
The Combined Effects of an External Field and Novel Functional Groups on the Structural and Electronic Properties of TMDs/Ti3C2 Heterostructures: A First-Principles Study

Abstract
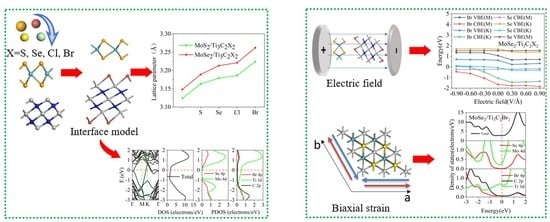
:
1. Introduction
2. Calculation Method
3. Results and Discussions
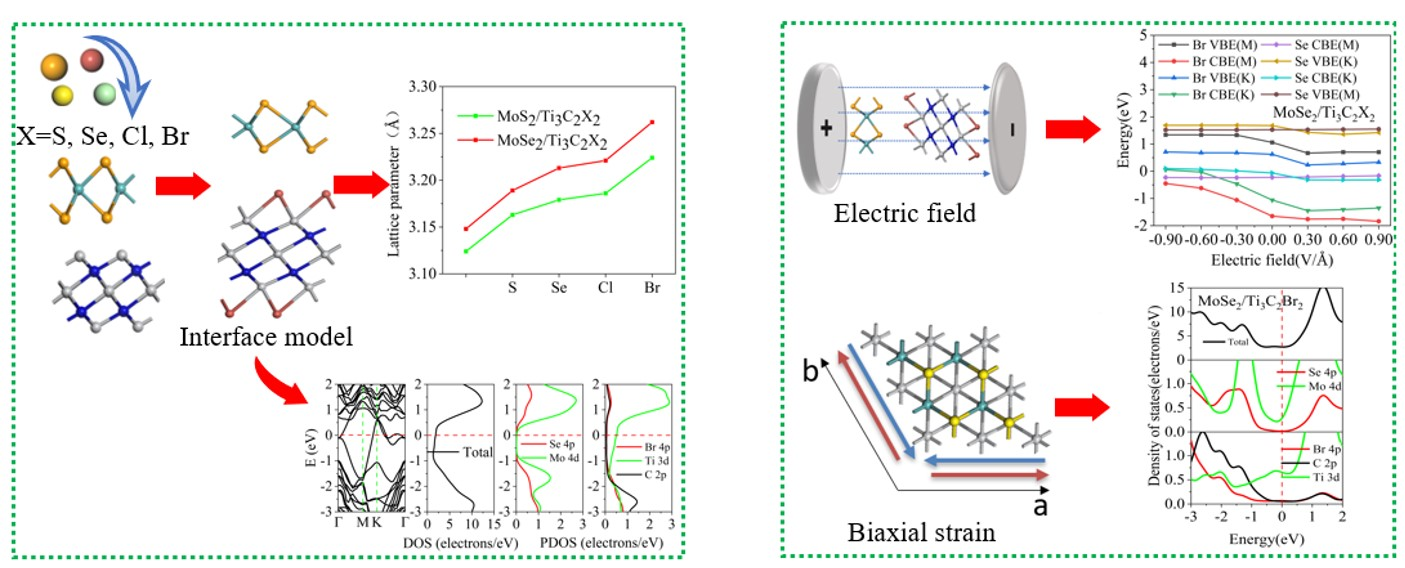
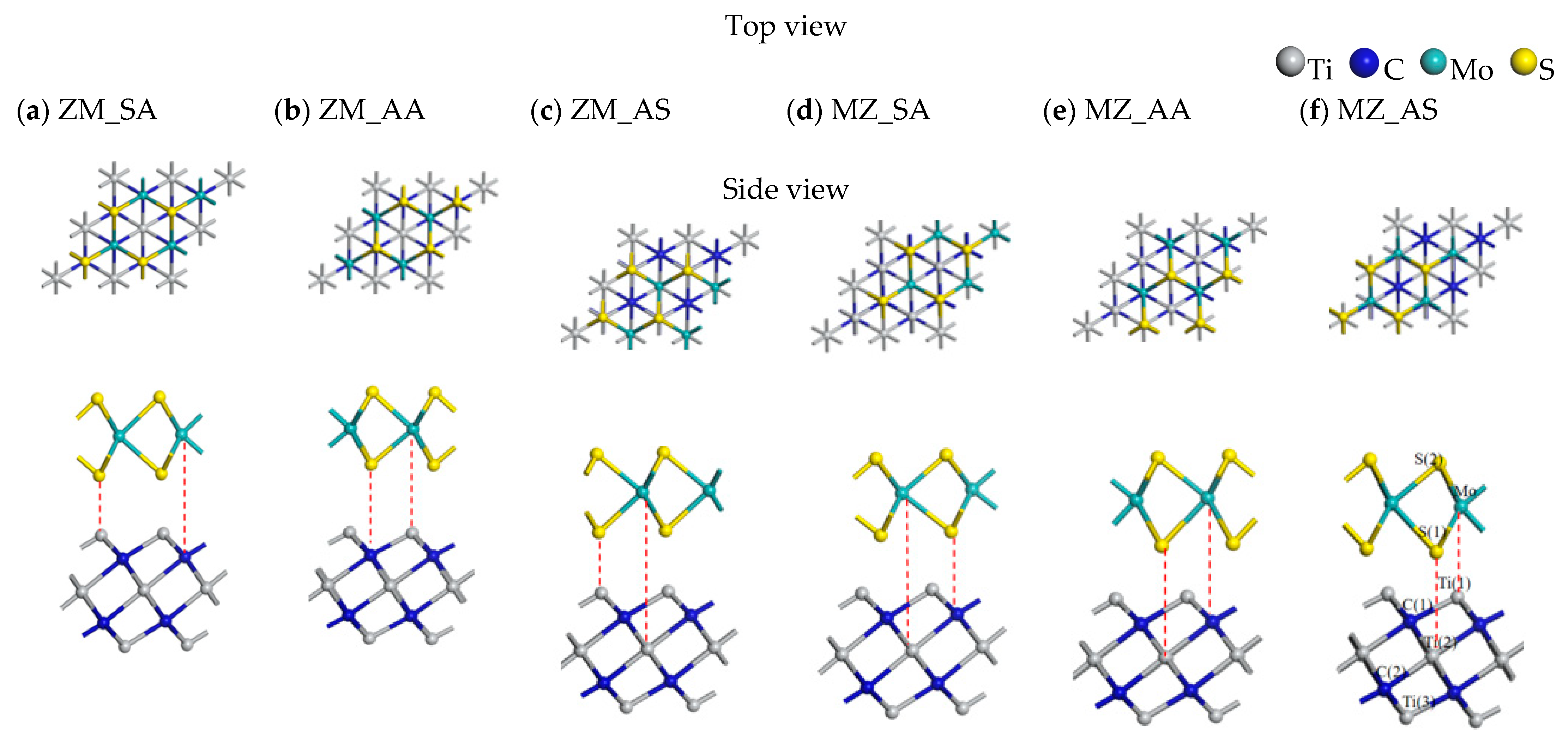
3.1. Structural Properties of the TMDs/Ti3C2 Heterostructures
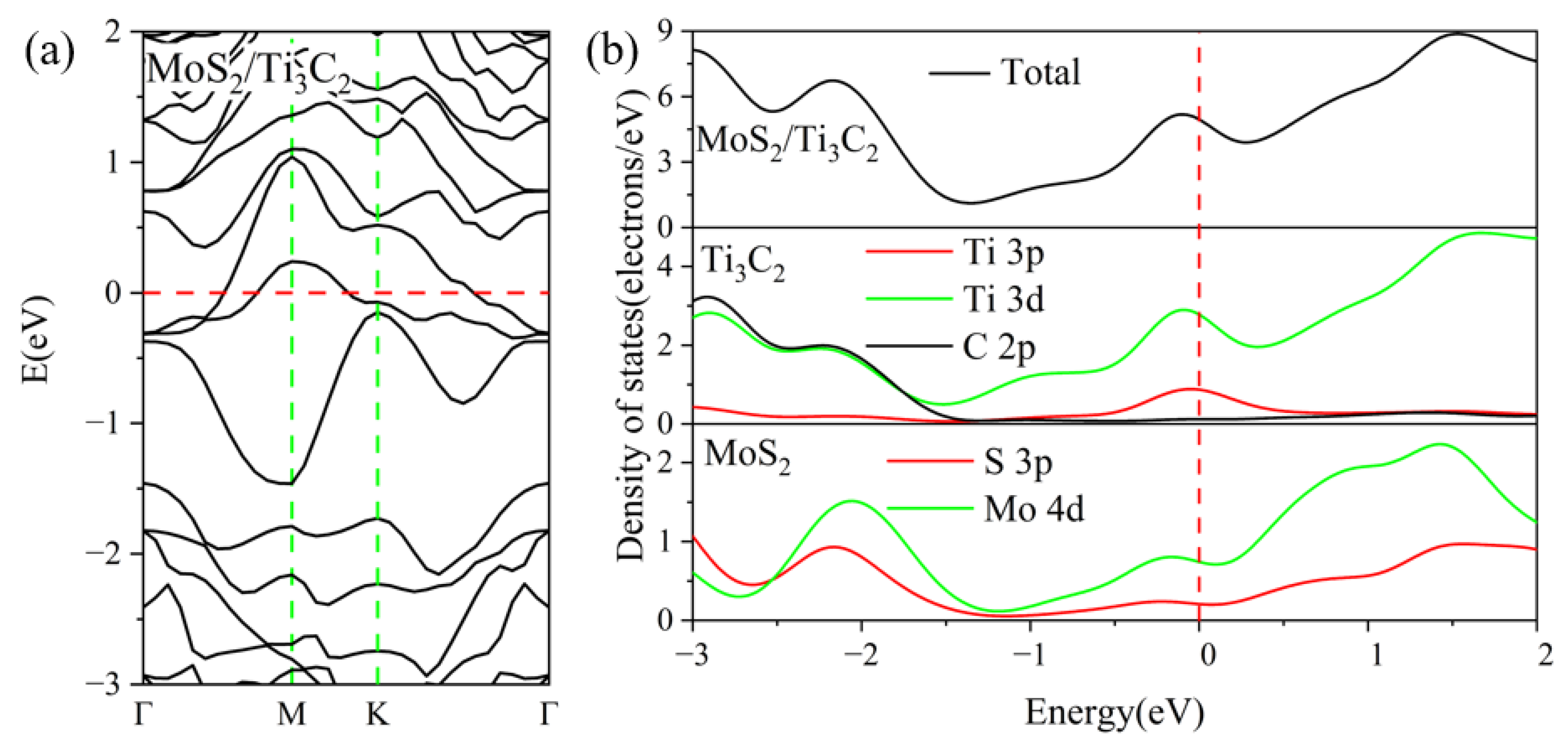
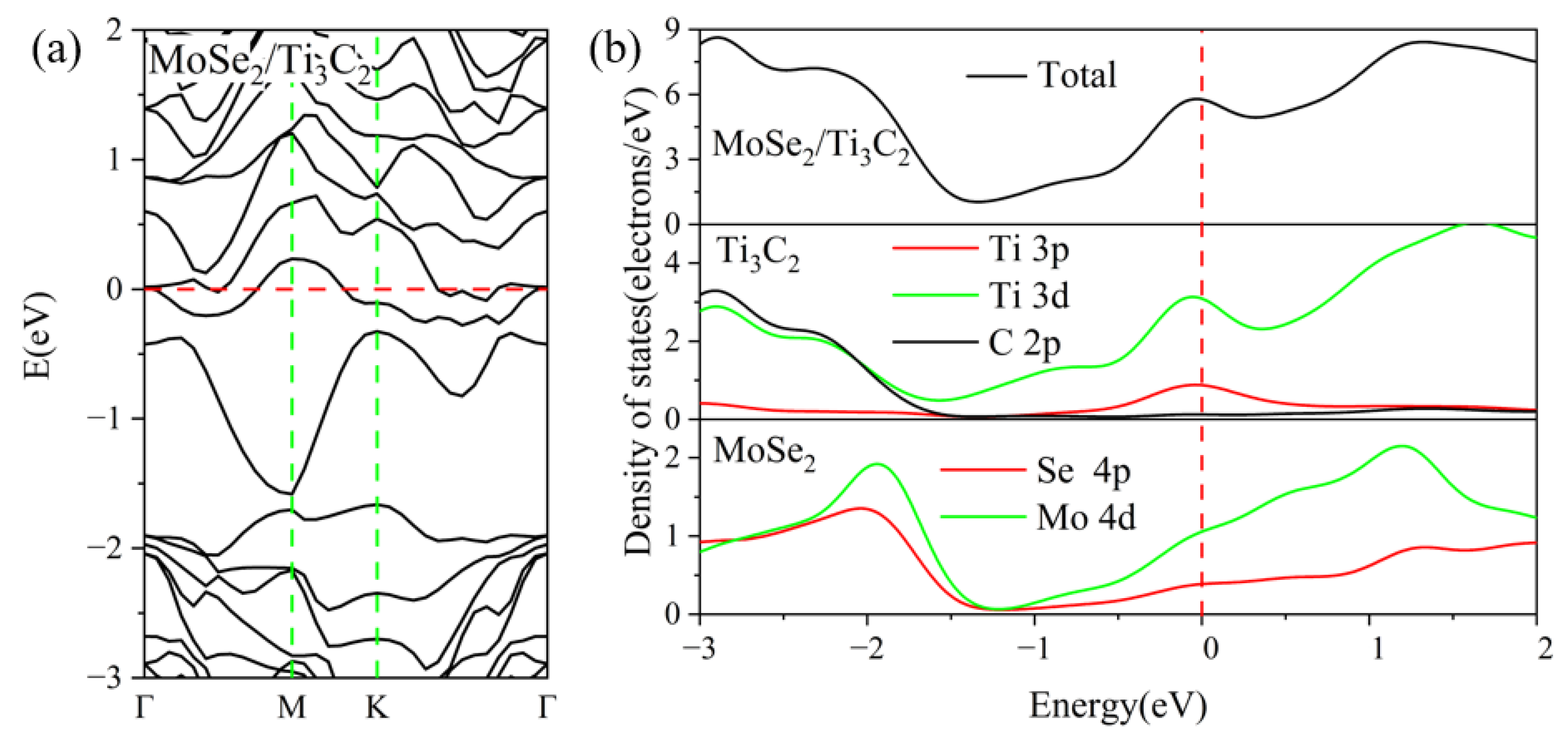
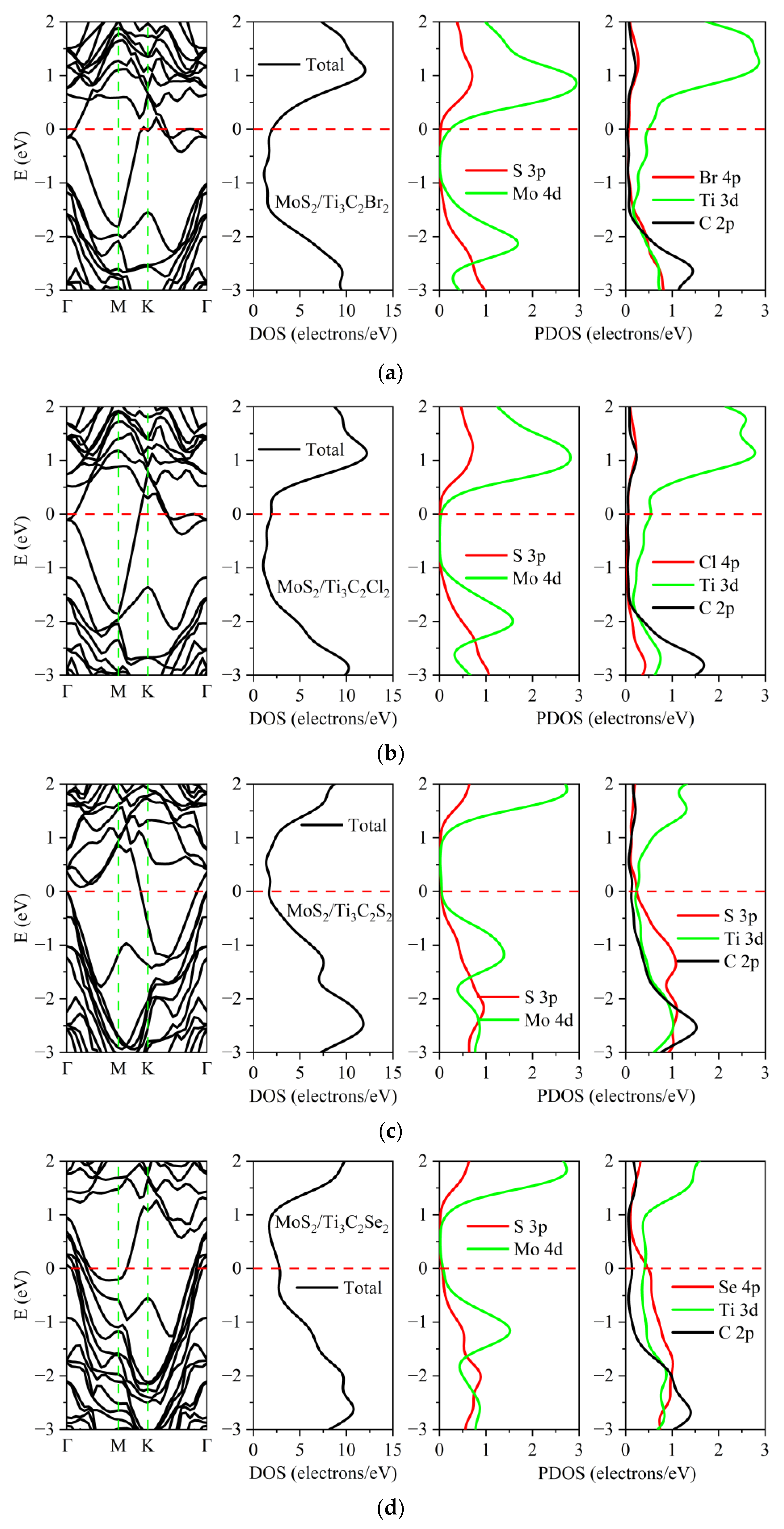
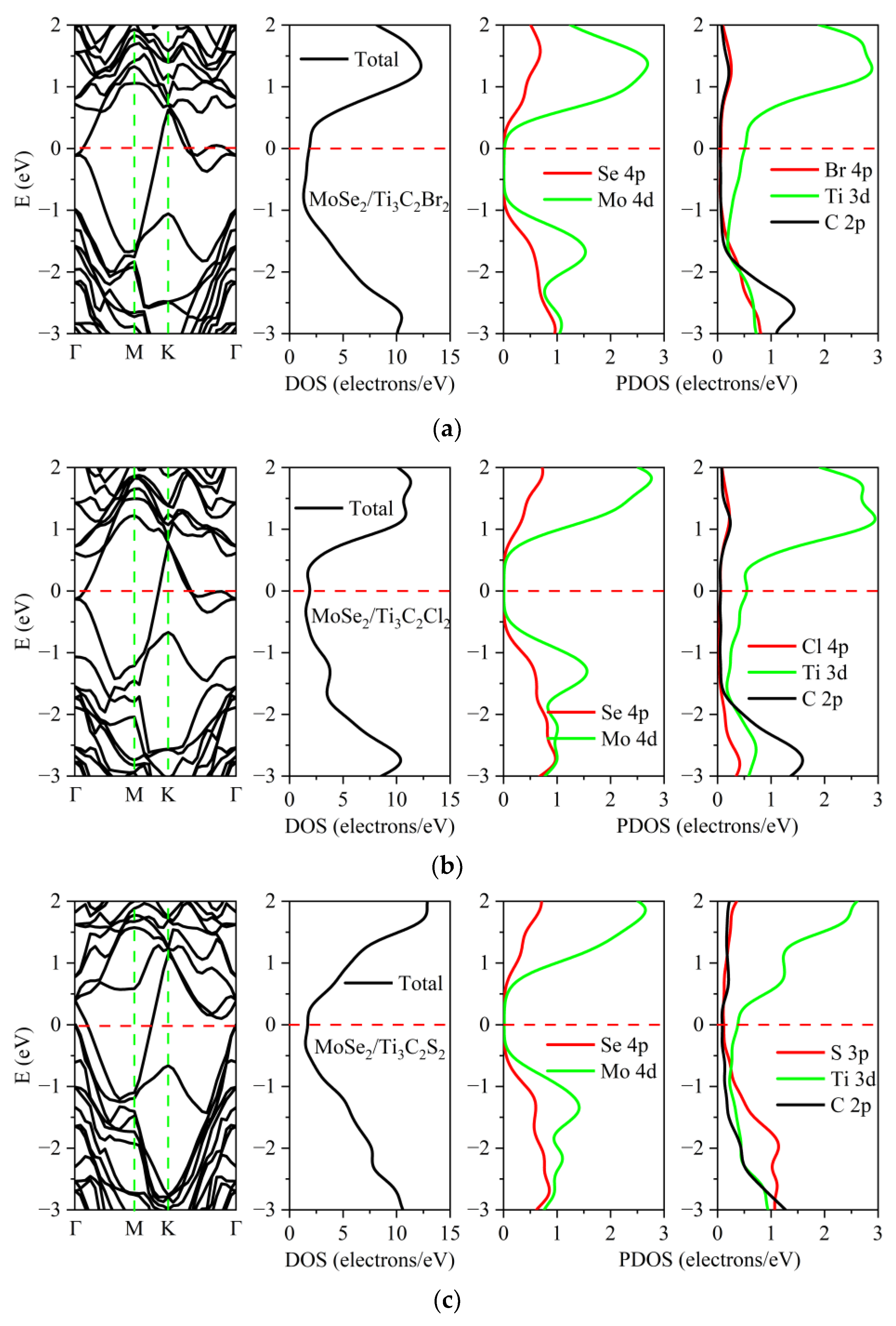
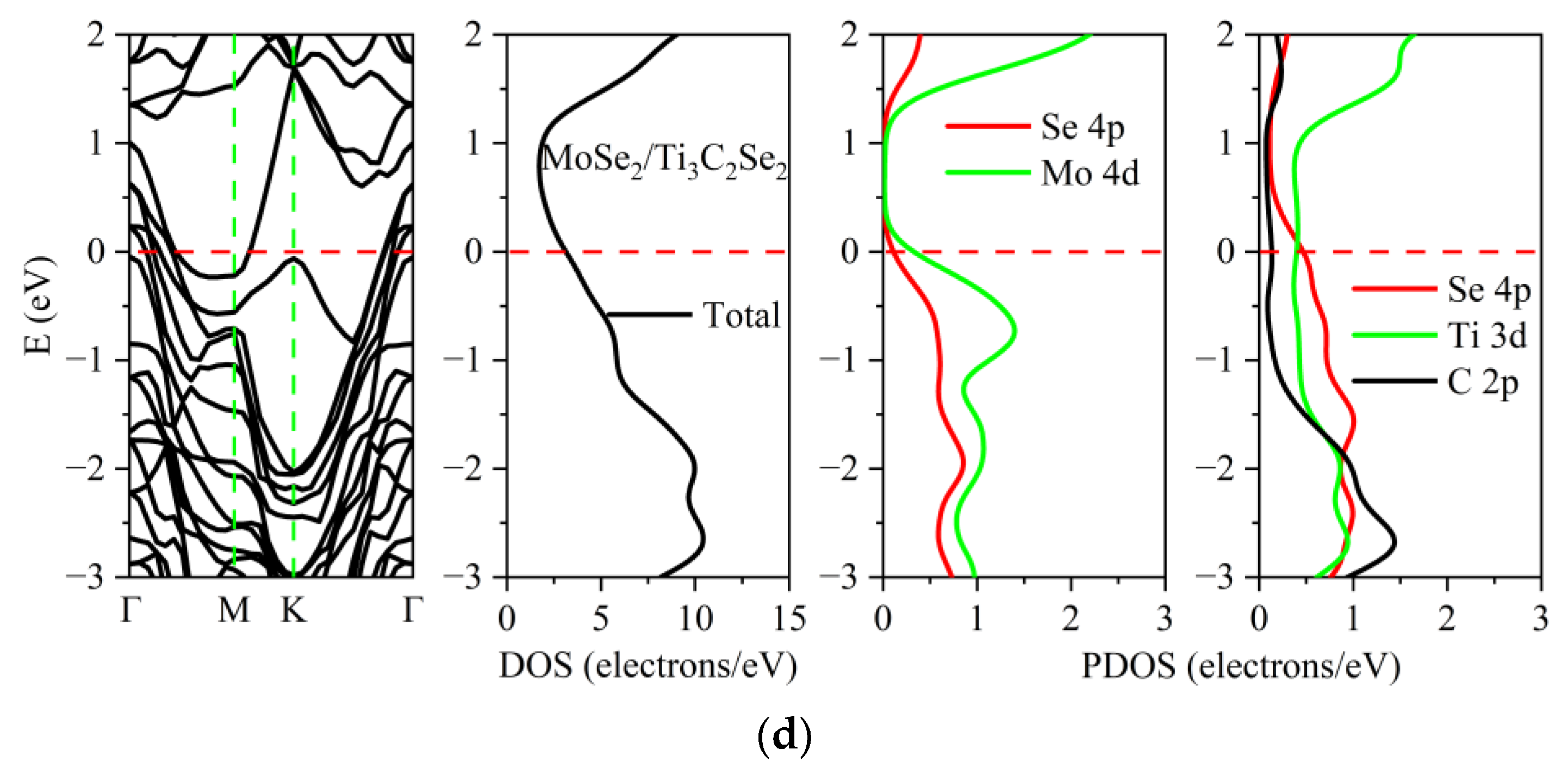
3.2. Electronic Properties of the TMDs/Ti3C2X2 Heterostructures
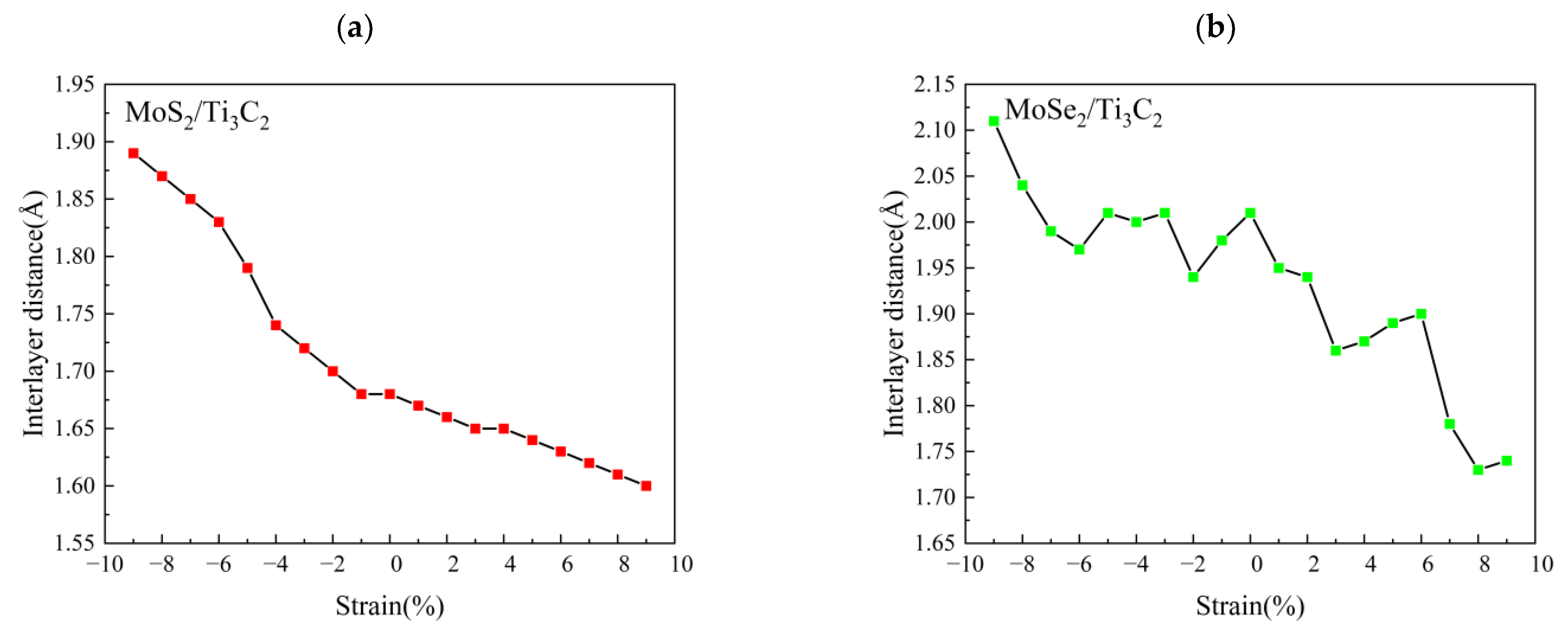
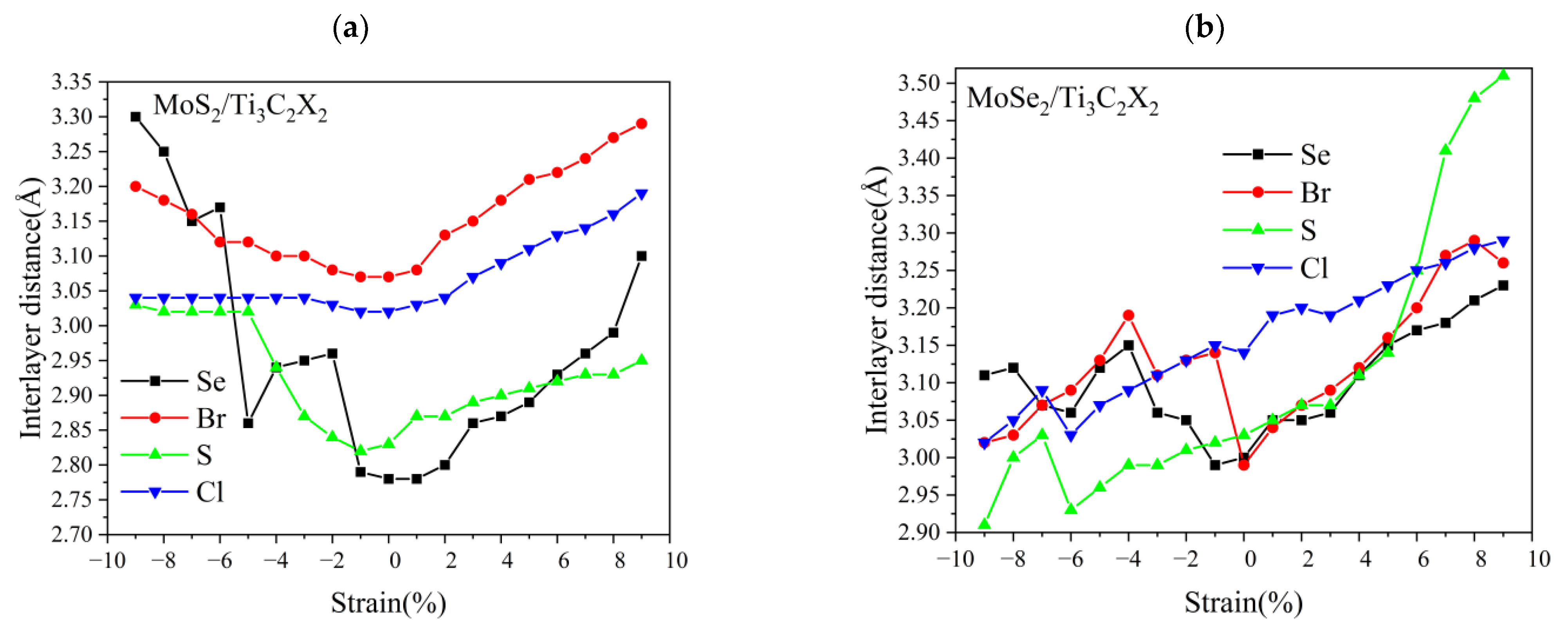
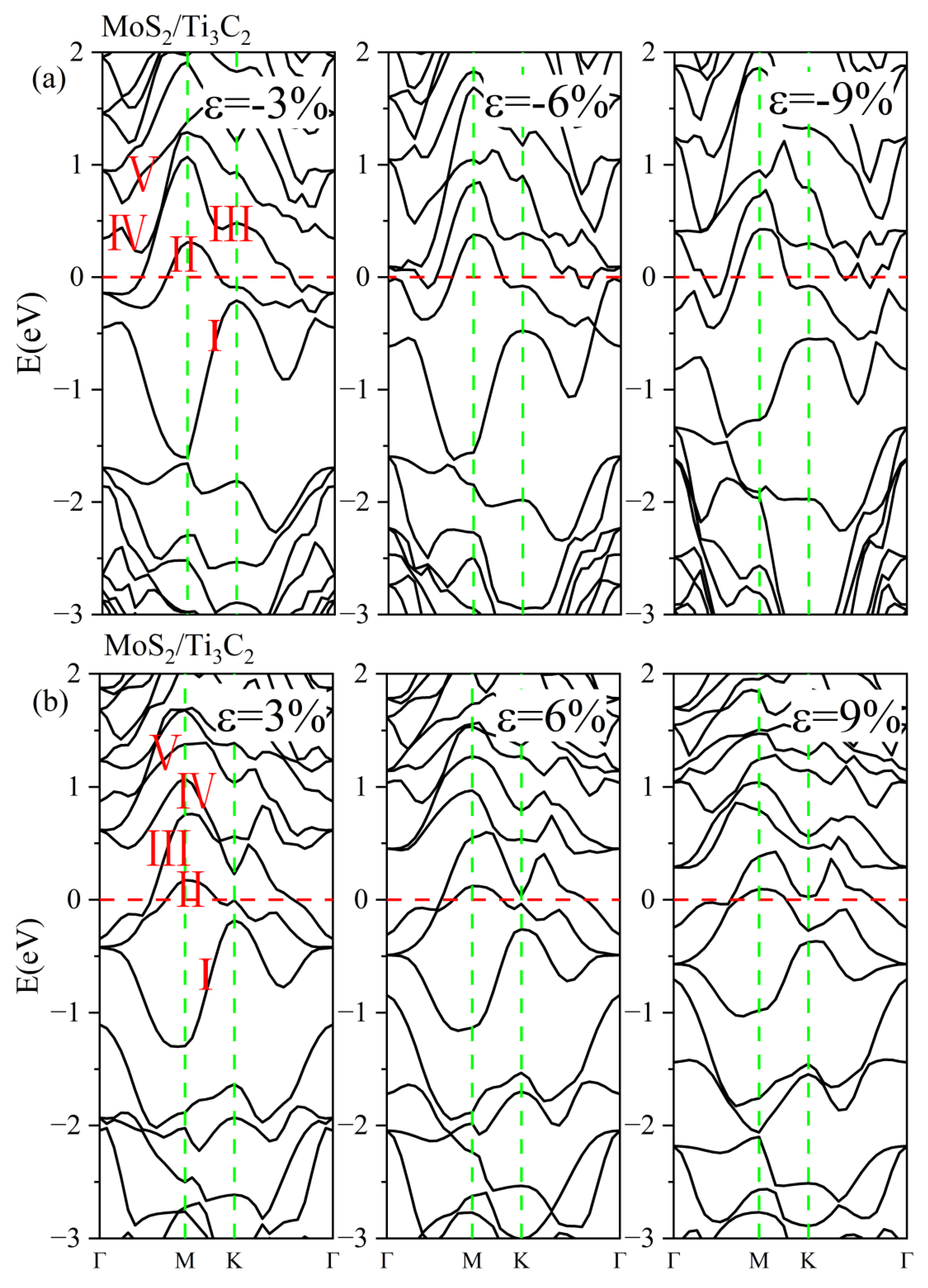
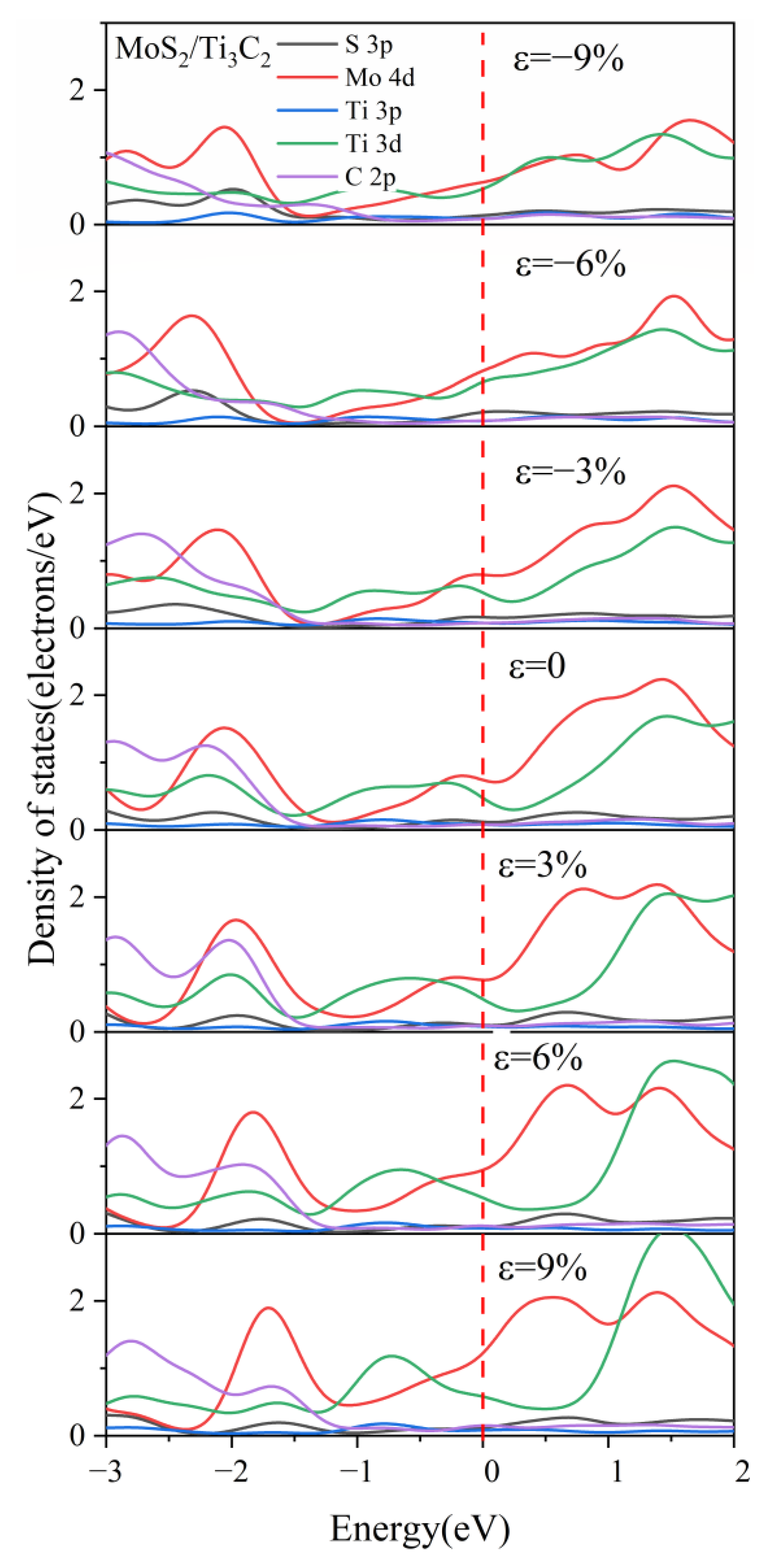
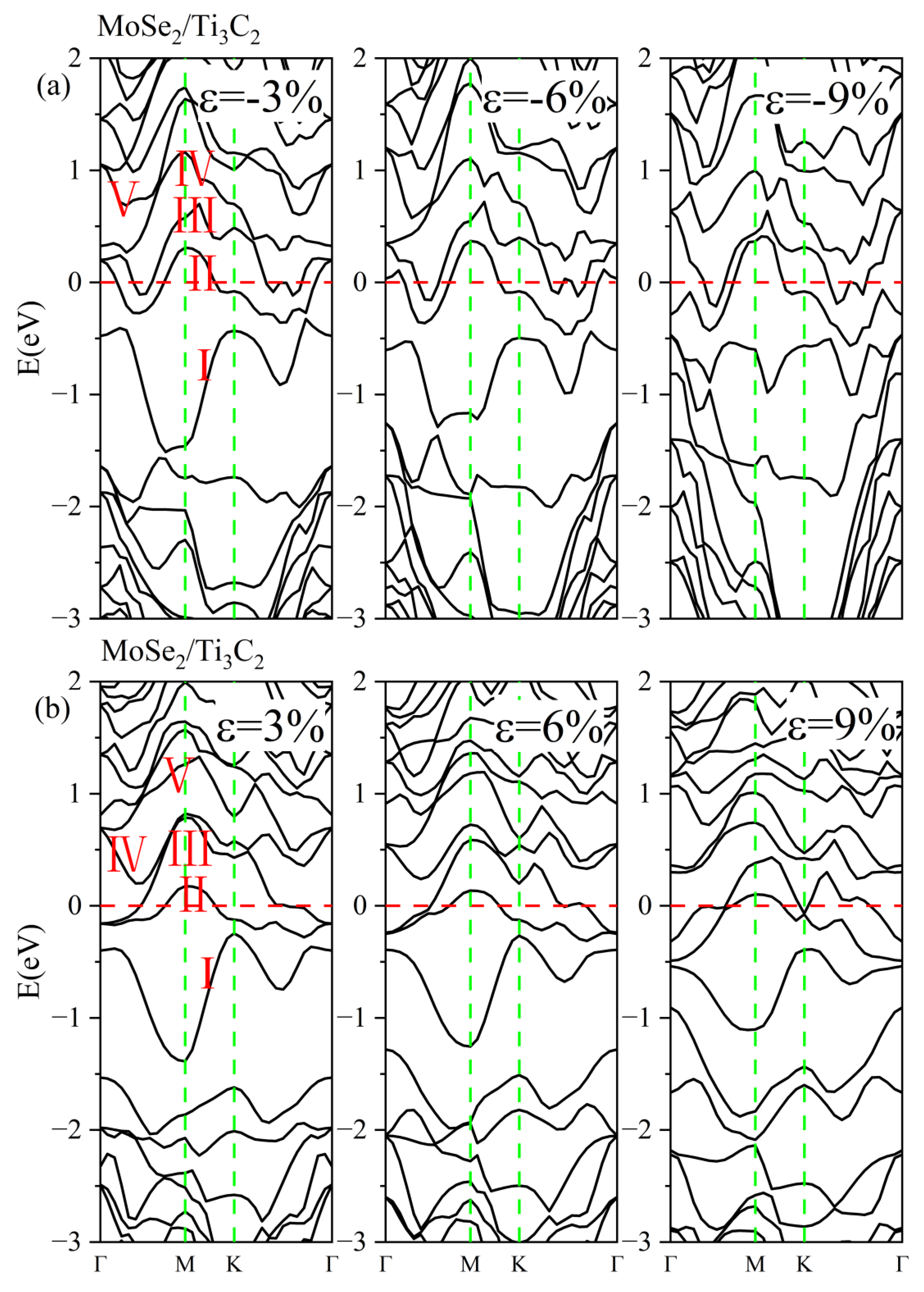
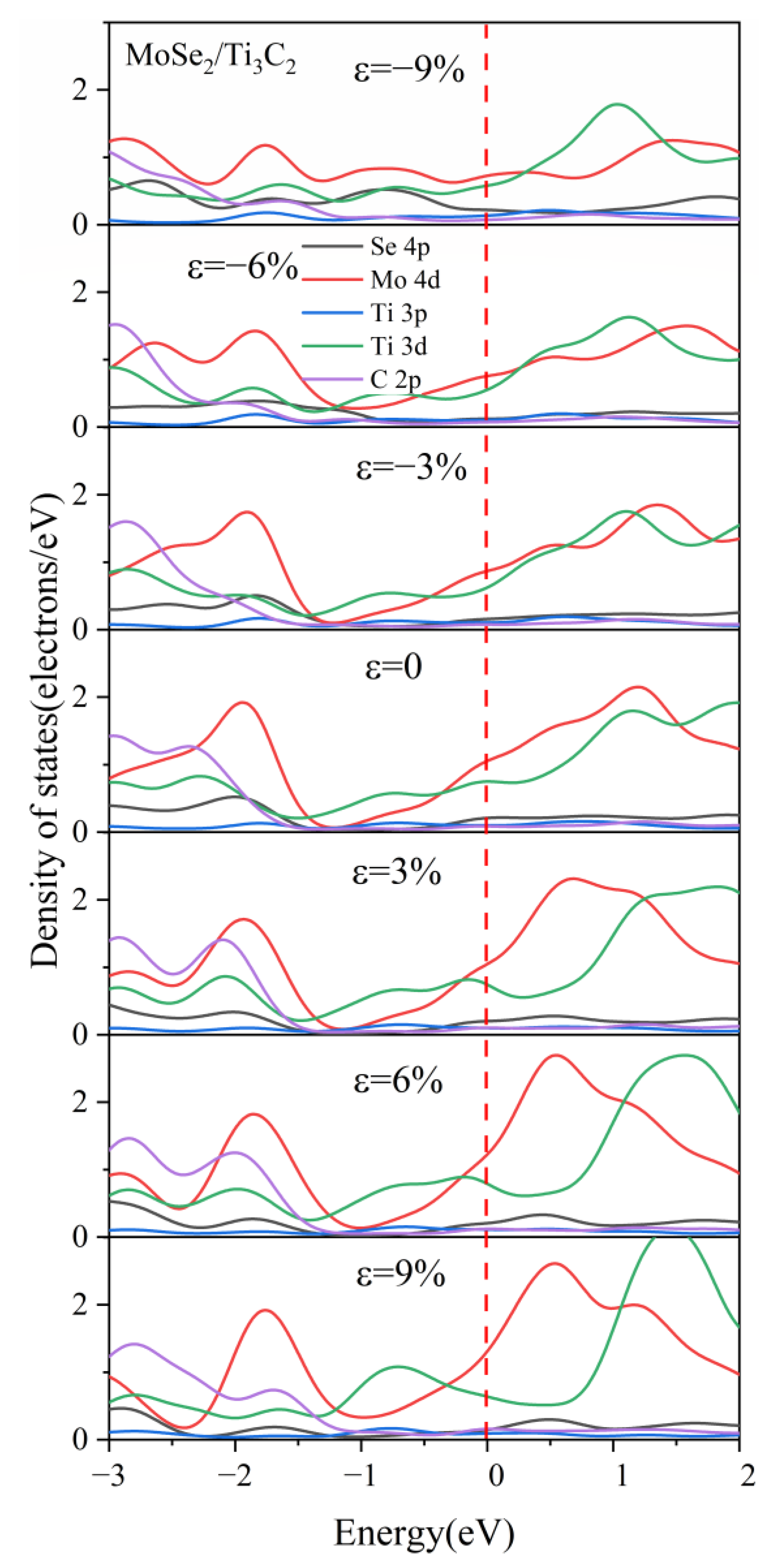
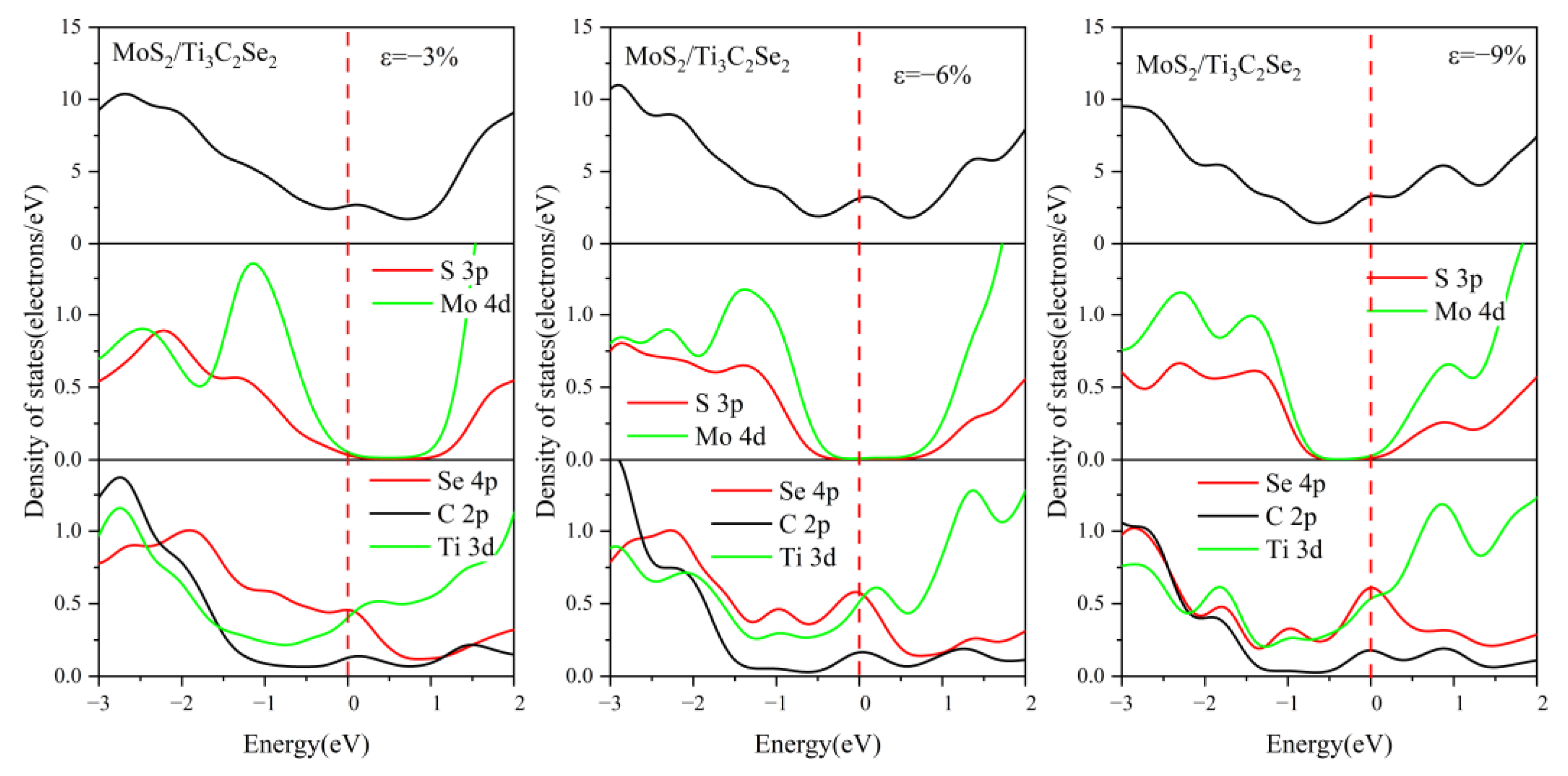
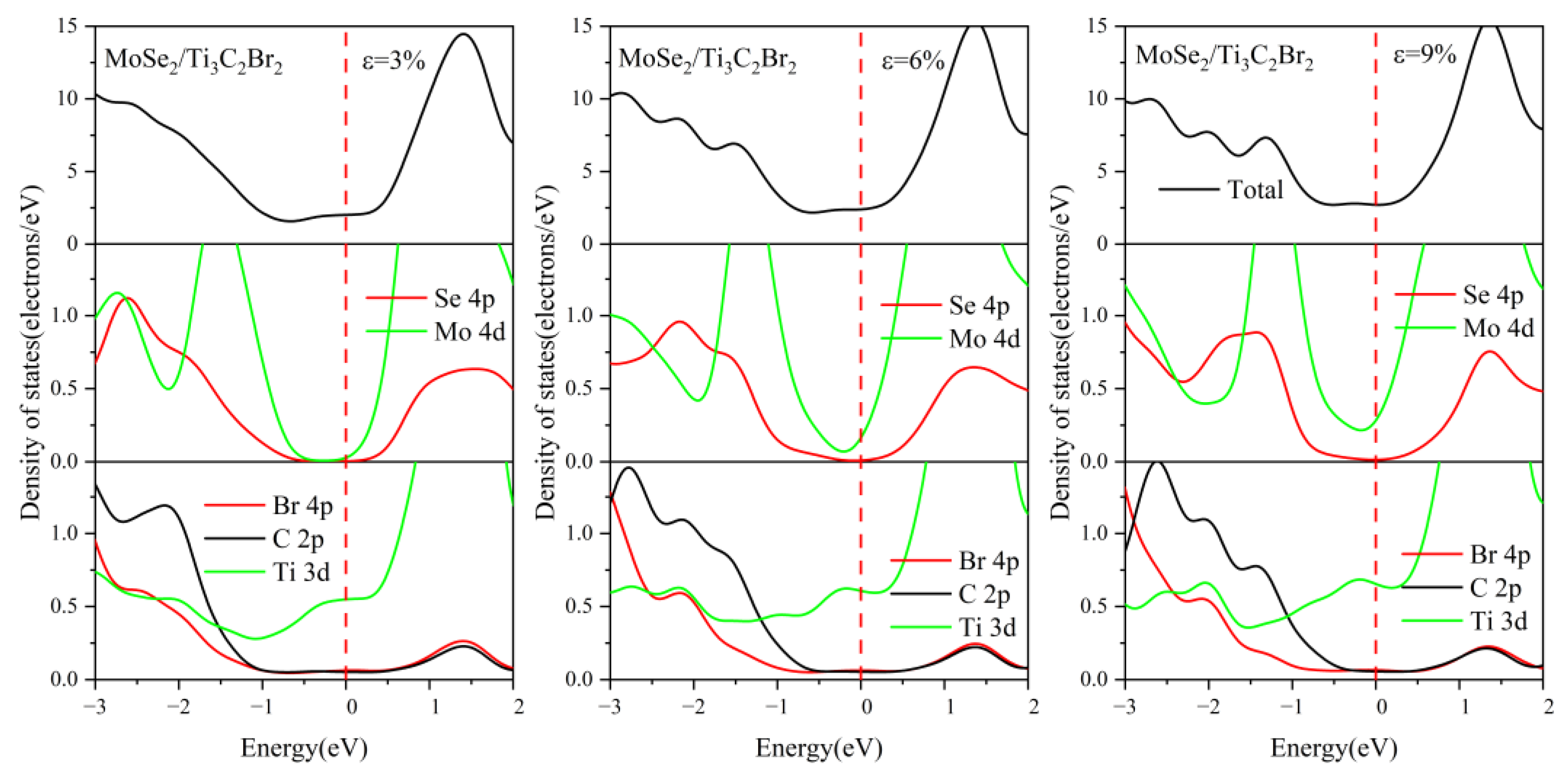
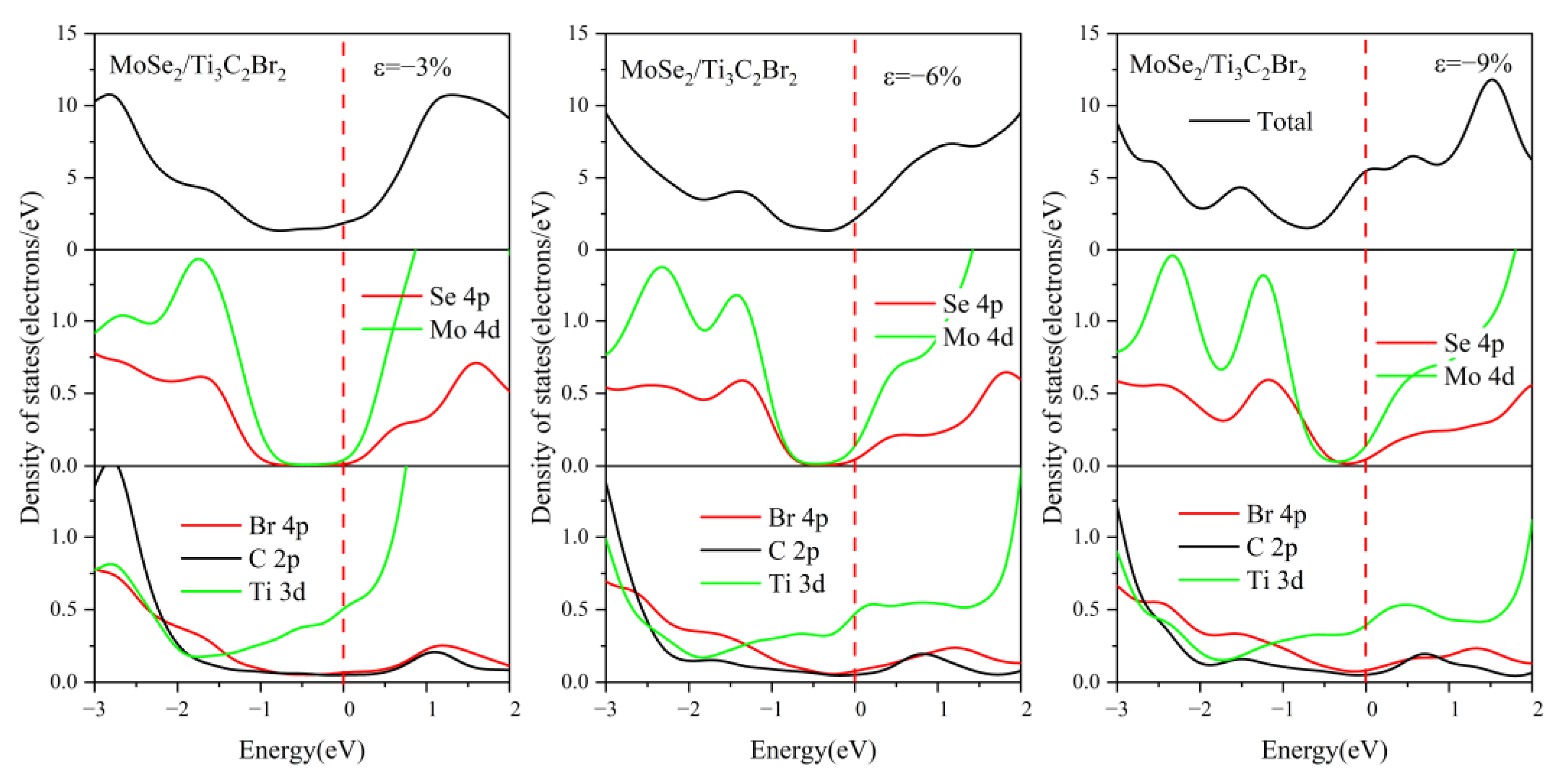
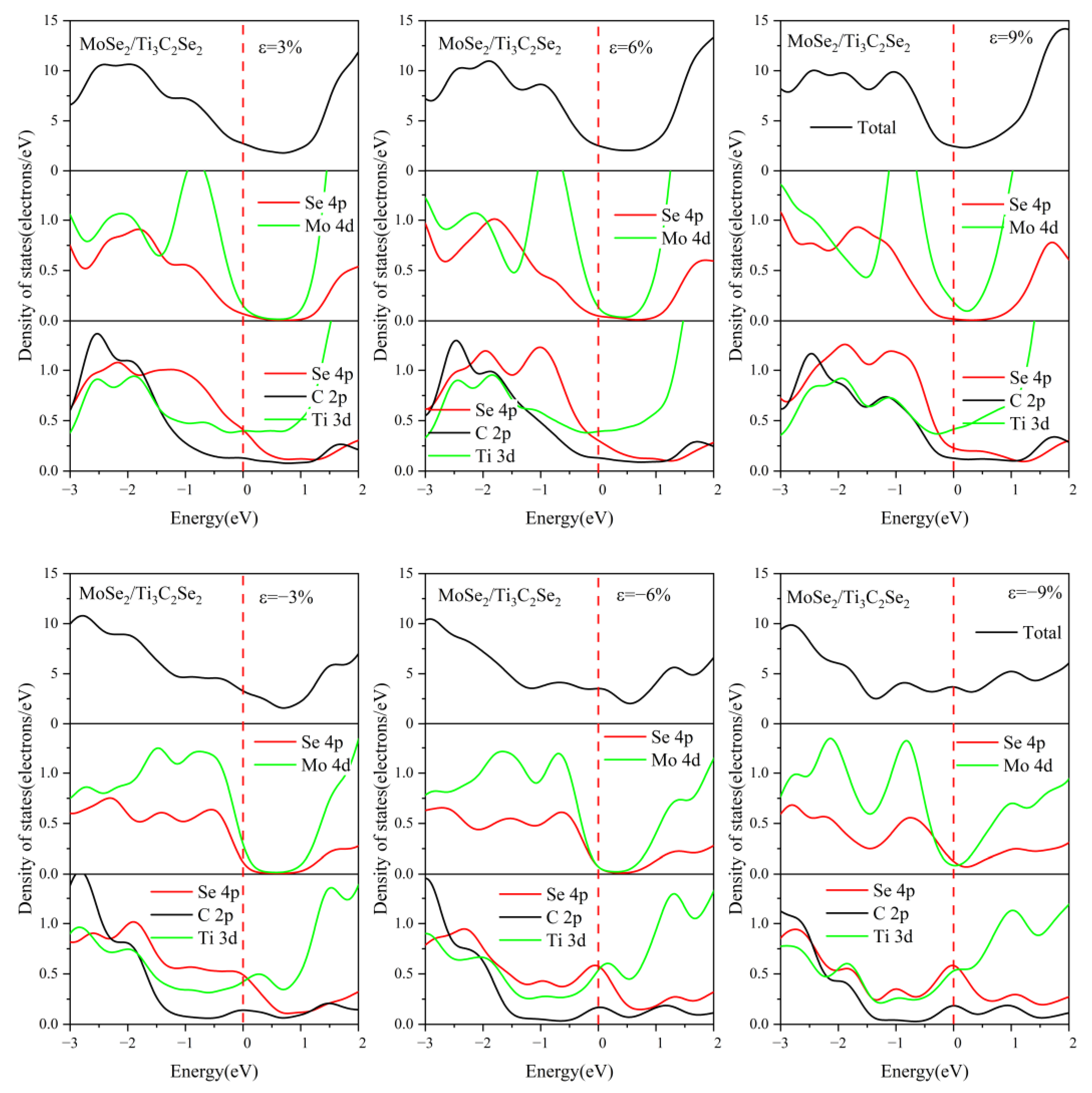
3.3. Effect of Biaxial Strain on the Structural and Electronic Properties of the TMDs/Ti3C2 Heterostructures
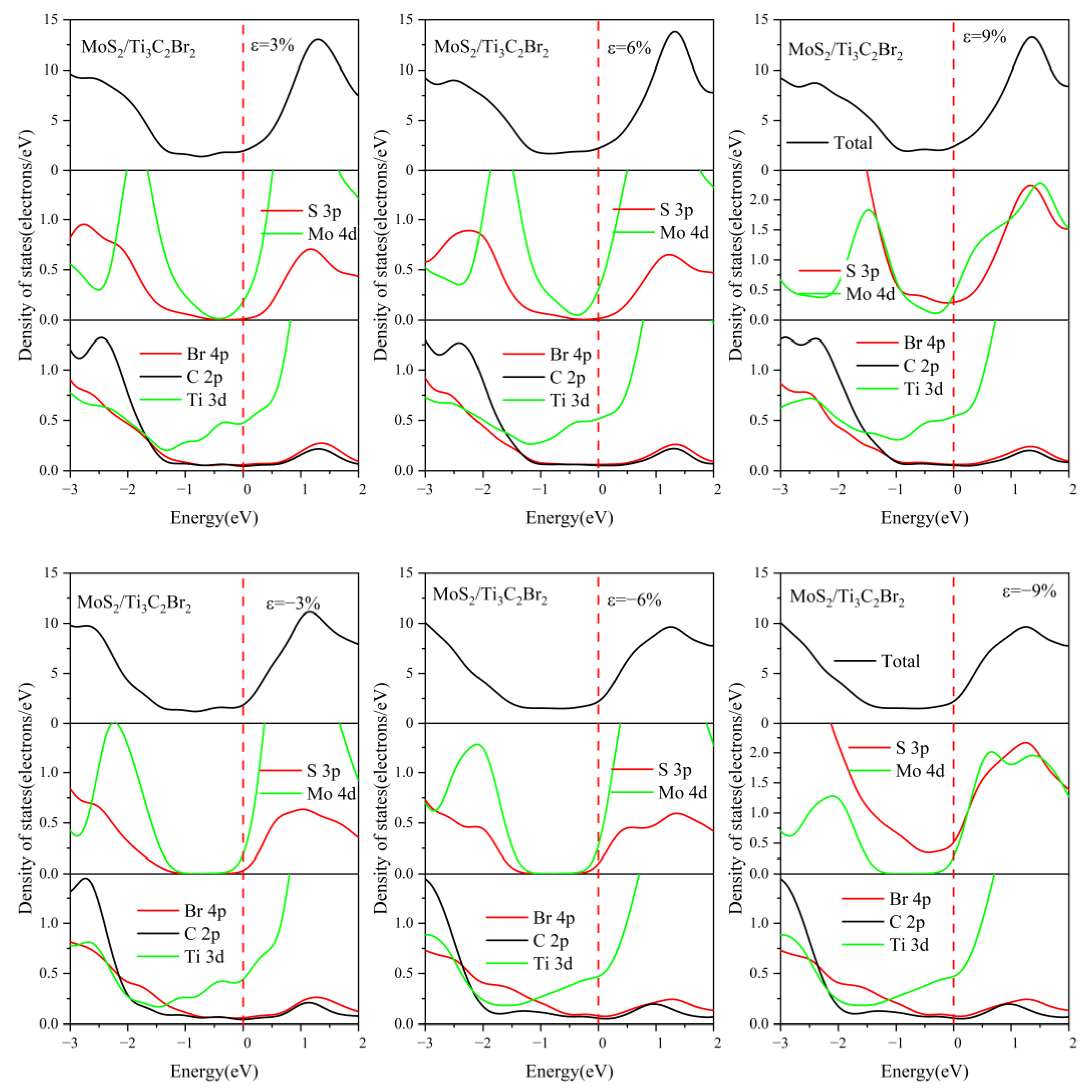
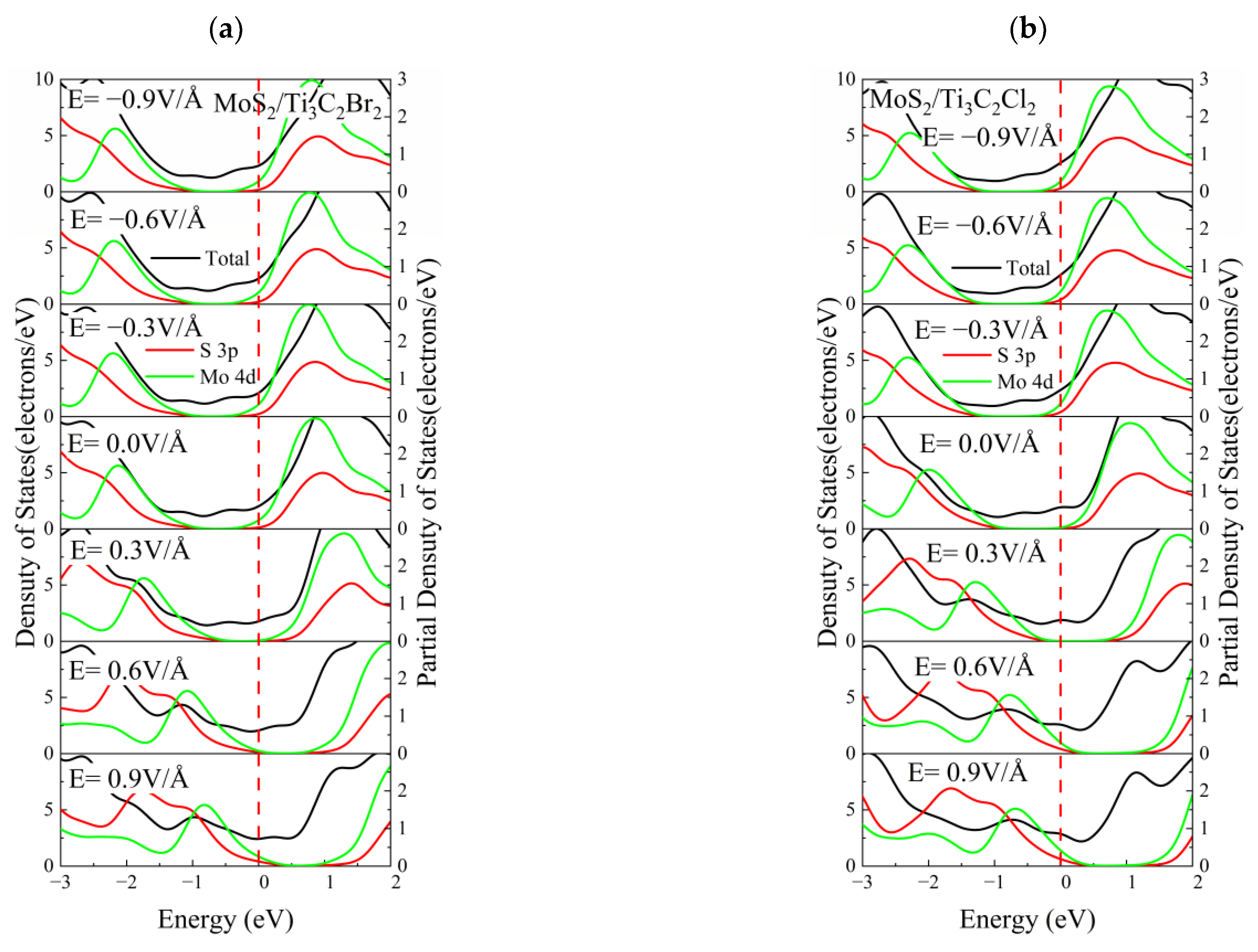
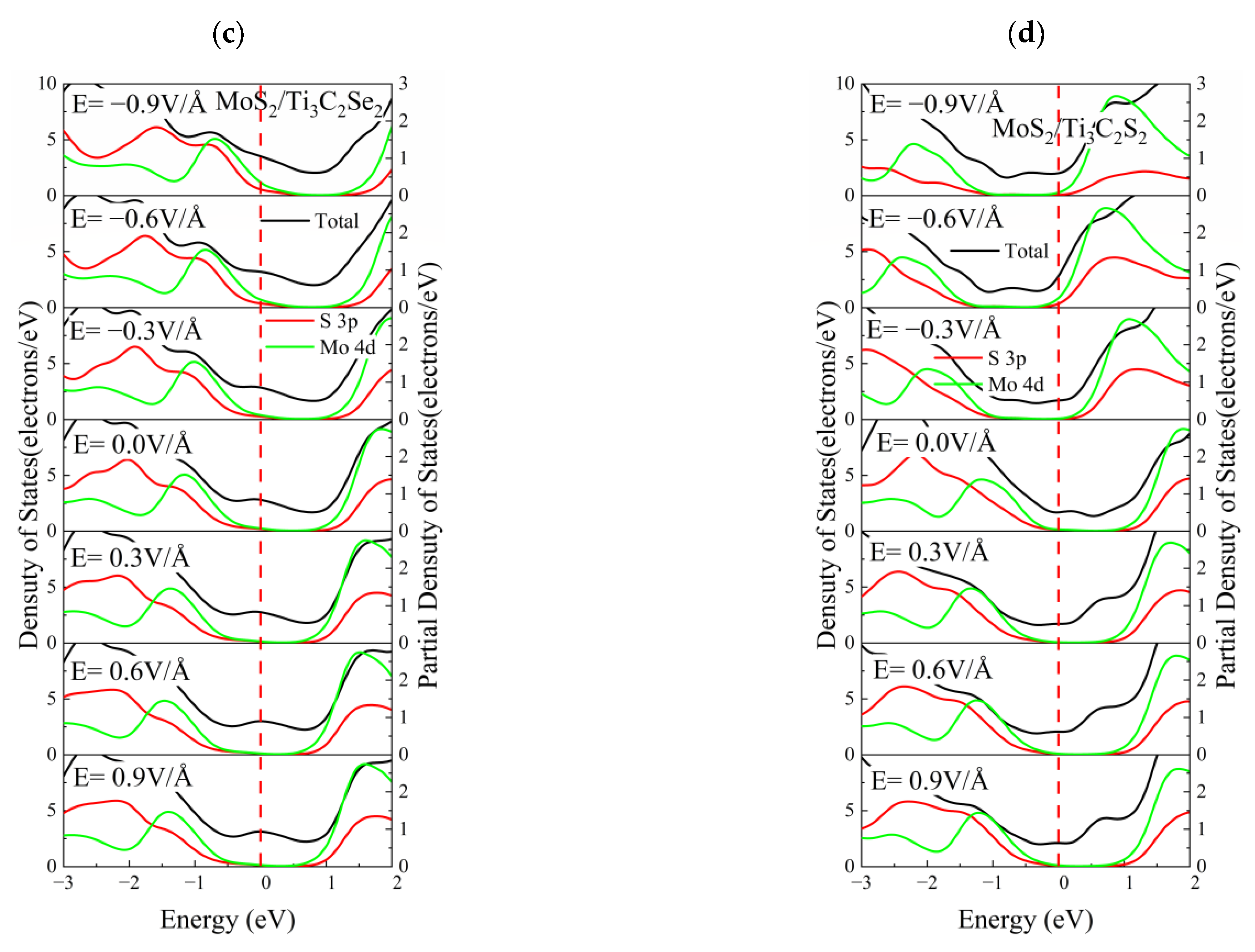
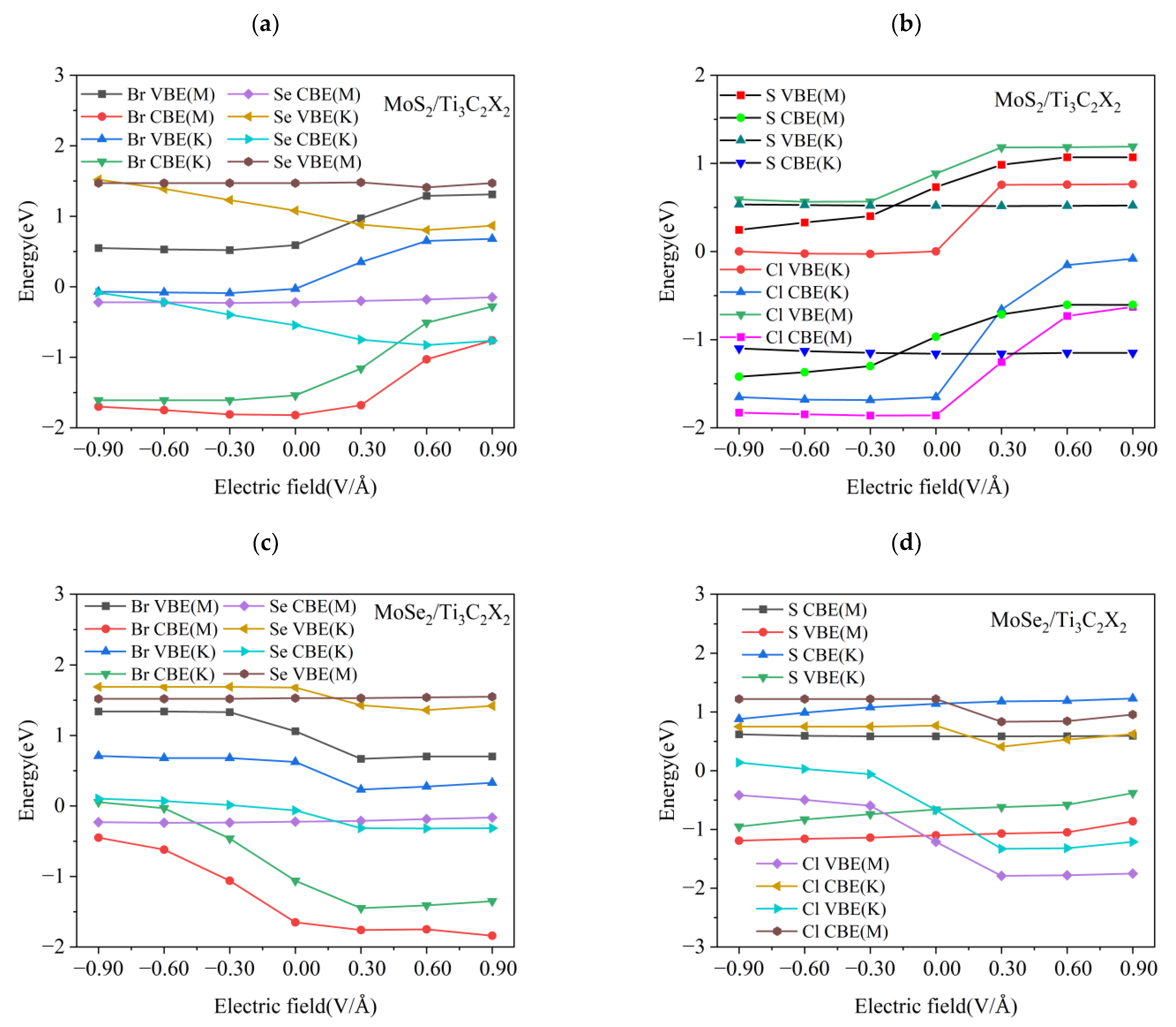
3.4. Effect of Electric Field on the Electronic Properties of the TMDs/Ti3C2X2 Heterostructures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chen, H.L.; Han, J.N.; Deng, X.Q.; Fan, Z.Q.; Sun, L.; Zhang, Z.H. Vertical Strain and Twist Induced Tunability on Electronic and Optical Properties of Janus HfSSe/SnC Van der Waals Heterostructure. Appl. Surf. Sci. 2022, 598, 153756. [Google Scholar] [CrossRef]
- Wang, J.L.; Zhao, X.W.; Hu, G.C.; Ren, J.F.; Yuan, X.B. Manipulable Electronic and Optical Properties of Two-Dimensional MoSTe/MoGe2N4 van der Waals Heterostructures. Nanomaterials 2022, 11, 3338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.S.; Chen, J.; Wang, X.D.; Huang, H.; Yao, M. First-Principles Study of Transition Metal Ti-Based MXenes (Ti2MC2Tx and M2TiC2Tx) as Anode Materials for Sodium-Ion Batteries. ACS Appl. Nano Mater. 2022, 5, 2358–2366. [Google Scholar] [CrossRef]
