
Exploring the Role of Miniemulsion Nanodroplet Confinement on the Crystallization of MoO3: Morphology Control and Insight on Crystal Formation by In Situ Time-Resolved SAXS/WAXS


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Miniemulsion (ME) Approach
2.3. Batch Approach
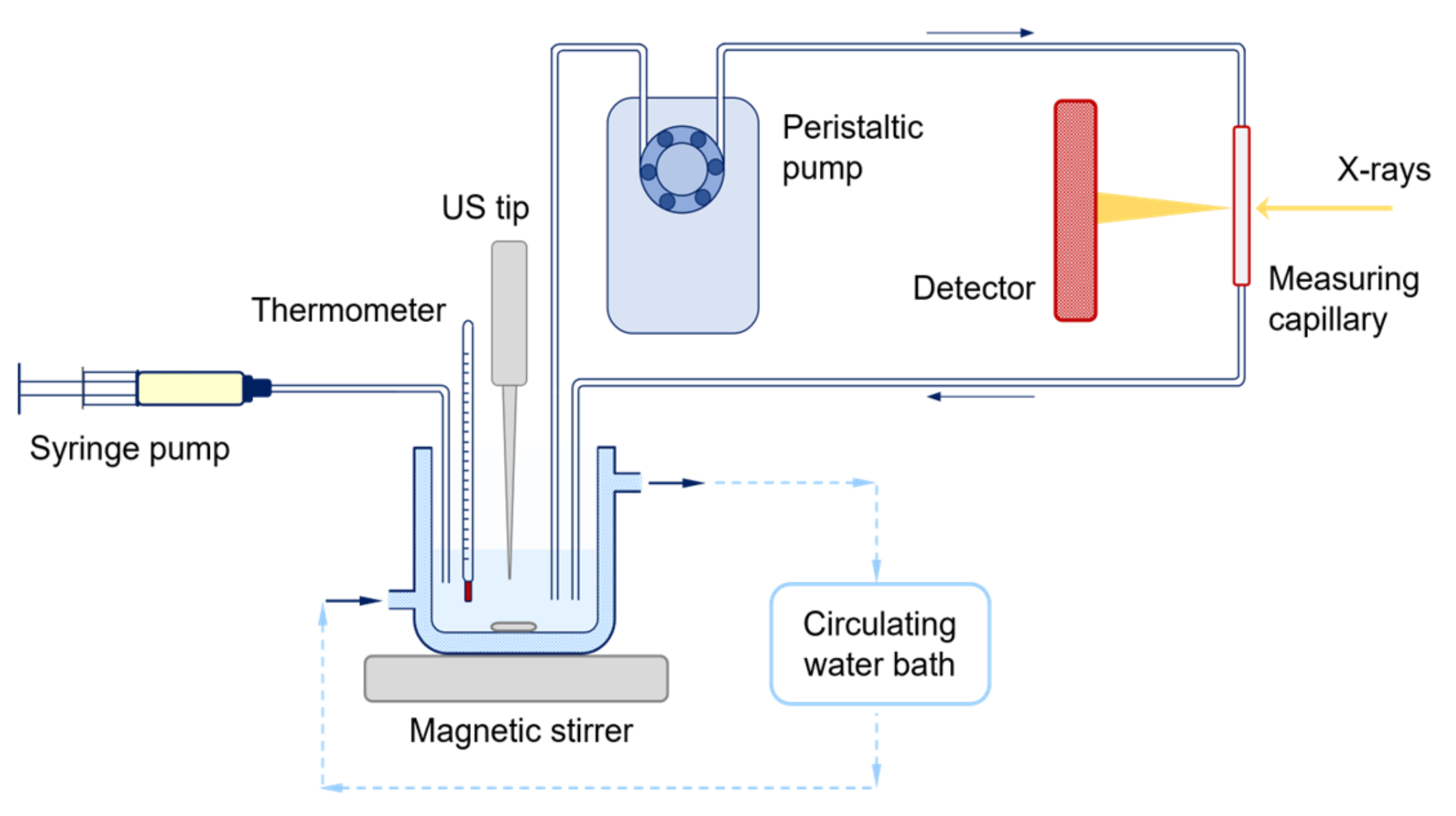
2.4. Experimental Setup for the In Situ Time-Resolved SAXS/WAXS Study
2.5. Characterization Methods
3. Results and Discussion
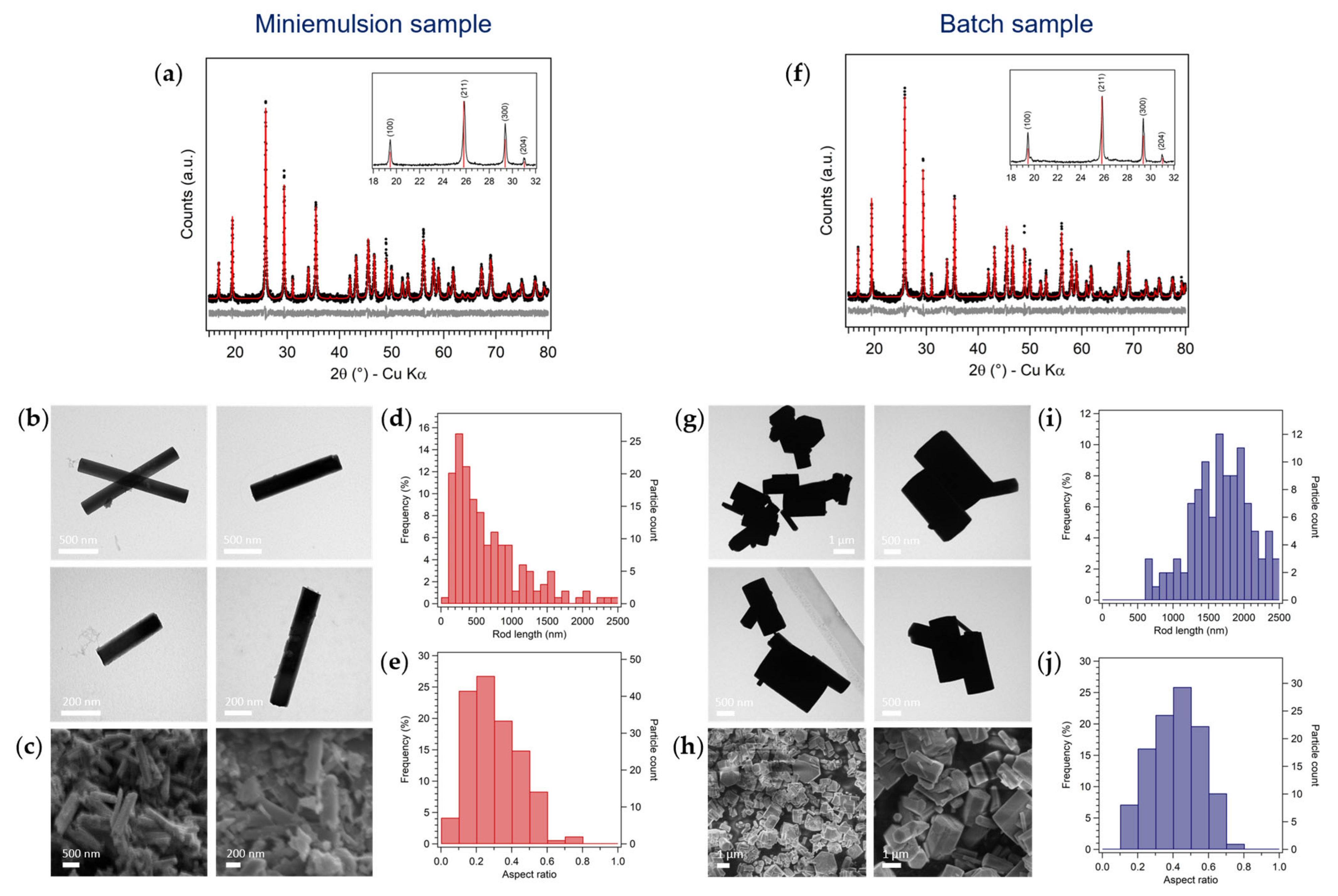
3.1. Synthesis of Hexagonal MoO3 by Inverse Miniemulsion and Batch Approaches
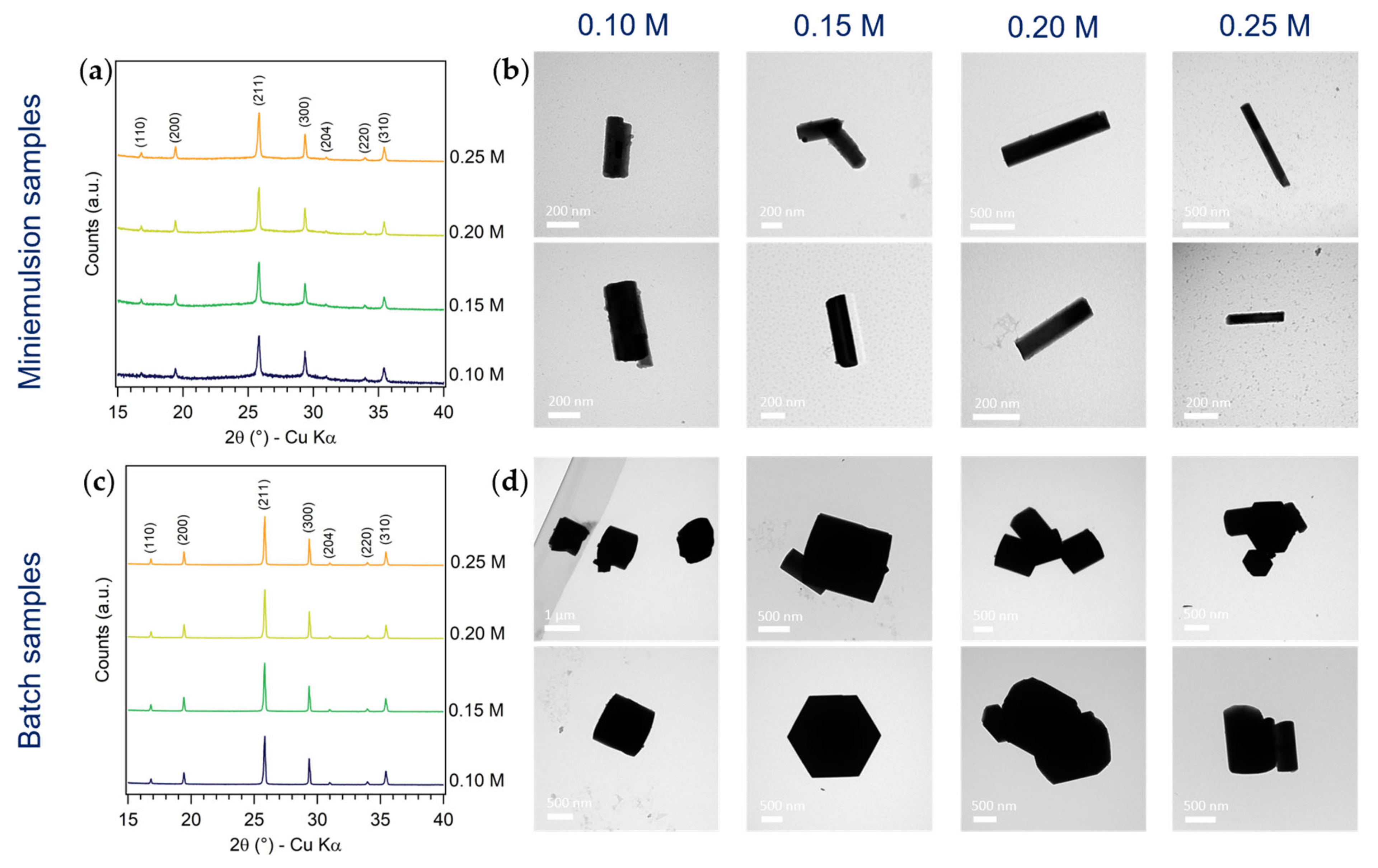
3.1.1. Effect of AHM Concentration
3.1.2. Effect of AHM:HNO3 Molar Ratio
3.1.3. Effect of Ultrasounds on HNO3 Diffusion into Miniemulsion Droplets
3.1.4. Effect of Reaction Time
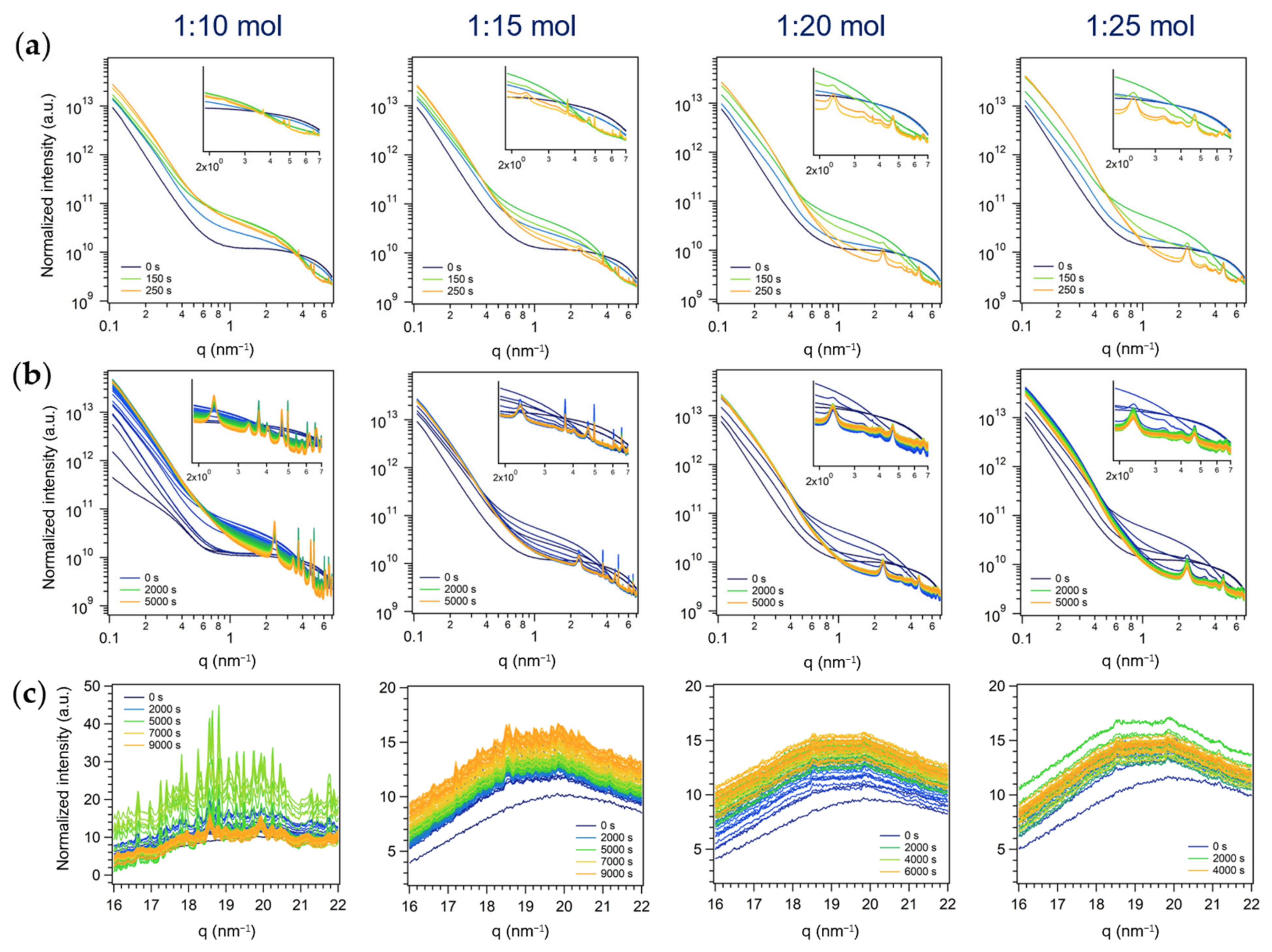
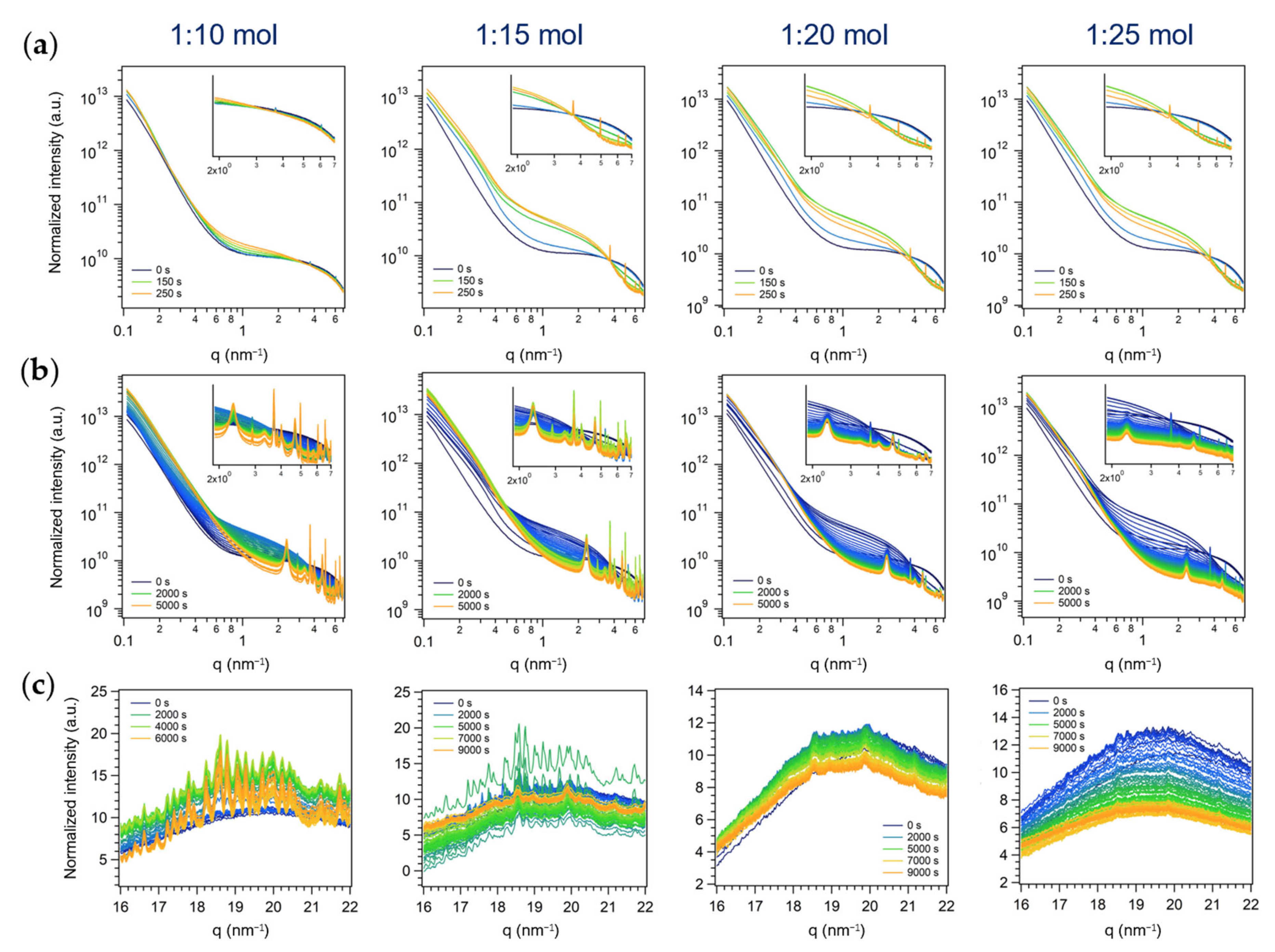
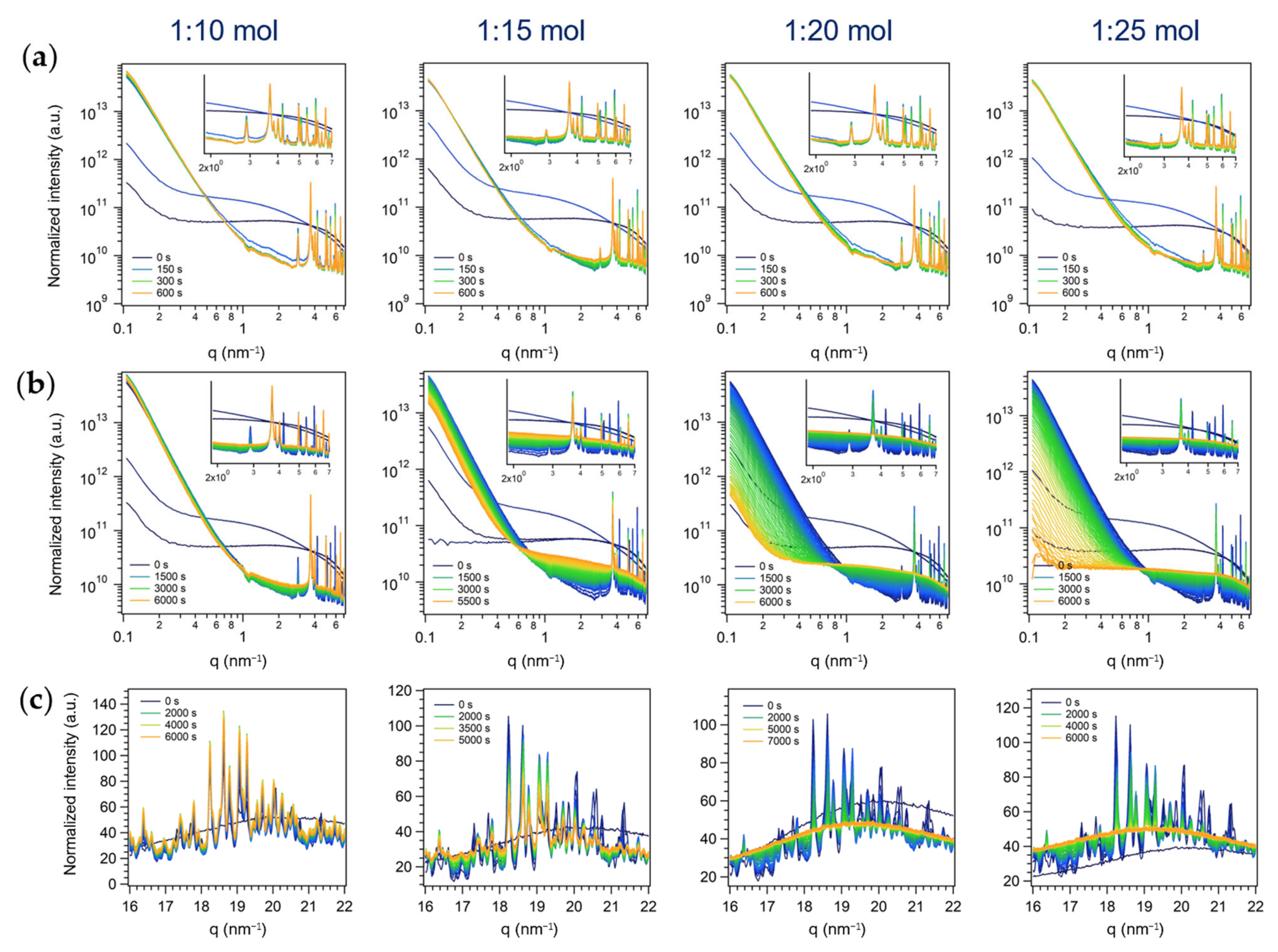
3.2. Time-Resolved in Situ SAXS/WAXS Study of MoO3 Crystallization by Inverse Miniemulsion and Batch Approaches
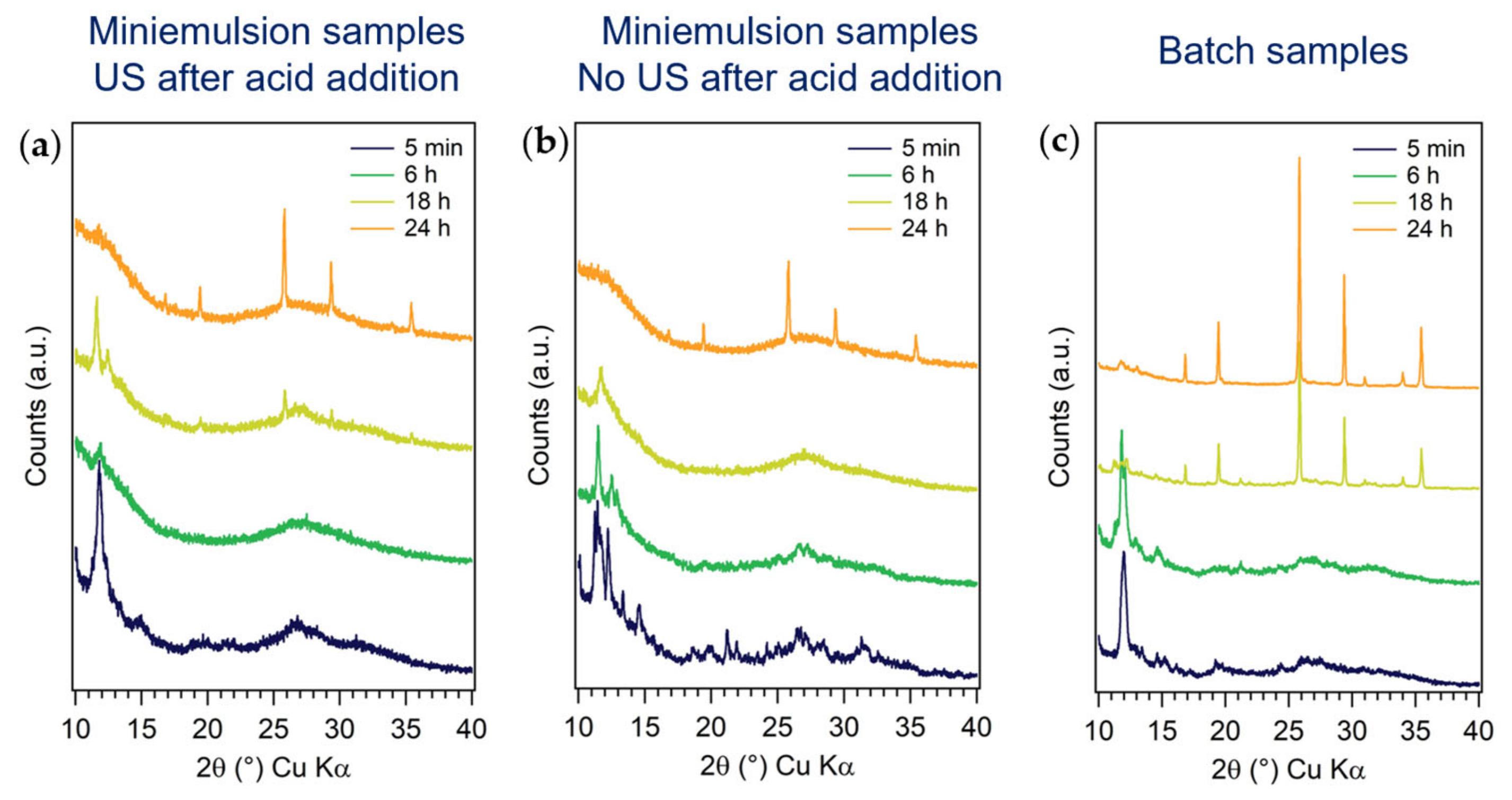
3.2.1. First Series: Miniemulsion Syntheses
3.2.2. Second Series: Miniemulsion Syntheses without US Step after Acid Addition
3.2.3. Third Series: Batch Syntheses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Christenson, H.K. Confinement Effects on Freezing and Melting. J. Phys. Condens. Matter 2001, 13, R95–R133. [Google Scholar] [CrossRef]
- Miners, S.A.; Rance, G.A.; Khlobystov, A.N. Chemical Reactions Confined within Carbon Nanotubes. Chem. Soc. Rev. 2016, 45, 4727–4746. [Google Scholar] [CrossRef] [PubMed]
- Kohler, F.; Pierre-Louis, O.; Dysthe, D.K. Crystal Growth in Confinement. Nat. Commun. 2022, 13, 6990. [Google Scholar] [CrossRef] [PubMed]
