
A DFT Study of Ruthenium fcc Nano-Dots: Size-Dependent Induced Magnetic Moments

Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
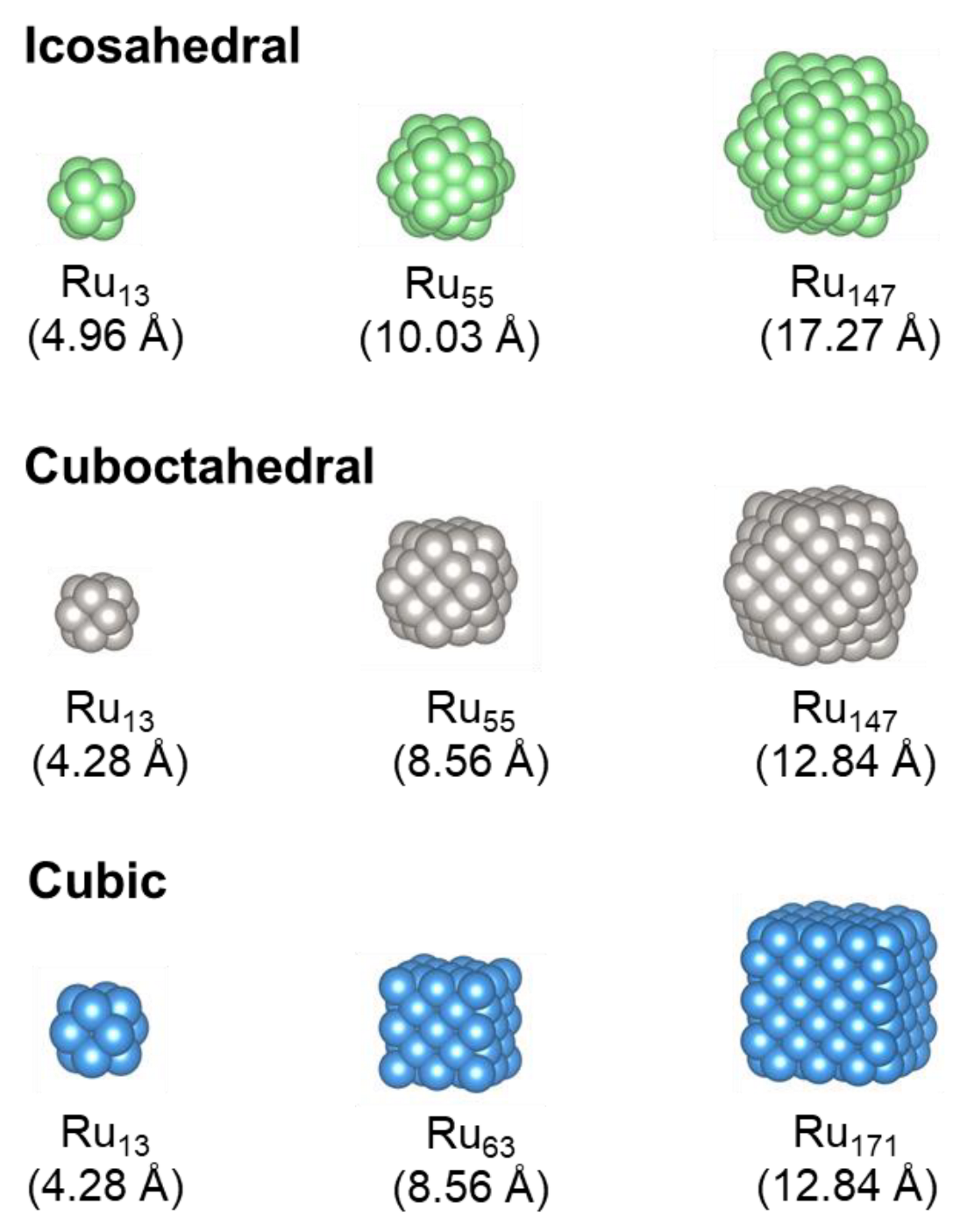
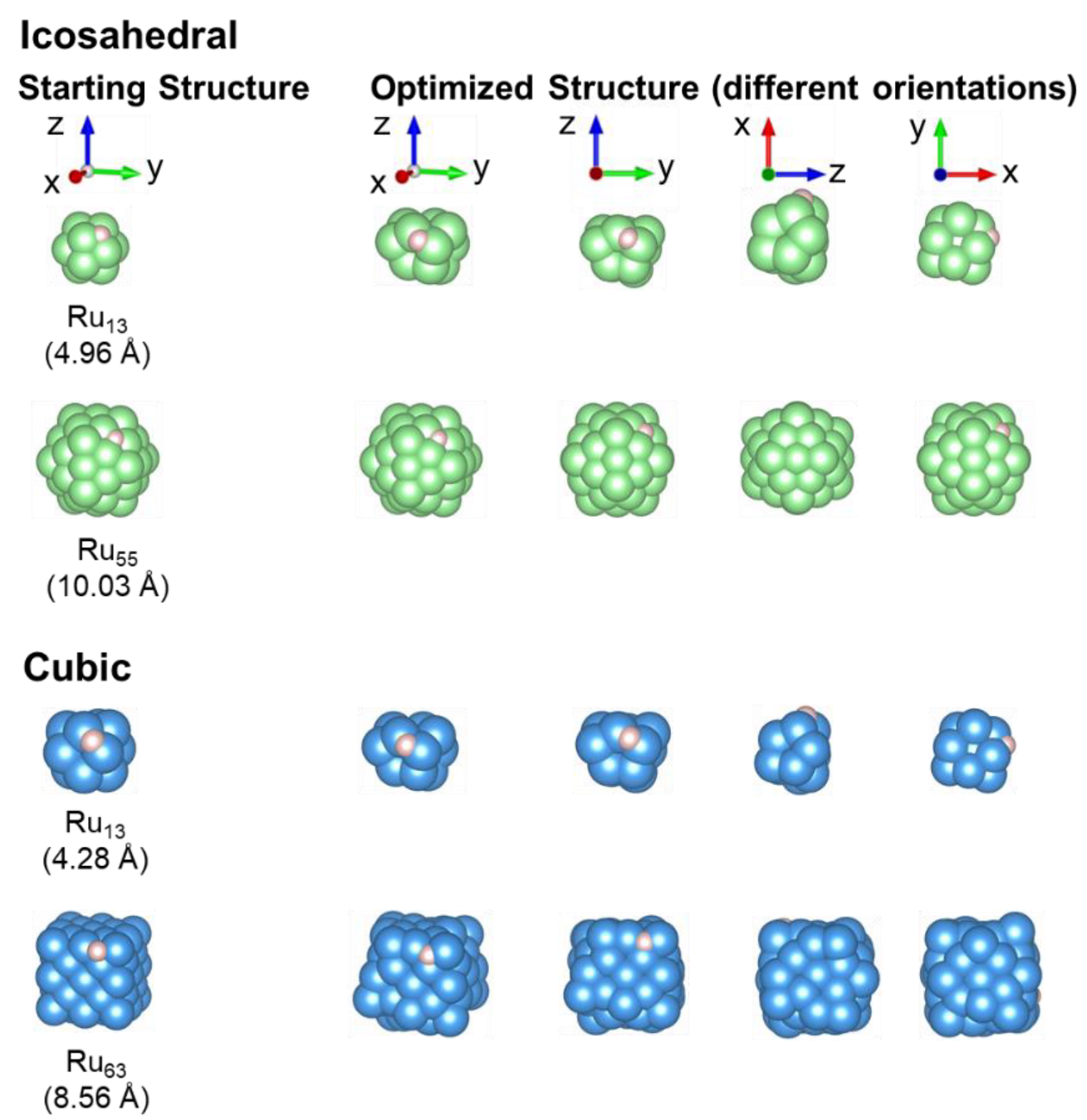
3.1. Ruthenium Nano-Dot Geometry
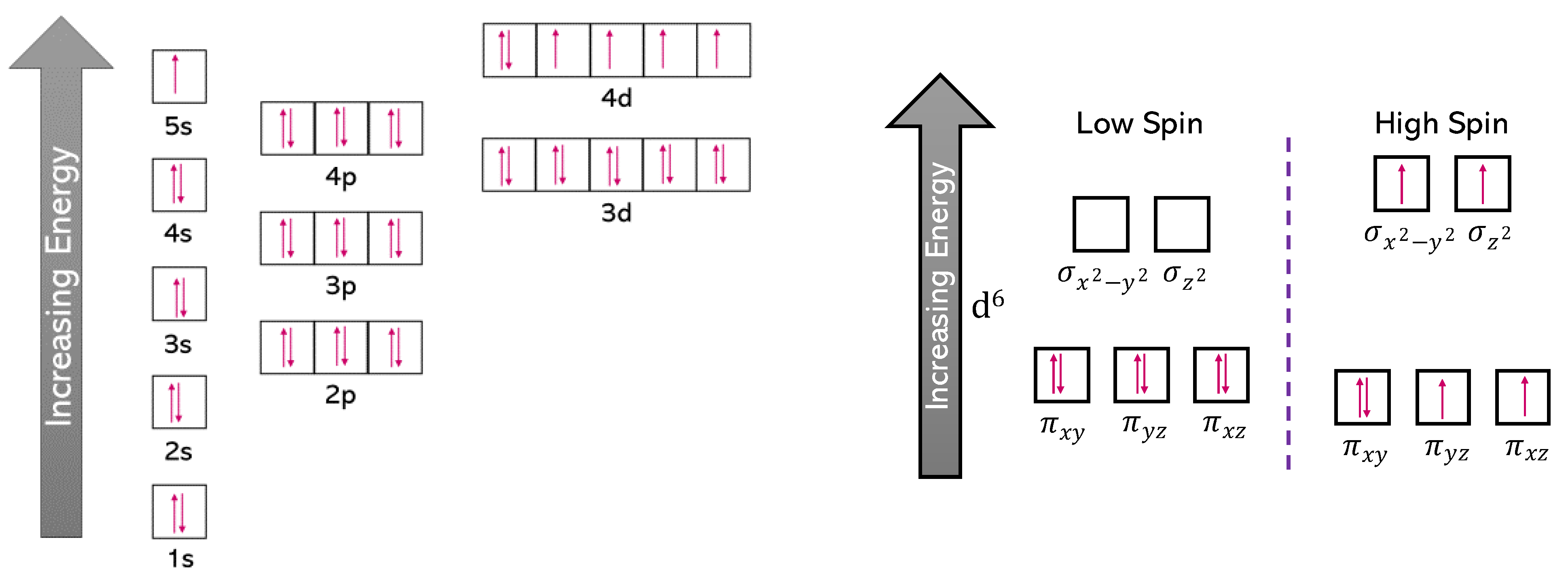
3.1.1. Spin-Polarised Data
3.1.2. Non-Spin-Polarised Data
3.2. Hydrogen Adsorption
3.3. Gaussian Correlation
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Park, Y.R.; Jeong, H.Y.; Seo, Y.S.; Choi, W.K.; Hong, Y.J. Quantum-Dot Light-Emitting Diodes with Nitrogen-Doped Carbon Nanodot Hole Transport and Electronic Energy Transfer Layer. Sci. Rep. 2017, 7, 46422. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Tang, S.; Wu, X.; Boulanger, N.; Gracia-Espino, E.; Wågberg, T.; Edman, L.; Wang, J. Carbon Nanodots: A Metal-Free, Easy-to-Synthesize, and Benign Emitter for Light-Emitting Electrochemical Cells. Nano. Res. 2022, 15, 5610–5618. [Google Scholar] [CrossRef]
- Galisteo-López, J.F.; Lozano, G. Nanophotonics for Current and Future White Light-Emitting Devices. J. Appl. Phys. 2021, 130, 200901. [Google Scholar] [CrossRef]
- Hong, A.J.; Liu, C.-C.; Wang, Y.; Kim, J.; Xiu, F.; Ji, S.; Zou, J.; Nealey, P.F.; Wang, K.L. Metal Nanodot Memory by Self-Assembled Block Copolymer Lift-Off. Nano. Lett. 2010, 10, 224–229. [Google Scholar] [CrossRef]
- Miyazaki, S.; Morisama, N.; Nakanishi, S.; Ikeda, M.; Makihara, K. Charge Storage and Optical Response of Hybrid Nanodots Floating Gate for Functional Memories. MRS Proc. 2009, 1208, 1205. [Google Scholar] [CrossRef]
