
Low Overpotential Electrochemical Reduction of CO2 to Ethanol Enabled by Cu/CuxO Nanoparticles Embedded in Nitrogen-Doped Carbon Cuboids


 ,
,  ,
,  ,
,
Abstract
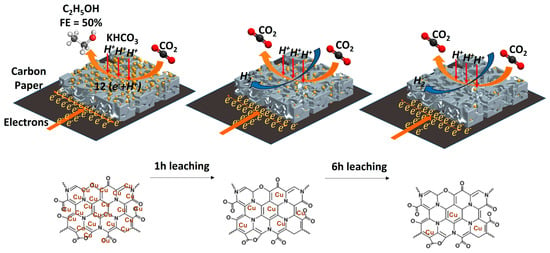
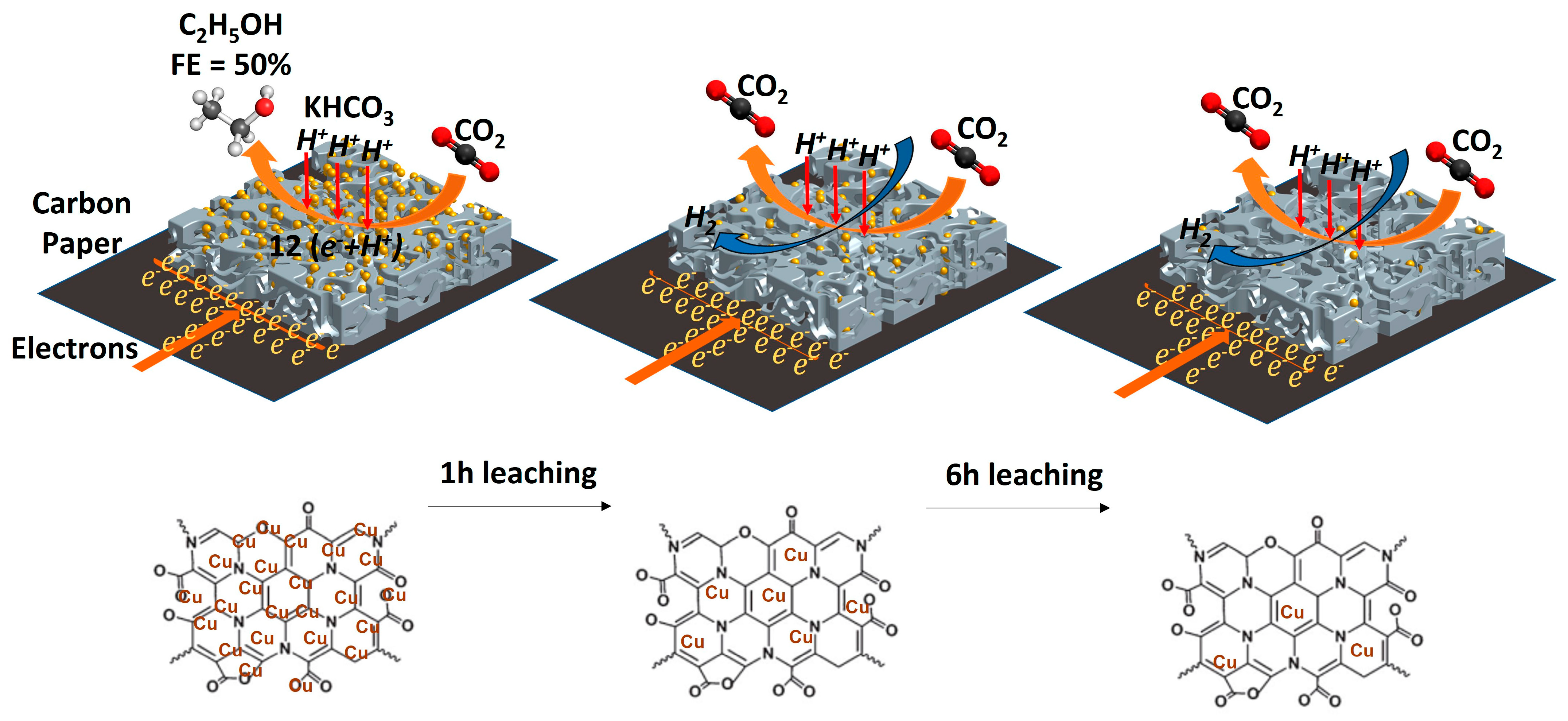