- Landfester, K. Miniemulsions for Nanoparticle Synthesis. In Colloid Chemistry II; Springer: Berlin/Heidelberg, Germany, 2003; pp. 75–123. [Google Scholar]
- Muñoz-Espí, R.; Weiss, C.K.; Landfester, K. Inorganic Nanoparticles Prepared in Miniemulsion. Curr. Opin. Colloid Interface Sci. 2012, 17, 212–224. [Google Scholar] [CrossRef]
- Willert, M.; Rothe, R.; Landfester, K.; Antonietti, M. Synthesis of Inorganic and Metallic Nanoparticles by Miniemulsification of Molten Salts and Metals. Chem. Mater. 2001, 13, 4681–4685. [Google Scholar] [CrossRef]
- Rossmanith, R.; Weiss, C.K.; Geserick, J.; Hüsing, N.; Hörmann, U.; Kaiser, U.; Landfester, K. Porous Anatase Nanoparticles with High Specific Surface Area Prepared by Miniemulsion Technique. Chem. Mater. 2008, 20, 5768–5780. [Google Scholar] [CrossRef]
- Nabih, N.; Schiller, R.; Lieberwirth, I.; Kockrick, E.; Frind, R.; Kaskel, S.; Weiss, C.K.; Landfester, K. Mesoporous CeO2 Nanoparticles Synthesized by an Inverse Miniemulsion Technique and Their Catalytic Properties in Methane Oxidation. Nanotechnology 2011, 22, 135606. [Google Scholar] [CrossRef]
- Varol, H.S.; Álvarez-Bermúdez, O.; Dolcet, P.; Kuerbanjiang, B.; Gross, S.; Landfester, K.; Muñoz-Espí, R. Crystallization at Nanodroplet Interfaces in Emulsion Systems: A Soft-Template Strategy for Preparing Porous and Hollow Nanoparticles. Langmuir 2016, 32, 13116–13123. [Google Scholar] [CrossRef]
- Dolcet, P.; Casarin, M.; Maccato, C.; Bovo, L.; Ischia, G.; Gialanella, S.; Mancin, F.; Tondello, E.; Gross, S. Miniemulsions as Chemical Nanoreactors for the Room Temperature Synthesis of Inorganic Crystalline Nanostructures: ZnO Colloids. J. Mater. Chem. 2012, 22, 1620–1626. [Google Scholar] [CrossRef]