- Sudsom, D.; Ehrmann, A. Micromagnetic Simulations of Fe and Ni Nanodot Arrays Surrounded by Magnetic or Non-Magnetic Matrices. Nanomaterials 2021, 11, 349. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Niu, Y.; Li, J.; Yang, Z.; Gao, J.; Ding, L.; Ni, H.; Zhu, P.; Liu, Y.; Tang, Y.; et al. Recent Progress of Quantum Dots for Energy Storage Applications. Carbon Neutrality 2022, 1, 13. [Google Scholar] [CrossRef]
- Ansari, S.A. Graphene Quantum Dots: Novel Properties and Their Applications for Energy Storage Devices. Nanomaterials 2022, 12, 3814. [Google Scholar] [CrossRef]
- Mu, S.; Liu, Q.; Kidkhunthod, P.; Zhou, X.; Wang, W.; Tang, Y. Molecular Grafting towards High-Fraction Active Nanodots Implanted in N-Doped Carbon for Sodium Dual-Ion Batteries. Natl. Sci. Rev. 2020, 8, nwaa178. [Google Scholar] [CrossRef]
- Natterer, F.D.; Yang, K.; Paul, W.; Willke, P.; Choi, T.; Greber, T.; Heinrich, A.J.; Lutz, C.P. Reading and Writing Single-Atom Magnets. Nature 2017, 543, 226–228. [Google Scholar] [CrossRef] [Green Version]
- Oviedo-Casado, S.; Urbina, A.; Prior, J. Magnetic Field Enhancement of Organic Photovoltaic Cells Performance. Sci. Rep. 2017, 7, 4297. [Google Scholar] [CrossRef] [Green Version]
- Sani, E.; Martina, M.; Salez, T.; Nakamae, S.; Dubois, E.; Peyre, V. Multifunctional Magnetic Nanocolloids for Hybrid Solar-Thermoelectric Energy Harvesting. Nanomaterials 2021, 11, 1031. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.; Youplao, P.; Pornsuwancharoen, N.; Jalil, M.A.; Chiangga, S.; Amiri, I.S.; Punthawanunt, S.; Aziz, M.S.; Singh, G.; Yupapin, P.; et al. Nano-Capacitor-like Model Using Light Trapping in Plasmonic Island Embedded Microring System. Results Phys. 2018, 10, 727–730. [Google Scholar] [CrossRef]