- Huang, H.X.; Zha, J.J.; Li, S.S.; Tan, C.L. Two-dimensional alloyed transition metal dichalcogenide nanosheets: Synthesis and applications. Chin. Chem. Lett. 2022, 33, 163–176. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, P.; Wang, F.; Ye, J.F.; He, T.; Wu, F.; Peng, M.; Wu, P.; Chen, Y.; Zhong, F.; et al. A Noble Metal Dichalcogenide for High-Performance Field-Effect Transistors and Broadband Photodetectors. Adv. Funct. Mater. 2020, 30, 1907945. [Google Scholar] [CrossRef]
- Na, J.; Park, C.; Lee, C.H.; Choi, W.R.; Choi, S.; Lee, J.U.; Yang, W.; Cheong, H.; Campbell, E.E.B.; Jhang, S.H. Indirect Band Gap in Scrolled MoS2 Monolayers. Nanomaterials 2022, 12, 3353. [Google Scholar] [CrossRef]
- Cai, Z.Y.; Liu, B.L.; Zou, X.L.; Cheng, H.-M. Chemical vapor deposition growth and applications of two-dimensional materials and their heterostructures. Chem. Rev. 2018, 118, 6091–6133. [Google Scholar] [CrossRef]
- Tan, T.; Jiang, X.T.; Wang, C.; Yao, B.C.; Zhang, H. 2D material optoelectronics for information functional device applications: Status and challenges. Adv. Sci. 2020, 7, 2000058. [Google Scholar] [CrossRef]
- Kuzel, P.; Nemec, H. Terahertz spectroscopy of nanomaterials: A close look at charge-carrier transport. Adv. Opti. Mater. 2020, 8, 1900623. [Google Scholar] [CrossRef]
- Bafekry, A.; Stampfl, C.; Ghergherehchi, M. Strain, electric-field and functionalization induced widely tunable electronic properties in MoS2/BC3, /C3N and /C3N4 van der Waals heterostructures. Nanotechnology 2020, 31, 295202. [Google Scholar] [CrossRef]
- Biroju, R.K.; Das, D.; Sharma, R.; Pal, S.; Mawlong, L.P.L.; Bhorkar, K.; Giri, P.K.; Singh, A.K.; Narayanan, T.N. Hydrogen evolution reaction activity of graphene-MoS2 van der Waals heterostructures. ACS Energy. Lett. 2017, 2, 1355–1361. [Google Scholar] [CrossRef]
- Li, J.T.; Zhou, X.L. First principles calculations of electrical and optical properties of Cu3N/MoS2 heterostructure with tunable bandgaps. Appl. Phys. A 2021, 127, 693. [Google Scholar] [CrossRef]
- Yelgel, C. First-principles modeling of GaN/MoSe2 van der Waals heterobilayer. Turk. J. Phys. 2017, 41, 463–468. [Google Scholar] [CrossRef]