- Dolcet, P.; Latini, F.; Casarin, M.; Speghini, A.; Tondello, E.; Foss, C.; Diodati, S.; Verin, L.; Motta, A.; Gross, S. Inorganic Chemistry in a Nanoreactor: Doped ZnO Nanostructures by Miniemulsion. Eur. J. Inorg. Chem. 2013, 2013, 2291–2300. [Google Scholar] [CrossRef]
- Singh, I.; Landfester, K.; Chandra, A.; Muñoz-Espí, R. A New Approach for Crystallization of Copper(II) Oxide Hollow Nanostructures with Superior Catalytic and Magnetic Response. Nanoscale 2015, 7, 19250–19258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butturini, E.; Dolcet, P.; Casarin, M.; Speghini, A.; Pedroni, M.; Benetti, F.; Motta, A.; Badocco, D.; Pastore, P.; Diodati, S.; et al. Simple, Common but Functional: Biocompatible and Luminescent Rare-Earth Doped Magnesium and Calcium Hydroxides from Miniemulsion. J. Mater. Chem. B 2014, 2, 6639–6651. [Google Scholar] [CrossRef]
- Dolcet, P.; Maurizio, C.; Casarin, M.; Pandolfo, L.; Gialanella, S.; Badocco, D.; Pastore, P.; Speghini, A.; Gross, S. An Effective Two-Emulsion Approach to the Synthesis of Doped ZnS Crystalline Nanostructures. Eur. J. Inorg. Chem. 2015, 2015, 706–714. [Google Scholar] [CrossRef]
- Dolcet, P.; Mambrini, A.; Pedroni, M.; Speghini, A.; Gialanella, S.; Casarin, M.; Gross, S. Room Temperature Crystallization of Highly Luminescent Lanthanide-Doped CaF2 in Nanosized Droplets: First Example of the Synthesis of Metal Halogenide in Miniemulsion with Effective Doping and Size Control. RSC Adv. 2015, 5, 16302–16310. [Google Scholar] [CrossRef]
- Chithambararaj, A.; Sanjini, N.S.; Bose, A.C.; Velmathi, S. Flower-like Hierarchical h-MoO3: New Findings of Efficient Visible Light Driven Nano Photocatalyst for Methylene Blue Degradation. Catal. Sci. Technol. 2013, 3, 1405–1414. [Google Scholar] [CrossRef]
