrates for Neurons during Neural Activity: A Critical Review of the Astrocyte-Neuron Lactate Shuttle Hypothesis. Br. J. Pharmacol. 2003, 23, 1263–1281. [Google Scholar] [CrossRef] [Green Version]
- Gladden, L.B. Lactate metabolism: A new paradigm for the third millennium. J. Physiol. 2004, 558, 5–30. [Google Scholar] [CrossRef]
- Mächler, P.; Wyss, M.T.; Elsayed, M.; Stobart, J.; Gutierrez, R.; Von Faber-Castell, A.; Kaelin, V.; Zuend, M.; Martín, A.S.; Romero-Gómez, I.; et al. In Vivo Evidence for a Lactate Gradient from Astrocytes to Neurons. Cell Metab. 2016, 23, 94–102. [Google Scholar] [CrossRef] [Green Version]
- Walz, W.; Mukerji, S. Lactate production and release in cultured astrocytes. Neurosci. Lett. 1988, 86, 296–300. [Google Scholar] [CrossRef]
- Walz, W.; Mukerji, S. Lactate release from cultured astrocytes and neurons: A comparison. Glia 1988, 1, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Chih, C.-P.; Lipton, P.; Roberts, E.L. Do active cerebral neurons really use lactate rather than glucose? Trends Neurosci. 2001, 24, 573–578. [Google Scholar] [CrossRef]
- Whatley, S.A.; Hall, C.; Lim, L. Hypothalamic Neurons in Dissociated Cell Culture: The Mechanism of Increased Survival Times in the Presence of Non-Neuronal Cells. J. Neurochem. 1981, 36, 2052–2056. [Google Scholar] [CrossRef]
- Swanson, R.A.; Choi, D.W. Glial Glycogen Stores Affect Neuronal Survival during Glucose Deprivation in vitro. Br. J. Pharmacol. 1993, 13, 162–169. [Google Scholar] [CrossRef] [Green Version]
- Champe, P.C.; Harvey, R.A. Biochemistry, 4th ed.; Lippincott Williams & Wilkins: Baltimore, MD, USA, 2008. [Google Scholar]
- Roach, P.J. Glycogen and its Metabolism. Curr. Mol. Med. 2002, 2, 101–120. [Google Scholar] [CrossRef]
- Messonnier, L.A.; Emhoff, C.-A.W.; Fattor, J.A.; Horning, M.A.; Carlson, T.J.; Brooks, G.A. Lactate kinetics at the lactate threshold in trained and untrained men. J. Appl. Physiol. 2013, 114, 1593–1602. [Google Scholar] [CrossRef]
- Brown, A.M. Brain glycogen re-awakened. J. Neurochem. 2004, 89, 537–552. [Google Scholar] [CrossRef]
- Tsacopoulos, M.; Veuthey, A.; Saravelos, S.; Perrottet, P.; Tsoupras, G. Glial cells transform glucose to alanine, which fuels the neurons in the honeybee retina. J. Neurosci. 1994, 14, 1339–1351. [Google Scholar] [CrossRef]
- Navarrete, M.; Araque, A. The Cajal school and the physiological role of astrocytes: A way of thinking. Front. Neuroanat. 2014, 8, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucci, S.J.; Maher, F.; Simpson, I.A. Glucose transporter proteins in brain: Delivery of glucose to neurons and glia. Glia 1997, 21, 2–21. [Google Scholar] [CrossRef]
- Gordon, G.R.J.; Choi, H.B.; Rungta, R.L.; Ellis-Davies, G.C.R.; MacVicar, B.A. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nat. Cell Biol. 2008, 456, 745–749. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P. The monocarboxylate transporter family-Structure and functional characterization. IUBMB Life 2012, 64, 1–9. [Google Scholar] [CrossRef]
- Stys, P.K.; Ransom, B.R.; Waxman, S.G. Compound action potential of nerve recorded by suction electrode: A theoretical and experimental analysis. Brain Res. 1991, 546, 18–32. [Google Scholar] [CrossRef]
- Brown, A.M.; Evans, R.D.; Black, J.; Ransom, B.R. Schwann cell glycogen selectively supports myelinated axon function. Ann. Neurol. 2012, 72, 406–418. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.M.; Tekkök, S.B.; Ransom, B.R. Glycogen Regulation and Functional Role in Mouse White Matter. J. Physiol. 2003, 549, 501–512. [Google Scholar] [CrossRef]