- Pogorielov, M.; Smyrnova, K.; Kyrylenko, S.; Gogotsi, O.; Zahorodna, V.; Pogrebnjak, A. MXenes-A New Class of Two-Dimensional Materials: Structure, Properties and Potential Applications. Nanomaterials 2022, 11, 3412. [Google Scholar] [CrossRef]
- Fleischmann, S.; Mitchell, J.B.; Wang, R.; Zhan, C.; Jiang, D.-E.; Presser, V.; Augustyn, V. Pseudocapacitance: From fundamental understanding to high power energy storage materials. Chem. Rev. 2020, 120, 6738–6782. [Google Scholar] [CrossRef]
- Hasan, M.M.; Hossain, M.M.; Chowdhury, H.K. Two-dimensional MXene-based flexible nanostructures for functional nanodevices: A review. J. Mater. Chem A 2021, 9, 3231–3269. [Google Scholar] [CrossRef]
- Jiang, Q.; Lei, Y.J.; Liang, H.F.; Xi, K.; Xia, C.; Alshareef, H.N. Alshareef, Review of MXene electrochemical microsupercapacitors. Energy Storage Mater. 2020, 27, 78–95. [Google Scholar] [CrossRef]
- Xiong, D.B.; Li, X.F.; Bai, Z.M.; Lu, S.G. Recent advances in layered Ti3C2Tx MXene for electrochemical energy storage. Small 2018, 14, 1703419. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Cao, H.H.; Liang, Z.S.; Cheng, Y.F.; Yin, T.T.; Liu, Z.Y.; Yan, S.W.; Jia, S.F.; Li, L.Y.; Wang, J.B.; et al. First-principles study of Ti-deficient Ti3C2 MXene nanosheets as NH3 gas sensors. ACS Appl. Nano Mater. 2022, 5, 2470–2475. [Google Scholar] [CrossRef]
- Li, D.Q.; Chen, X.F.; Xiang, P.; Du, H.Y.; Xiao, B.B. Chalcogenated-Ti3C2X2 MXene (X = O, S, Se and Te) as a high-performance anode material for Li-ion batteries. Appl. Surf. Sci. 2020, 501, 144221. [Google Scholar] [CrossRef]
- Mathis, T.S.; Maleski, K.; Goad, A.; Sarycheva, A.; Anayee, M.; Foucher, A.C.; Hantanasirisakul, K.; Shuck, C.E.; Stach, E.A.; Gogotsi, Y. Modified MAX phase synthesis for environmentally stable and highly conductive Ti3C2 MXene. ACS Nano 2021, 15, 6420–6429. [Google Scholar] [CrossRef] [PubMed]
- Li, C.L.; Guo, J.; Wang, C.Y.; Ma, D.C.; Wang, B.L. Design of MXene contacts for high-performance WS2 transistors. Appl. Surf. Sci. 2020, 527, 146701. [Google Scholar] [CrossRef]
- Liang, P.H.; Xu, T.F.; Zhu, K.J.; Rao, Y.; Zheng, H.J.; Wu, M.; Chen, J.T.; Liu, J.S.; Yan, K.; Wang, J. Heterogeneous interface-boosted zinc storage of H2V3O8 nanowire/Ti3C2Tx MXene composite toward high-rate and long cycle lifespan aqueous zinc-ion batteries. Energy Storage Mater. 2022, 50, 63–74. [Google Scholar] [CrossRef]