- Chithambararaj, A.; Sanjini, N.S.; Velmathi, S.; Chandra Bose, A. Preparation of h-MoO3 and α-MoO3 Nanocrystals: Comparative Study on Photocatalytic Degradation of Methylene Blue under Visible Light Irradiation. Phys. Chem. Chem. Phys. 2013, 15, 14761–14769. [Google Scholar] [CrossRef]
- Huang, L.; Fang, W.; Yang, Y.; Wu, J.; Yu, H.; Dong, X.; Wang, T.; Liu, Z.; Zhao, B. Three-Dimensional MoO3 Nanoflowers Assembled with Nanosheets for Rhodamine B Degradation under Visible Light. Mater. Res. Bull. 2018, 108, 38–45. [Google Scholar] [CrossRef]
- Jittiarporn, P.; Sikong, L.; Kooptarnond, K.; Taweepreda, W. Effects of Precipitation Temperature on the Photochromic Properties of H-MoO3. Ceram. Int. 2014, 40, 13487–13495. [Google Scholar] [CrossRef]
- Jittiarporn, P.; Sikong, L.; Kooptarnond, K.; Taweepreda, W. Influence of Calcination Temperature on the Structural and Photochromic Properties of Nanocrystalline MoO3. Dig. J. Nanomater. Biostructures 2015, 10, 1237–1248. [Google Scholar]
- Zheng, L.; Xu, Y.; Jin, D.; Xie, Y. Novel Metastable Hexagonal MoO3 Nanobelts: Synthesis, Photochromic, and Electrochromic Properties. Chem. Mater. 2009, 21, 5681–5690. [Google Scholar] [CrossRef]
- Barazzouk, S.; Tandon, R.P.; Hotchandani, S. MoO3-Based Sensor for NO, NO2 and CH4 Detection. Sens. Actuators B Chem. 2006, 119, 691–694. [Google Scholar] [CrossRef]
- Comini, E.; Yubao, L.; Brando, Y.; Sberveglieri, G. Gas Sensing Properties of MoO3 Nanorods to CO and CH3OH. Chem. Phys. Lett. 2005, 407, 368–371. [Google Scholar] [CrossRef]
- Kihlborg, L. Least Squares Refinement of Crystal Structure of Molybdenum Trioxide. Ark. Kemi 1963, 21, 357–364. [Google Scholar]
- McCarron, E.M. β-MoO3: A Metastable Analogue of WO3. J. Chem. Soc. Chem. Commun. 1986, 4, 336–338. [Google Scholar] [CrossRef]