- Wang, K.; Pei, P.; Zuo, Y.; Wei, M.; Wang, H.; Zhang, P.; Chen, Z.; Shang, N. Magnetic Zinc-Air Batteries for Storing Wind and Solar Energy. iScience 2022, 25, 103837. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.M.; Merazzo, K.J.; Gonçalves, R.; Amos, C.; Lanceros-Méndez, S. Magnetically Active Lithium-Ion Batteries towards Battery Performance Improvement. iScience 2021, 24, 102691. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.A.; Peng, S.; Cheng, K.; Sun, S. Magnetic Nanoparticles: Synthesis, Functionalization, and Applications in Bioimaging and Magnetic Energy Storage. Chem. Soc. Rev. 2009, 38, 2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Spek, M.; Banet, C.; Bauer, C.; Gabrielli, P.; Goldthorpe, W.; Mazzotti, M.; Munkejord, S.T.; Røkke, N.A.; Shah, N.; Sunny, N.; et al. Perspective on the Hydrogen Economy as a Pathway to Reach Net-Zero CO 2 Emissions in Europe. Energy Environ. Sci. 2022, 15, 1034–1077. [Google Scholar] [CrossRef]
- Ge, Z.; Fu, B.; Zhao, J.; Li, X.; Ma, B.; Chen, Y. A Review of the Electrocatalysts on Hydrogen Evolution Reaction with an Emphasis on Fe, Co and Ni-Based Phosphides. J. Mater. Sci. 2020, 55, 14081–14104. [Google Scholar] [CrossRef]
- Bhalothia, D.; Krishnia, L.; Yang, S.-S.; Yan, C.; Hsiung, W.-H.; Wang, K.-W.; Chen, T.-Y. Recent Advancements and Future Prospects of Noble Metal-Based Heterogeneous Nanocatalysts for Oxygen Reduction and Hydrogen Evolution Reactions. Appl. Sci. 2020, 10, 7708. [Google Scholar] [CrossRef]