- Ariga, K. Don’t forget Langmuir-Blodgett films 2020: Interfacial nanoarchitectonics with molecules, materials, and living objects. Langmuir 2020, 36, 7158–7180. [Google Scholar] [CrossRef] [PubMed]
- Nahirniak, S.; Ray, A.; Saruhan, B. Challenges and Future Prospects of the MXene-Based Materials for Energy Storage Applications. Batteries 2023, 9, 126. [Google Scholar] [CrossRef]
- Yu, L.; Hu, L.F.; Anasori, B.; Liu, Y.-T.; Zhu, Q.Z.; Zhang, P.; Gogotsi, Y.; Xu, B. MXene-Bonded activated carbon as a flexible electrode for high-performance supercapacitors. ACS Energy Lett. 2018, 3, 1597–1603. [Google Scholar] [CrossRef]
- Shen, C.J.; Wang, L.B.; Zhou, A.G.; Wang, B.; Wang, X.L.; Lian, W.W.; Hu, Q.K.; Qin, G.; Liu, X.Q. Synthesis and Electrochemical Properties of Two-Dimensional RGO/Ti3C2Tx Nanocomposites. Nanomaterials 2018, 8, 80. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.L.; Zhao, C.H.; Niu, D.J.; Zhu, J.F.; Wei, D.; Wang, C.W.; Wang, L.; Yang, L.Q. Ultrathin N-doped Ti3C2-MXene decorated with NiCo2S4 nanosheets as advanced electrodes for supercapacitors. Appl. Surf. Sci. 2021, 539, 148272. [Google Scholar] [CrossRef]
- Debow, S.; Zhang, T.; Liu, X.S.; Song, F.Z.; Qian, Y.Q.; Han, J.; Maleski, K.; Zander, Z.B.; Creasy, W.R.; Kuhn, D.L. Charge dynamics in TiO2/MXene composites. J. Phys. Chem. C 2021, 125, 10473–10482. [Google Scholar] [CrossRef]
- Jing, H.R.; Ling, F.L.; Liu, X.Q.; Chen, Y.K.; Zeng, W.; Zhang, Y.X.; Fang, L.; Zhou, M. Strain-engineered robust and Schottky-barrier-free contact in 2D metal-semiconductor heterostructure. Electron. Struct. 2019, 1, 015010. [Google Scholar] [CrossRef]
- Guan, Q.Y.; Yan, H.J.; Cai, Y.Q. Flatten the Li-ion Activation in Perfectly Lattice-Matched MXene and 1T-MoS2 Heterostructures via Chemical Functionalization. Adv. Mater. Interfaces 2022, 9, 2101838. [Google Scholar] [CrossRef]
- Li, C.L.; Wu, G.X.; Wang, C.Y.; Fu, Y.; Wang, B.L. Tuning electronic and transport properties of MoS2/Ti2C heterostructure by external strain and electric field. Comput. Mater. Sci. 2018, 153, 417–423. [Google Scholar] [CrossRef]
- Xu, E.Z.; Zhang, Y.; Wang, H.; Zhu, Z.F.; Quan, J.J.; Chang, Y.J.; Li, P.C.; Yu, D.B.; Jiang, Y. Ultrafast kinetics net electrode assembled via MoSe2/MXene heterostructure for high-performance sodium-ion batteries. Chem. Eng. J. 2020, 385, 123839. [Google Scholar] [CrossRef]