- McCarron, E.M.; Thomas, D.M.; Calabrese, J.C. Hexagonal Molybdates: Crystal Structure of (Na.2H2O)Mo5.22[H4.5]0.67O18. Inorg. Chem. 1987, 26, 370–373. [Google Scholar] [CrossRef]
- Pan, W.; Tian, R.; Jin, H.; Guo, Y.; Zhang, L.; Wu, X.; Zhang, L.; Han, Z.; Liu, G.; Li, J.; et al. Structure, Optical, and Catalytic Properties of Novel Hexagonal Metastable h-MoO3 Nano- and Microrods Synthesized with Modified Liquid-Phase Processes. Chem. Mater. 2010, 22, 6202–6208. [Google Scholar] [CrossRef]
- Xia, T.; Li, Q.; Liu, X.; Meng, J.; Cao, X. Morphology-Controllable Synthesis and Characterization of Single-Crystal Molybdenum Trioxide. J. Phys. Chem. B 2006, 110, 2006–2012. [Google Scholar] [CrossRef]
- Lunk, H.J.; Hartl, H.; Hartl, M.A.; Fait, M.J.G.; Shenderovich, I.G.; Feist, M.; Frisk, T.A.; Daemen, L.L.; Mauder, D.; Eckelt, R.; et al. “Hexagonal Molybdenum Trioxide”—Known for 100 Years and Still a Fount of New Discoveries. Inorg. Chem. 2010, 49, 9400–9408. [Google Scholar] [CrossRef]
- Song, J.; Ni, X.; Gao, L.; Zheng, H. Synthesis of Metastable H-MoO3 by Simple Chemical Precipitation. Mater. Chem. Phys. 2007, 102, 245–248. [Google Scholar] [CrossRef]
- Ramana, C.V.; Atuchin, V.V.; Troitskaia, I.B.; Gromilov, S.A.; Kostrovsky, V.G.; Saupe, G.B. Low-Temperature Synthesis of Morphology Controlled Metastable Hexagonal Molybdenum Trioxide (MoO3). Solid State Commun. 2009, 149, 6–9. [Google Scholar] [CrossRef]
- Muñoz-Espí, R.; Burger, C.; Krishnan, C.V.; Chu, B. Polymer-Controlled Crystallization of Molybdenum Oxides from Peroxomolybdates: Structural Diversity and Application to Catalytic Epoxidation. Chem. Mater. 2008, 20, 7301–7311. [Google Scholar] [CrossRef]