- Zaman, N.; Noor, T.; Iqbal, N. Recent Advances in the Metal-Organic Framework-Based Electrocatalysts for the Hydrogen Evolution Reaction in Water Splitting: A Review. RSC Adv. 2021, 11, 21904–21925. [Google Scholar] [CrossRef]
- Wang, J.; Xu, F.; Jin, H.; Chen, Y.; Wang, Y. Non-Noble Metal-Based Carbon Composites in Hydrogen Evolution Reaction: Fundamentals to Applications. Adv. Mater. 2017, 29, 1605838. [Google Scholar] [CrossRef]
- Cai, J.; Javed, R.; Ye, D.; Zhao, H.; Zhang, J. Recent Progress in Noble Metal Nanocluster and Single Atom Electrocatalysts for the Hydrogen Evolution Reaction. J. Mater. Chem. A Mater. 2020, 8, 22467–22487. [Google Scholar] [CrossRef]
- Lu, B.; Guo, L.; Wu, F.; Peng, Y.; Lu, J.E.; Smart, T.J.; Wang, N.; Finfrock, Y.Z.; Morris, D.; Zhang, P.; et al. Ruthenium Atomically Dispersed in Carbon Outperforms Platinum toward Hydrogen Evolution in Alkaline Media. Nat. Commun. 2019, 10, 631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Wei, Z.; Wang, B.; Liu, Y.; Song, H.; Tang, Z.; Yang, B.; Lu, S. Carbon Quantum Dots Enhanced the Activity for the Hydrogen Evolution Reaction in Ruthenium-Based Electrocatalysts. Mater. Chem. Front. 2020, 4, 277–284. [Google Scholar] [CrossRef]
- Xu, J.; Zhong, W.; Gao, D.; Wang, X.; Wang, P.; Yu, H. Phosphorus-Enriched Platinum Diphosphide Nanodots as a Highly Efficient Cocatalyst for Photocatalytic H2 Evolution of CdS. Chem. Eng. J. 2022, 439, 135758. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Niu, H.-J.; Ma, X.; Feng, J.