- Ling, F.L.; Kang, W.; Jing, H.R.; Zeng, W.; Chen, Y.K.; Liu, X.Q.; Zhang, Y.X.; Zhou, M. Enhancing hydrogen evolution on the basal plane of transition metal dichacolgenide van der Waals heterostructures. NPJ Comput. Mater. 2019, 5, 20. [Google Scholar] [CrossRef] [Green Version]
- Kamysbayev, V.; Filatov, A.S.; Hu, H.C.; Rui, X.; Lagunas, F.; Wang, D.; Klie, R.F.; Talapin, D.V. Covalent surface modifications and superconductivity of two-dimensional metal carbide MXenes. Science 2020, 369, 979–983. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP, Z. Krist-Cryst. Materials 2005, 220, 567–570. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- McNellis, E.R.; Meyer, J.; Reuter, K. Azobenzene at coinage metal surfaces: Role of dispersive van der Waals interactions. Phys. Rev. B 2009, 80, 205414. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.C.; Xiao, B.; Li, J.L.; Deng, Q.; Xu, Y.H.; Wang, H.F.; Rao, D.W. Improved transport properties and novel Li diffusion dynamics in van der waals C2N/graphene heterostructure as anode materials for lithium-ion batteries: A first-principles investigation. J. Phys. Chem. C 2019, 123, 3353–3367. [Google Scholar] [CrossRef]
- Ibragimova, R.; Lv, Z.P.; Komsa, H.P. First principles study of the stability of MXenes under an electron beam. Nanoscale Adv. 2021, 3, 1934–1941. [Google Scholar] [CrossRef]
- Yu, Y.X. Can all nitrogen-doped defects improve the performance of graphene anode materials for lithium-ion batteries? Phys. Chem. Chem. Phys. 2013, 15, 16819–16827. [Google Scholar] [CrossRef] [PubMed]
- Froyen, S. Brillouin-zone integration by Fourier quadrature: Special points for superlattice and supercell calculations. Phys. Rev. B 1989, 39, 3168–3172. [Google Scholar] [CrossRef]
- Zeng, L.H.; Wu, D.; Jie, J.S.; Ren, X.Y.; Hu, X.; Lau, S.P.; Chai, Y.; Tsang, Y.H. Van der Waals epitaxial growth of mosaic-like 2D platinum ditelluride layers for room-temperature mid-infrared photodetection up to 10.6 µm. Adv. Mater. 2020, 32, 2004412. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.; Kang, J.; Park, J.; Han, B. First-principles study on thermodynamic stability of the hybrid interfacial structure of LiMn2O4 cathode and carbonate electrolyte in Li-ion batteries. Phys. Chem. Chem. Phys. 2018, 20, 11592–11597. [Google Scholar] [CrossRef] [PubMed]