- Li, Z.; Ma, J.; Zhang, B.; Song, C.; Wang, D. Crystal Phase- and Morphology-Controlled Synthesis of MoO3 Materials. CrystEngComm 2017, 19, 1479–1485. [Google Scholar] [CrossRef]
- Guo, J.; Zavalij, P.; Whittingham, M.S. Metastable Hexagonal Molybdates: Hydrothermal Preparation, Structure, and Reactivity. J. Solid State Chem. 1995, 117, 323–332. [Google Scholar] [CrossRef]
- Coelho, A.A. TOPAS and TOPAS-Academic: An Optimization Program Integrating Computer Algebra and Crystallographic Objects Written in C++. J. Appl. Crystallogr. 2018, 51, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Long, D.L.; Burkholder, E.; Cronin, L. Polyoxometalate Clusters, Nanostructures and Materials: From Self Assembly to Designer Materials and Devices. Chem. Soc. Rev. 2007, 36, 105–121. [Google Scholar] [CrossRef]
- Bretos, I.; Diodati, S.; Jiménez, R.; Tajoli, F.; Ricote, J.; Bragaggia, G.; Franca, M.; Calzada, M.L.; Gross, S. Low-Temperature Solution Crystallization of Nanostructured Oxides and Thin Films. Chem. A Eur. J. 2020, 26, 9157–9179. [Google Scholar] [CrossRef]
- Dhage, S.R.; Hassan, M.S.; Yang, O.B. Low Temperature Fabrication of Hexagon Shaped H-MoO3 Nanorods and Its Phase Transformation. Mater. Chem. Phys. 2009, 114, 511–514. [Google Scholar] [CrossRef]
- Moura, J.V.B.; Silveira, J.V.; da Silva Filho, J.G.; Souza Filho, A.G.; Luz-Lima, C.; Freire, P.T.C. Temperature-Induced Phase Transition in h-MoO3: Stability Loss Mechanism Uncovered by Raman Spectroscopy and DFT Calculations. Vib. Spectrosc. 2018, 98, 98–104. [Google Scholar] [CrossRef]
- Silveira, J.V.; Moura, J.V.B.; Luz-Lima, C.; Freire, P.T.C.; Souza Filho, A.G. Laser-Induced Thermal Effects in Hexagonal MoO3 Nanorods. Vib. Spectrosc. 2018, 98, 145–151. [Google Scholar] [CrossRef]