-J.; Weng, X.; Huang, H.; Wang, A.-J. Flower-like Platinum-Cobalt-Ruthenium Alloy Nanoassemblies as Robust and Highly Efficient Electrocatalyst for Hydrogen Evolution Reaction. J. Colloid. Interface Sci. 2020, 561, 372–378. [Google Scholar] [CrossRef]
- Kusada, K.; Kobayashi, H.; Yamamoto, T.; Matsumura, S.; Sumi, N.; Sato, K.; Nagaoka, K.; Kubota, Y.; Kitagawa, H. Discovery of Face-Centered-Cubic Ruthenium Nanoparticles: Facile Size-Controlled Synthesis Using the Chemical Reduction Method. J. Am. Chem. Soc. 2013, 135, 5493–5496. [Google Scholar] [CrossRef]
- Zhao, M.; Xia, Y. Crystal-Phase and Surface-Structure Engineering of Ruthenium Nanocrystals. Nat. Rev. Mater. 2020, 5, 440–459. [Google Scholar] [CrossRef]
- Rakhtsaum, G. Platinum Alloys: A Selective Review of the Available Literature. Platin. Met. Rev. 2013, 57, 202–213. [Google Scholar] [CrossRef]
- Darolia, R. Development of Strong, Oxidation and Corrosion Resistant Nickel-Based Superalloys: Critical Review of Challenges, Progress and Prospects. Int. Mater. Rev. 2019, 64, 355–380. [Google Scholar] [CrossRef]
- Ungerer, M.J.; de Leeuw, N.H. Computational Insights into Ru, Pd and Pt Fcc Nano-Catalysts from Density Functional Theory Calculations: The Influence of Long-Range Dispersion Corrections. Catalysts 2022, 12, 1287. [Google Scholar] [CrossRef]
- Bhattarai, H.; Drisko, C.; Duraes, A.D.S.; Lin, T.; Vardeman, C.F., II; Christopher, J.; Fennell, M.A.M.; Louden, P.; Neidhart, S.; Kuang, S.; et al. OpenMD: Molecular Dynamibs in the Open, 2019. Available online: http://OpenMD.org (accessed on 25 February 2021).