- Wender, R.; Brown, A.M.; Fern, R.; Swanson, R.A.; Farrell, K.; Ransom, B.R. Astrocytic Glycogen Influences Axon Function and Survival during Glucose Deprivation in Central White Matter. J. Neurosci. 2000, 20, 6804–6810. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.B.; Sickmann, H.M.; Brown, A.; Bouman, S.D.; Ransom, B.; Schousboe, A.; Waagepetersen, H.S. Characterization of 1,4-dideoxy-1,4-imino-d-arabinitol (DAB) as an inhibitor of brain glycogen shunt activity. J. Neurochem. 2008, 105, 1462–1470. [Google Scholar] [CrossRef]
- Brown, A.M.; Sickmann, H.M.; Fosgerau, K.; Lund, T.M.; Schousboe, A.; Waagepetersen, H.S.; Ransom, B.R. Astrocyte glycogen metabolism is required for neural activity during aglycemia or intense stimulation in mouse white matter. J. Neurosci. Res. 2004, 79, 74–80. [Google Scholar] [CrossRef]
- Tekkök, S.B.; Brown, A.M.; Westenbroek, R.; Pellerin, L.; Ransom, B.R. Transfer of glycogen-derived lactate from astrocytes to axons via specific monocarboxylate transporters supports mouse optic nerve activity. J. Neurosci. Res. 2005, 81, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Hamner, M.A.; Brown, A.M.; Evans, R.D.; Ye, Z.-C.; Chen, S.; Ransom, B.R. Novel hypoglycemic injury mechanism: N-methyl-D-aspartate receptor-mediated white matter damage. Ann. Neurol. 2014, 75, 492–507. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-Neuron Lactate Transport Is Required for Long-Term Memory Formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Descalzi, G.; Gao, V.; Steinman, M.Q.; Suzuki, A.; Alberini, C.M. Lactate from astrocytes fuels learning-induced mRNA translation in excitatory and inhibitory neurons. Commun. Biol. 2019, 2, 1–11. [Google Scholar] [CrossRef]
- Harris, R.A.; Lone, A.; Lim, H.; Martinez, F.; Frame, A.K.; Scholl, T.J.; Cumming, R.C. Aerobic Glycolysis Is Required for Spatial Memory Acquisition But Not Memory Retrieval in Mice. Eneuro 2019, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Netzahualcoyotzi, C.; Pellerin, L. Neuronal and astroglial monocarboxylate transporters play key but distinct roles in hippocampus-dependent learning and memory formation. Prog. Neurobiol. 2020, 194, 101888. [Google Scholar] [CrossRef]
- Yang, J.; Ruchti, E.; Petit, J.-M.; Jourdain, P.; Grenningloh, G.; Allaman, I.; Magistretti, P.J. Lactate promotes plasticity gene expression by potentiating NMDA signaling in neurons. Proc. Natl. Acad. Sci. USA 2014, 111, 12228–12233. [Google Scholar] [CrossRef] [Green Version]
- Vezzoli, E.; Calì, C.; De Roo, M.; Ponzoni, L.; Sogne, E.; Gagnon, N.; Francolini, M.; Braida, D.; Sala, M.; Muller, D.; et al. Ultrastructural Evidence for a Role of Astrocytes and Glycogen-Derived Lactate in Learning-Dependent Synaptic Stabilization. Cereb. Cortex 2020, 30, 2114–2127. [Google Scholar] [CrossRef]
- Bingul, D.; Kalra, K.; Murata, E.; Belser, A.; Dash, M. Persistent changes in extracellular lactate dynamics following synaptic potentiation. Neurobiol. Learn. Mem. 2020, 175, 107314. [Google Scholar] [CrossRef]
- Gibbs, M.E.; O’Dowd, B.S.; Hertz, E.; Hertz, L. Astrocytic energy metabolism consolidates memory in young chicks. Neuroscience 2006, 141, 9–13. [Google Scholar] [CrossRef]
- Gibbs, M.E.; Anderson, D.G.; Hertz, L. Inhibition of glycogenolysis in astrocytes interrupts memory consolidation in young chickens. Glia 2006, 54, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.E. Role of Glycogenolysis in Memory and Learning: Regulation by Noradrenaline, Serotonin and ATP. Front. Integr. Neurosci. 2016, 9, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shram, N.; Netchiporouk, L.; Cespuglio, R. Lactate in the brain of the freely moving rat: Voltammetric monitoring of the changes related to the sleep-wake states. Eur. J. Neurosci. 2002, 16, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Scharf, M.T.; Naidoo, N.; Zimmerman, J.E.; Pack, A.I. The energy hypothesis of sleep revisited. Prog. Neurobiol. 2008, 86, 264–280. [Google Scholar] [CrossRef] [Green Version]