- Atuchin, V.V.; Gavrilova, T.A.; Kostrovsky, V.G.; Pokrovsky, L.D.; Troitskaia, I.B. Morphology and Structure of Hexagonal MoO3 Nanorods. Inorg. Mater. 2008, 44, 622–627. [Google Scholar] [CrossRef]
- Cölfen, H.; Mann, S. Higher-Order Organization by Mesoscale Self-Assembly and Transformation of Hybrid Nanostructures. Angew. Chemie Int. Ed. 2003, 42, 2350–2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cölfen, H.; Antonietti, M. Mesocrystals and Nonclassical Crystallization; John Wiley & Sons: Chichester, UK, 2008; ISBN 9780470994603. [Google Scholar]
- Nakamoto, K. Belowred Spectra of Inorganic and Coordination Compounds; John Wiley & Sons: New York, NY, USA, 1963. [Google Scholar]
- LaMer, V.K. Nucleation in Phase Transitions. Ind. Eng. Chem. 1952, 44, 1270–1277. [Google Scholar] [CrossRef]
- Sugimoto, T. Spontaneous Nucleation of Monodisperse Silver Halide Particles from Homogeneous Gelatin Solution I: Silver Chloride. Colloids Surf. A Physicochem. Eng. Asp. 2000, 164, 183–203. [Google Scholar] [CrossRef]
- Chu, D.B.K.; Owen, J.S.; Peters, B. Nucleation and Growth Kinetics from LaMer Burst Data. J. Phys. Chem. A 2017, 121, 7511–7517. [Google Scholar] [CrossRef]
- Jehn, H.; Kurtz, W.; Schneider, D.; Trobisch, U.; Wagner, J. Gmelin Handbook of Inorganic Chemistry: Mo Molybdenum; Katscher, H., Kurtz, W., Schröder, F., Eds.; Springer: Berlin/Heidelberg, Germany, 1989; Volume B5, ISBN 978-3-662-06329-3. [Google Scholar]
- Pope, M.T.; Müller, A. (Eds.) Polyoxometalate Chemistry from Topology via Self-Assembly to Applications; Springer: Dordrecht, The Netherlands, 2001. [Google Scholar]
- Ostwald, W. Studies on the formation and transformation of solid bodies. 1. Treatise: Supersaturation and Overcooling. Z. Phys. Chem. 1897, 22, 289–330. [Google Scholar] [CrossRef]