- Sutton, A.P.; Chen, J. Long-Range Finnis-Sinclair Potentials. Philos. Mag. Lett. 1990, 61, 139–146. [Google Scholar] [CrossRef]
- Prasai, B.; Ren, Y.; Shan, S.; Zhao, Y.; Cronk, H.; Luo, J.; Zhong, C.-J.; Petkov, V. Synthesis-Atomic Structure-Properties Relationships in Metallic Nanoparticles by Total Scattering Experiments and 3D Computer Simulations: Case of Pt-Ru Nanoalloy Catalysts. Nanoscale 2015, 7, 8122–8134. [Google Scholar] [CrossRef] [PubMed]
- Prasai, B.; Ren, Y.; Shan, S.; Zhao, Y.; Cronk, H.; Luo, J.; Zhong, C.-J.; Petkov, V. Correction: Synthesis-Atomic Structure-Properties Relationships in Metallic Nanoparticles by Total Scattering Experiments and 3D Computer Simulations: Case of Pt-Ru Nanoalloy Catalysts. Nanoscale 2015, 7, 10279. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular-Dynamics Simulation of the Liquid-Metalamorphous-Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple- ERRATA. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High-Precision Sampling for Brillouin-Zone Integration in Metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zon Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved Tetrahedron Method for Brillouin-Zone Integrations. Phys. Rev. B 1994, 49, 16223–16233. [Google Scholar] [CrossRef]
- Huang, B.; Kobayashi, H.; Yamamoto, T.; Toriyama, T.; Matsumura, S.; Nishida, Y.; Sato, K.; Nagaoka, K.; Haneda, M.; Xie, W.; et al. A CO Adsorption Site Change Induced by Copper Substitution in a Ruthenium Catalyst for Enhanced CO Oxidation Activity. Angew. Chem.—Int. Ed. 2019, 58, 2230–2235. [Google Scholar] [CrossRef]
- Song, C.; Sakata, O.; Kumara, L.S.R.; Kohara, S.; Yang, A.; Kusada, K.; Kobayashi, H.; Kitagawa, H. Size Dependence of Structural Parameters in Fcc and Hcp Ru Nanoparticles, Revealed by Rietveld Refinement Analysis of High-Energy X-ray Diffraction Data. Sci. Rep. 2016, 6, 31400. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, V.B.G.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision. D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XXIII. A Polarization-Type Basis Set for Second-Row Elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef] [Green Version]
- Farkaš, B.; de Leeuw, N.H. Towards a Morphology of Cobalt Nanoparticles: Size and Strain Effects. Nanotechnology 2020, 31, 195711. [Google Scholar] [CrossRef] [PubMed]
- Ungerer, M.J.; Santos-Carballal, D.; Cadi-Essadek, A.; van Sittert, C.G.C.E.; de Leeuw, N.H. Interaction of H2O with the Platinum Pt (001), (011), and (111) Surfaces: A Density Functional Theory Study with Long-Range Dispersion Corrections. J. Phys. Chem. C 2019, 123, 27465–27476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungerer, M.J.; Santos-Carballal, D.; Cadi-Essadek, A.; van Sittert, C.G.C.E.; de Leeuw, N.H. Interaction of SO2 with the Platinum (001), (011), and (111) Surfaces: A DFT Study. Catalysts 2020, 10, 558. [Google Scholar] [CrossRef]
- Ungerer, M.J.; Santos-Carballal, D.; van Sittert, C.G.C.E.; de Leeuw, N.H. Competitive Adsorption of H2O and SO2 on Catalytic Platinum Surfaces: A Density Functional Theory Study. S. Afr. J. Chem. 2021, 74, 57–68. [Google Scholar] [CrossRef]