- Bandeira, N.S.; da Costa, D.R.; Chaves, A.; Farias, G.A.; Filho, R.N.C. Gap opening in graphene nanoribbons by application of simple shear strain and in-plane electric field. J. Phys. Condens. Matter 2020, 33, 065503. [Google Scholar] [CrossRef]
- Hu, T.; Wang, J.M.; Zhang, H.; Li, Z.J.; Hu, M.M.; Wang, X.H. Vibrational properties of Ti3C2 and Ti3C2T2(T = O, F, OH) monosheets by first-principles calculations: A comparative study. Phys. Chem. Chem. Phys. 2015, 17, 9997–10003. [Google Scholar] [CrossRef]
- Lindroth, D.O.; Erhart, P. Thermal transport in van der Waals solids from first-principles calculations. Phys. Rev. B 2016, 94, 115205. [Google Scholar] [CrossRef] [Green Version]
- Frechette, L.B.; Dellago, C.; Geissler, P.L. Consequences of lattice mismatch for phase equilibrium in heterostructured solids. Phys. Rev. Lett. 2019, 123, 135701. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.H.; Zhang, Y.; Xie, Z.F.; Wei, X.; Guo, T.T.; Fan, J.B.; Ni, L.; Tian, Y.; Liu, J.; Duan, L. Tunable electronic properties of an Sb/InSe van der Waals heterostructure by electric field effects. Phys. Chem. Chem. Phys. 2019, 10, 5627–5633. [Google Scholar] [CrossRef]
- Hanbicki, A.T.; Chuang, H.J.; Rosenberger, M.R.; Hellberg, C.S.; Sivaram, S.V.; McCreary, K.M.; Mazin, I.I.; Jonker, B.T. Double indirect interlayer exciton in a MoSe2/WSe2 van der Waals heterostructure. ACS Nano 2018, 5, 4719–4726. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Fan, J. Computational insights into modulating the performance of MXene based electrode materials for rechargeable batteries. Nanotechnology 2021, 32, 252001. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Han, B.; Kapuscinski, P.; Delhomme, A.; Faugeras, C.; Amand, T.; Molas, M.R.; Marie, X. Measurement of the spin-forbidden dark excitons in MoS2 and MoSe2 monolayers. Nat. Commun. 2020, 11, 4037. [Google Scholar] [CrossRef] [PubMed]
- Ahn, G.H.; Amani, M.; Rasool, H.; Lien, D.-H.; Mastandrea, J.P.; Iii, J.W.A.; Dubey, M.; Chrzan, D.C.; Minor, A.M.; Javey, A. Strain-engineered growth of two-dimensional materials. Nat. Commun. 2017, 8, 608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.X.; Chen, Y.J.; Jiang, C.B. Strain engineering of two-dimensional materials: Methods, properties, and applications. InfoMat 2021, 3, 397–420. [Google Scholar] [CrossRef]