- Meldrum, F.C.; Cölfen, H. Controlling Mineral Morphologies and Structures in Biological and Synthetic Systems. Chem. Rev. 2008, 108, 4332–4432. [Google Scholar] [CrossRef]
- Feigin, L.A.; Svergun, D.I. Structure Analysis by Small-Angle X-ray and Neutron Scattering; Springer: New York, NY, USA, 2013. [Google Scholar]
- Niederberger, M.; Cölfen, H. Oriented Attachment and Mesocrystals: Non-Classical Crystallization Mechanisms Based on Nanoparticle Assembly. Phys. Chem. Chem. Phys. 2006, 8, 3271–3287. [Google Scholar] [CrossRef]
- Cölfen, H.; Antonietti, M. Mesocrystals: Inorganic Superstructures Made by Highly Parallel Crystallization and Controlled Alignment. Angew. Chemie Int. Ed. 2005, 44, 5576–5591. [Google Scholar] [CrossRef] [Green Version]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tajoli, F.; Massagrande, M.V.; Muñoz-Espí, R.; Gross, S. Exploring the Role of Miniemulsion Nanodroplet Confinement on the Crystallization of MoO3: Morphology Control and Insight on Crystal Formation by In Situ Time-Resolved SAXS/WAXS. Nanomaterials 2023, 13, 1046. https://doi.org/10.3390/nano13061046
Tajoli F, Massagrande MV, Muñoz-Espí R, Gross S. Exploring the Role of Miniemulsion Nanodroplet Confinement on the Crystallization of MoO3: Morphology Control and Insight on Crystal Formation by In Situ Time-Resolved SAXS/WAXS. Nanomaterials. 2023; 13(6):1046. https://doi.org/10.3390/nano13061046
Chicago/Turabian StyleTajoli, Francesca, Maria Vittoria Massagrande, Rafael Muñoz-Espí, and Silvia Gross. 2023. "Exploring the Role of Miniemulsion Nanodroplet Confinement on the Crystallization of MoO3: Morphology Control and Insight on Crystal Formation by In Situ Time-Resolved SAXS/WAXS" Nanomaterials 13, no. 6: 1046. https://doi.org/10.3390/nano13061046