- Ungerer, M.J.; van Sittert, C.G.C.E.; de Leeuw, N.H. Behavior of S, SO, and SO 3 on Pt (001), (011), and (111) Surfaces: A DFT Study. J. Chem. Phys. 2021, 154, 194701. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A Fast and Robust Algorithm for Bader Decomposition of Charge Density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Sanville, E.; Kenny, S.D.; Smith, R.; Henkelman, G. Improved Grid-Based Algorithm for Bader Charge Allocation. J. Comput. Chem. 2007, 28, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Sanville, E.; Henkelman, G. A Grid-Based Bader Analysis Algorithm without Lattice Bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Trinkle, D.R. Accurate and Efficient Algorithm for Bader Charge Integration. J. Chem. Phys. 2011, 134, 064111. [Google Scholar] [CrossRef] [Green Version]
- Momma, K.; Izumi, F. VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Mackay, A.L. A Dense Non-Crystallographic Packing of Equal Spheres. Acta Crystallogr. 1962, 15, 916–918. [Google Scholar] [CrossRef]
- Coxeter, H.S.M. 2.3 Quasi-Regular Polyhedra. In Regular Polytopes; Dover Publications: Dover, UK, 1973; pp. 18–19. ISBN 0-486-61480-8. [Google Scholar]
- Hafner, J. Ab-Initio Simulations of Materials Using VASP: Density-Functional Theory and Beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef]
- Cann, P. Ionization Energies, Parallel Spins, and the Stability of Half-Filled Shells. J. Chem. Educ. 2000, 77, 1056. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A Materials Genome Approach to Accelerating Materials Innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef] [Green Version]
- Ge, G.-X.; Yan, H.-X.; Jing, Q.; Luo, Y.-H. Theoretical Study of Hydrogen Adsorption on Ruthenium Clusters. J. Clust. Sci. 2011, 22, 473–489. [Google Scholar] [CrossRef] [Green Version]
- Andzelm, J.; Radzio, E.; Salahub, D.R. Model Potential Calculations for Second-row Transition Metal Molecules within the Local-spin-density Method. J. Chem. Phys. 1985, 83, 4573–4580. [Google Scholar] [CrossRef]
- Chase, M. NIST-JANAF Thermochemical Tables, 4th ed.; Vol. Monograph; American Institute of Physics: College Park, MD, USA, 1998; ISBN 1563968312. [Google Scholar]
- Onwudinanti, C.; Brocks, G.; Koelman, V.; Morgan, T.; Tao, S. Hydrogen Diffusion out of Ruthenium—An Ab Initio Study of the Role of Adsorbates. Phys. Chem. Chem. Phys. 2020, 22, 7935–7941. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| DFT-D3 | DFT-D3(BJ) | |||||
|---|---|---|---|---|---|---|
| E0 (eV) | Ecoh (eV) | μB | E0 (eV) | Ecoh (eV) | μB | |
| Icosahedral | ||||||
| Ru13 (4.96 Å) | −87.670 | 2.893 | 1.60 | −88.307 | 2.948 | 0.94 |
| Ru55 (10.03 Å) | −435.576 | 1.718 | 0.28 | −439.517 | 1.750 | 0.28 |
| Ru147 (17.27 Å) | −1241.226 | 1.193 | 0.00 | −1251.887 | 1.225 | 0.00 |
| Cuboctahedral | ||||||
| Ru13 (4.28 Å) | −86.745 | 2.964 | 1.39 | −89.956 | 2.821 | 0.31 |
| Ru55 (8.56 Å) | −434.176 | 1.743 | 0.00 | −438.140 | 1.775 | 0.00 |
| Ru147 (12.84 Å) | −1227.905 | 1.284 | 0.00 | −1238.298 | 1.317 | 0.00 |
| Cubic | ||||||
| Ru13 (4.28 Å) | −86.745 | 2.964 | 1.39 | −90.268 | 2.797 | 0.32 |
| Ru63 (8.56 Å) | −491.376 | 1.837 | 0.15 | −502.370 | 1.767 | 0.03 |