- Saha, S.; Samanta, P.; Murmu, N.C.; Kuila, T. A review on the heterostructure nanomaterials for supercapacitor application. J. Energy Storage 2018, 17, 181–202. [Google Scholar] [CrossRef]
- Ouyang, W.X.; Teng, F.; Fang, X.S. High performance BiOCl nanosheets/TiO2 nanotube arrays heterojunction UV photodetector: The influences of self-induced inner electric fields in the BiOCl nanosheets. Adv. Funct. Mater. 2018, 28, 1707178. [Google Scholar] [CrossRef]
- Chen, X.J.; Wang, J.; Chai, Y.Q.; Zhang, Z.J.; Zhu, Y.F. Efficient photocatalytic overall water splitting induced by the giant internal electric field of a g-C3N4/rGO/PDIP Z-scheme heterojunction. Adv. Mater. 2021, 33, 2007479. [Google Scholar] [CrossRef]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Configurations | Eb (meV/Å2) | d (Å) | d12 (Å) | dTi(3)–C(2) (Å) | dTi(1)–C(1) (Å) |
|---|---|---|---|---|---|
| (a) ZM_SA | −1.79 | 1.68 | 0.069 | 2.059 | 2.125 |
| (b) ZM_AA | −1.21 | 1.69 | 0.078 | 2.057 | 2.152 |
| (c) ZM_AS | −1.42 | 2.47 | 0.031 | 2.058 | 2.069 |
| (d) MZ_SA | −1.27 | 2.03 | 0.109 | 2.055 | 2.094 |
| (e) MZ_AA | −1.48 | 1.92 | 0.134 | 2.057 | 2.081 |
| (f) MZ_AS | −1.55 | 2.46 | 0.021 | 2.063 | 2.067 |
| Configurations | Eb (meVÅ2) | d (Å) | d34 (Å) | dTi(3)–C(2) (Å) | dTi(1)–C(1) (Å) |
|---|---|---|---|---|---|
| (g) SA_ZM | −1.03 | 1.89 | 0.051 | 2.064 | 2.119 |
| (h) AA_ZM | −1.01 | 2.56 | 0.013 | 2.071 | 2.073 |
| (i) AS_ZM | −0.39 | 2.51 | 0.021 | 2.067 | 2.074 |
| (j) SA_MZ | −0.76 | 2.12 | 0.101 | 2.061 | 2.096 |
| (k) AA_MZ | −0.96 | 2.09 | 0.107 | 2.068 | 2.083 |
| (m) AS_MZ | −0.59 | 2.01 | 0.063 | 2.069 | 2.122 |
| Configurations | Eb (meVÅ2) | d (Å) | d56 (Å) | dTi(1)–C (Å) | dTi(2)–X (Å) | dTi(1)–X (Å) |
|---|---|---|---|---|---|---|
| MoS2/Ti3C2S2 | −8.44 | 2.83 | 0.009 | 2.182 | 2.397 | 2.389 |
| MoS2/Ti3C2Se2 | −3.95 | 2.78 | 0.007 | 2.146 | 2.520 | 2.496 |
| MoS2/Ti3C2Cl2 | −3.17 | 3.02 | 0.003 | 2.104 | 2.503 | 2.492 |
| MoS2/Ti3C2Br2 | −8.42 | 3.07 | 0.004 | 2.117 | 2.626 | 2.621 |
| MoSe2/Ti3C2S2 | −3.55 | 3.03 | 0.003 | 2.193 | 2.501 | 2.403 |
| MoSe2/Ti3C2Se2 | −4.82 | 3.00 | 0.000 | 2.170 | 2.628 | 2.542 |