- Kilic, K.; Karatas, H.; Dönmez-Demir, B.; Eren-Kocak, E.; Gursoy-Ozdemir, Y.; Can, A.; Petit, J.-M.; Magistretti, P.J.; Dalkara, T. Inadequate brain glycogen or sleep increases spreading depression susceptibility. Ann. Neurol. 2018, 83, 61–73. [Google Scholar] [CrossRef]
- Wang, J.; Tu, J.; Cao, B.; Mu, L.; Yang, X.; Cong, M.; Ramkrishnan, A.S.; Chan, R.H.; Wang, L.; Li, Y. Astrocytic l -Lactate Signaling Facilitates Amygdala-Anterior Cingulate Cortex Synchrony and Decision Making in Rats. Cell Rep. 2017, 21, 2407–2418. [Google Scholar] [CrossRef]
- Jha, M.K.; Lee, Y.; Russell, K.A.; Yang, F.; Dastgheyb, R.M.; Deme, P.; Ament, X.H.; Chen, W.; Liu, Y.; Guan, Y.; et al. Monocarboxylate transporter 1 in Schwann cells contributes to maintenance of sensory nerve myelination during aging. Glia 2019, 68, 161–177. [Google Scholar] [CrossRef] [Green Version]
- Bouçanova, F.; Pollmeier, G.; Sandor, K.; Urbina, C.M.; Nijssen, J.; Médard, J.-J.; Bartesaghi, L.; Pellerin, L.; Svensson, C.I.; Hedlund, E.; et al. Disrupted function of lactate transporter MCT1, but not MCT4, in Schwann cells affects the maintenance of motor end-plate innervation. Glia 2020. [Google Scholar] [CrossRef]
- Ding, F.; Yao, J.; Rettberg, J.R.; Chen, S.; Brinton, R.D. Early Decline in Glucose Transport and Metabolism Precedes Shift to Ketogenic System in Female Aging and Alzheimer’s Mouse Brain: Implication for Bioenergetic Intervention. PLoS ONE 2013, 8, e79977. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Huang, J.; Sun, S.; Huang, S.; Gan, S.; Xu, J.; Yang, M.; Xu, S.; Jiang, X. Changes in lactate content and monocarboxylate transporter 2 expression in Ab25-35-treated rat model of Alzheimer’s disease. Neurol. Sci. 2015, 36, 871–876. [Google Scholar] [CrossRef]
- Zhang, M.; Cheng, X.; Dang, R.; Zhang, W.; Zhang, J.; Yao, Z. Lactate Deficit in an Alzheimer Disease Mouse Model: The Relationship With Neuronal Damage. J. Neuropathol. Exp. Neurol. 2018, 77, 1163–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraiuolo, L.; Higginbottom, A.; Heath, P.R.; Barber, S.; Greenald, D.; Kirby, J.; Shaw, P.J. Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain 2011, 134, 2627–2641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newington, I.T.; Harris, R.A.; Cumming, R.C. Reevaluating Alzheimer’s disease from the perspective of the astrocyte-neuron lactate shuttle model. J. Neurodegener. Dis. 2013, 2013, 234572. [Google Scholar] [CrossRef] [PubMed]
- Vlassenko, A.G.; Vaishnavi, S.N.; Couture, L.; Sacco, D.; Shannon, B.J.; Mach, R.H.; Morris, J.C.; Raichle, M.E.; Mintun, M.A. Spatial correlation between brain aerobic glycolysis and amyloid- aerobic glycolysis and amyloid-deposition. Proc. Natl. Acad. Sci. USA 2010, 107, 17763–17767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liguori, C.; Chiaravalloti, A.; Sancesario, G.; Stefani, A.; Sancesario, G.M.; Mercuri, N.B.; Schillaci, O.; Pierantozzi, M. Cerebrospinal fluid lactate levels and brain [18F]FDG PET hypometabolism within the default mode network in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 2040–2049. [Google Scholar] [CrossRef] [PubMed]
- Carrard, A.; Elsayed, M.; Margineanu, M.B.; Boury-Jamot, B.; Fragnière, L.; Meylan, E.M.; Petit, J.-M.; Fiumelli, H.; Magistretti, P.J.; Martin, J.-L. Peripheral administration of lactate produces antidepressant-like effects. Mol. Psychiatry 2018, 23, 392–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandoorne, T.; De Bock, K.; Van Den Bosch, L. Energy metabolism in ALS: An underappreciated opportunity? Acta Neuropathol. 2018, 135, 489–509. [Google Scholar] [CrossRef] [Green Version]