| Ru171 (12.84 Å) | −1423.041 | 1.315 | 0.05 | −1435.418 | 1.347 | 0.04 |
| Icosahedral | Cuboctahedral/Cubic | |||
|---|---|---|---|---|
| Atom # | DFT-D3 | DFT-D3(BJ) | DFT-D3 | DFT-D3(BJ) |
| 1 (core) | 0.269 | 0.404 | 0.290 | 0.267 |
| 2 | −0.055 | −0.011 | 0.040 | −0.014 |
| 3 | 0.010 | −0.066 | −0.152 | −0.030 |
| 4 | −0.055 | −0.060 | 0.102 | 0.008 |
| 5 | 0.010 | −0.017 | −0.023 | −0.047 |
| 6 | 0.010 | 0.013 | 0.102 | −0.019 |
| 7 | −0.055 | −0.064 | −0.151 | −0.002 |
| 8 | 0.010 | −0.061 | −0.089 | −0.084 |
| 9 | 0.010 | 0.012 | −0.087 | −0.036 |
| 10 | −0.055 | −0.072 | −0.023 | −0.065 |
| 11 | −0.055 | −0.063 | 0.040 | −0.018 |
| 12 | 0.010 | 0.002 | 0.102 | −0.051 |
| 13 | −0.055 | −0.017 | −0.152 | 0.089 |
| DFT-D3 | DFT-D3(BJ) | |||
|---|---|---|---|---|
| E0 (eV) | ∆Ediff (eV) | E0 (eV) | ∆Ediff (eV) | |
| Icosahedral | ||||
| Ru13 (4.96 Å) | −86.38 | 1.29 | −87.95 | 0.35 |
| ∆e↑↓ = 0 | −87.73 | −0.06 | −87.94 | 0.36 |
| Ru55 (10.03 Å) | −435.62 | −0.04 | −439.09 | 0.42 |
| Cubic | ||||
| Ru13 (4.28 Å) | −86.64 | 0.10 | −90.16 | 0.11 |
| ∆e↑↓ = 0 | −88.25 | −1.50 | −90.27 | 0.00 |
| Ru63 (8.56 Å) | −491.46 | −0.09 | −503.33 | −0.96 |
| DFT-D3 | DFT-D3(BJ) | ||||||
|---|---|---|---|---|---|---|---|
| E0 (eV) | Eads (eV) | μB,tot | E0 (eV) | Eads (eV) | μB,tot | ||
| Icosahedral | |||||||
| Ru13 + 1H | SP | −91.90 | −0.58 | 10.32 | −92.89 | −1.17 | 10.69 |
| NSP | −91.21 | −0.13 | 0.00 | −92.32 | −0.61 | 0.00 | |
| μi = +1 | −91.56 | −0.48 | 9.81 | −92.01 | −0.30 | 0.05 | |
| ∆e↑↓ = 0 | −91.20 | −0.13 | 0.00 | −92.02 | −0.31 | 0.00 | |
| ∆e↑↓ = 1 | −91.22 | −0.15 | 1.00 | −92.05 | −0.34 | 1.00 | |
| Ru55 + 1H | SP | −439.80 | −0.82 | 15.29 | −443.78 | −0.86 | 15.33 |
| NSP | −439.37 | −0.39 | 0.00 | −443.35 | −0.42 | 0.00 | |
| Cubic | |||||||
| Ru13 + 1H | SP | −91.36 | −1.21 | 10.79 | −94.29 | -0.61 | 6.01 |
| NSP | −92.75 | −2.60 | 0.00 | −94.51 | −0.84 | 0.00 | |
| ∆e↑↓ = 0 | −92.87 | −2.72 | 0.00 | −93.77 | −0.10 | 0.00 | |
| ∆e↑↓ = 1 | −92.13 | −1.98 | 1.00 | −93.86 | −0.18 | 1.00 | |
| Ru63 + 1H | SP | −502.22 | −7.44 | 1.47 | −510.54 | −4.77 | 0.00 |
| NSP | −503.38 | −8.60 | 0.00 | −510.60 | −4.83 | 0.00 | |
| Low Spin | High Spin | ||||
|---|---|---|---|---|---|
| E0 (eV) | Eads (eV) | E0 (eV) | Eads (eV) | ∆Ediff (eV) | |
| 1H | −13.60 | ||||
| H2 | −31.39 | −4.19 | |||
| Icosahedral | −33,219.29 | −33,219.97 | −0.68 | ||
| Ru13 + ½ H2 | −33,235.60 | −0.62 | −33,236.16 | −0.49 | −0.55 |
| Cubic | −33,220.46 | −33,219.51 | 0.95 | ||
| Ru13 + ½ H2 | −33,236.82 | −0.66 | −33,234.88 | 0.33 | −1.94 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ungerer, M.J.; de Leeuw, N.H. A DFT Study of Ruthenium fcc Nano-Dots: Size-Dependent Induced Magnetic Moments. Nanomaterials 2023, 13, 1118. https://doi.org/10.3390/nano13061118
Ungerer MJ, de Leeuw NH. A DFT Study of Ruthenium fcc Nano-Dots: Size-Dependent Induced Magnetic Moments. Nanomaterials. 2023; 13(6):1118. https://doi.org/10.3390/nano13061118
Chicago/Turabian StyleUngerer, Marietjie J., and Nora H. de Leeuw. 2023. "A DFT Study of Ruthenium fcc Nano-Dots: Size-Dependent Induced Magnetic Moments" Nanomaterials 13, no. 6: 1118. https://doi.org/10.3390/nano13061118