| MoSe2/Ti3C2Cl2 | −3.12 | 3.14 | 0.003 | 2.120 | 2.509 | 2.506 |
| MoSe2/Ti3C2Br2 | −8.41 | 2.99 | 0.001 | 2.127 | 2.638 | 2.617 |
| Species | Ion | Total | Charge | Total | Charge | Total | Charge |
|---|---|---|---|---|---|---|---|
| Compressive 9% | Strain Free | Tensile 9% | |||||
| C | 1 | 4.70 | −0.70 | 4.75 | −0.75 | 4.78 | −0.78 |
| C | 2 | 4.69 | −0.69 | 4.71 | −0.71 | 4.71 | −0.71 |
| S | 1 | 5.94 | 0.06 | 5.98 | 0.02 | 6.05 | −0.05 |
| S | 2 | 6.01 | −0.01 | 6.06 | −0.06 | 6.11 | −0.11 |
| Ti | 1 | 11.24 | 0.76 | 11.25 | 0.75 | 11.34 | 0.66 |
| Ti | 2 | 11.62 | 0.38 | 11.59 | 0.41 | 11.57 | 0.43 |
| Ti | 3 | 11.72 | 0.28 | 11.63 | 0.37 | 11.49 | 0.51 |
| Mo | 1 | 14.08 | −0.08 | 14.03 | −0.03 | 13.95 | 0.05 |
| Species | Ion | Total | Charge | Total | Charge | Total | Charge |
|---|---|---|---|---|---|---|---|
| Compressive 9% | Strain Free | Tensile 9% | |||||
| C | 1 | 4.69 | −0.69 | 4.71 | −0.71 | 4.71 | −0.71 |
| C | 2 | 4.70 | −0.70 | 4.75 | −0.75 | 4.78 | −0.78 |
| Se | 1 | 5.87 | 0.13 | 5.86 | 0.14 | 5.90 | 0.10 |
| Se | 2 | 5.68 | 0.32 | 5.77 | 0.23 | 5.74 | 0.26 |
| Ti | 1 | 11.27 | 0.73 | 11.27 | 0.73 | 11.36 | 0.64 |
| Ti | 2 | 11.71 | 0.29 | 11.62 | 0.38 | 11.49 | 0.51 |
| Ti | 3 | 11.71 | 0.29 | 11.67 | 0.33 | 11.69 | 0.31 |
| Mo | 1 | 14.37 | −0.37 | 14.35 | −0.35 | 14.33 | −0.33 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, S.; Li, C.; Wang, C.; Ma, D.; Wang, B. The Combined Effects of an External Field and Novel Functional Groups on the Structural and Electronic Properties of TMDs/Ti3C2 Heterostructures: A First-Principles Study. Nanomaterials 2023, 13, 1218. https://doi.org/10.3390/nano13071218
Zheng S, Li C, Wang C, Ma D, Wang B. The Combined Effects of an External Field and Novel Functional Groups on the Structural and Electronic Properties of TMDs/Ti3C2 Heterostructures: A First-Principles Study. Nanomaterials. 2023; 13(7):1218. https://doi.org/10.3390/nano13071218
Chicago/Turabian StyleZheng, Siyu, Chenliang Li, Chaoying Wang, Decai Ma, and Baolai Wang. 2023. "The Combined Effects of an External Field and Novel Functional Groups on the Structural and Electronic Properties of TMDs/Ti3C2 Heterostructures: A First-Principles Study" Nanomaterials 13, no. 7: 1218. https://doi.org/10.3390/nano13071218