- Ernst, J.; Hock, A.; Henning, A.; Seifritz, E.; Boeker, H.; Grimm, S. Increased pregenual anterior cingulate glucose and lactate concentrations in major depressive disorder. Mol. Psychiatry 2016, 22, 113–119. [Google Scholar] [CrossRef]
- Detka, J.; Kurek, A.; Kucharczyk, M.; Głombik, K.; Basta-Kaim, A.; Kubera, M.; Laso, W.; Budziszewska, B. Brain glucose metabolism in an animal model of depression. Neuroscience 2015, 295, 198–208. [Google Scholar] [CrossRef]
- Jouroukhin, Y.; Kageyama, Y.; Misheneva, V.; Shevelkin, A.; Andrabi, S.; Prandovszky, E.; Yolken, R.H.; Dawson, V.L.; Dawson, T.M.; Aja, S.; et al. DISC1 regulates lactate metabolism in astrocytes: Implications for psychiatric disorders. Transl. Psychiatry 2018, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, C.R.; Mielnik, C.A.; Funk, A.; O’Donovan, S.M.; Bentea, E.; Pletnikov, M.; Ramsey, A.J.; Wen, Z.; Rowland, L.M.; McCullumsmith, R.E. Measurement of lactate levels in postmortem brain, iPSCs, and animal models of schizophrenia. Sci. Rep. 2019, 9, 5087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy-Royal, C.; Johnston, A.D.; Boyce, A.K.J.; Diaz-Castro, B.; Institoris, A.; Peringod, G.; Zhang, O.; Stout, R.F.; Spray, D.C.; Thompson, R.J.; et al. Stress gates an astrocytic energy reservoir to impair synaptic plasticity. Nat. Commun. 2020, 11, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tertil, M.; Skupio, U.; Barut, J.; Dubovyk, V.; Wawrzczak-Bargiela, A.; Soltys, Z.; Golda, S.; Kudla, L.; Wiktorowska, L.; Szklarczyk, K.; et al. Glucocorticoid receptor signaling in astrocytes is required for aversive memory formation. Transl. Psychiatry 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Dong, J.-H.; Wang, Y.-J.; Cui, M.; Wang, X.-J.; Zheng, W.-S.; Ma, M.-L.; Yang, F.; He, D.-F.; Hu, Q.-X.; Zhang, D.-L.; et al. Adaptive Activation of a Stress Response Pathway Improves Learning and Memory Through Gs and β-Arrestin-1–Regulated Lactate Metabolism. Biol. Psychiatry 2017, 81, 654–670. [Google Scholar] [CrossRef] [PubMed]
- Yiu, G.; He, Z. Glial inhibition of CNS axon regeneration. Nat. Rev. Neurosci. 2006, 7, 617–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Sami, A.; Noristani, H.N.; Slattery, K.; Qiu, J.; Groves, T.; Wang, S.; Veerasammy, K.; Chen, Y.X.; Morales, J.; et al. Glial Metabolic Rewiring Promotes Axon Regeneration and Functional Recovery in the Central Nervous System. Cell Metab. 2020, 32, 767–785.e7. [Google Scholar] [CrossRef]
- Morrison, B.M.; Tsingalia, A.; Vidensky, S.; Lee, Y.; Youngjin, L.; Farah, M.H.; Lengacher, S.; Magistretti, P.J.; Pellerin, L.; Rothsteinb, J.D. Deficiency in monocarboxylate transporter 1 (MCT1) in mice delays regeneration of peripheral nerves following sciatic nerve crush. Exp. Neurol. 2015, 263, 325–338. [Google Scholar] [CrossRef] [Green Version]
- Babetto, E.; Wong, K.M.; Beirowski, B. A glycolytic shift in Schwann cells supports injured axons. Nat. Neurosci. 2020, 23, 1215–1228. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Powell, C.L.; Davidson, A.R.; Brown, A.M. Universal Glia to Neurone Lactate Transfer in the Nervous System: Physiological Functions and Pathological Consequences. Biosensors 2020, 10, 183. https://doi.org/10.3390/bios10110183
Powell CL, Davidson AR, Brown AM. Universal Glia to Neurone Lactate Transfer in the Nervous System: Physiological Functions and Pathological Consequences. Biosensors. 2020; 10(11):183. https://doi.org/10.3390/bios10110183
Chicago/Turabian StylePowell, Carolyn L., Anna R. Davidson, and Angus M. Brown. 2020. "Universal Glia to Neurone Lactate Transfer in the Nervous System: Physiological Functions and Pathological Consequences" Biosensors 10, no. 11: 183. https://doi.org/10.3390/bios10110183