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Material Synthesis
2.3. Material Characterization
2.4. Working Electrode Preparation
2.5. Electrochemical Measurements
2.6. Gaseous Product Quantification
2.7. Liquid Product Quantification
3. Results and Discussion
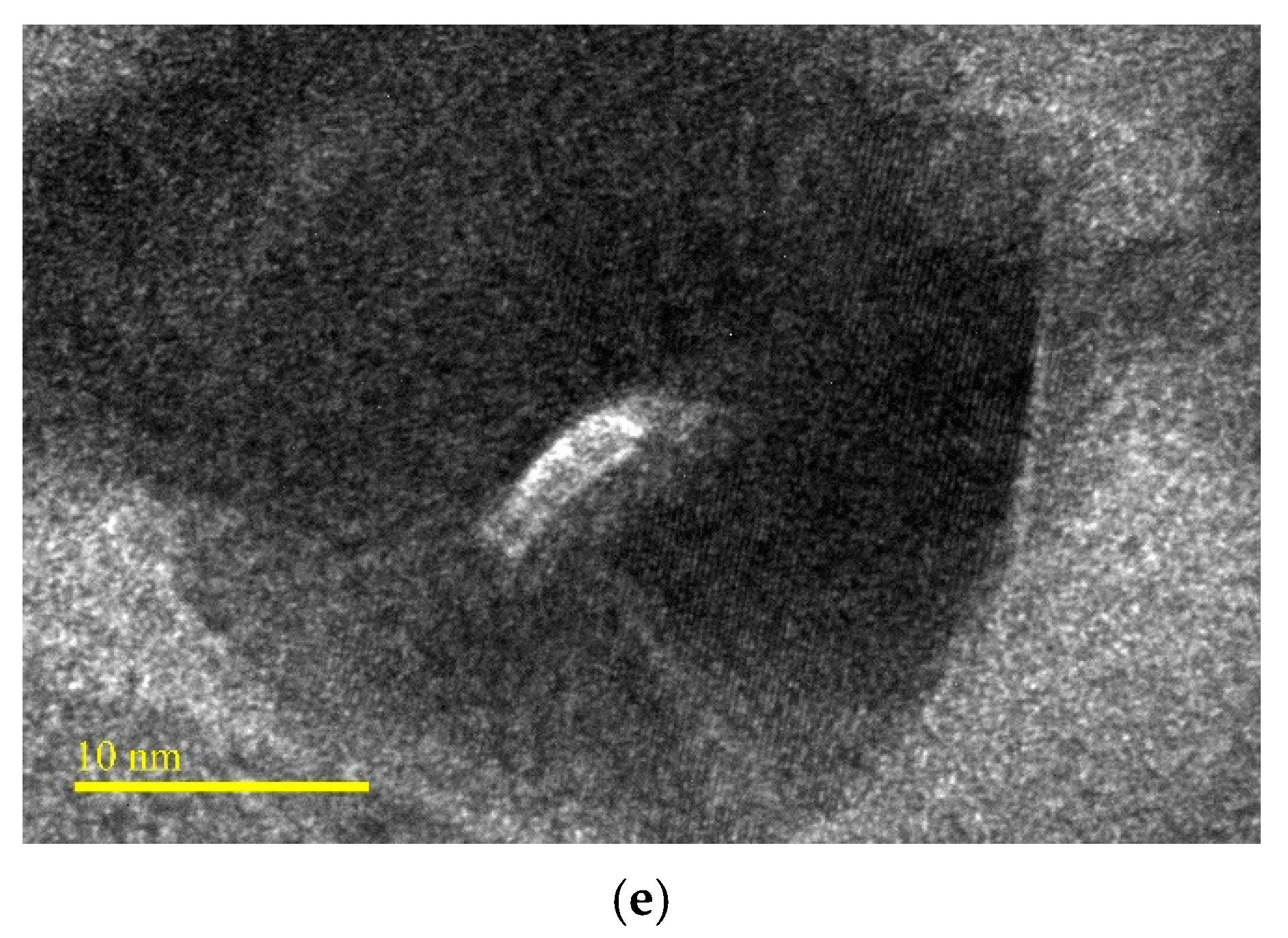
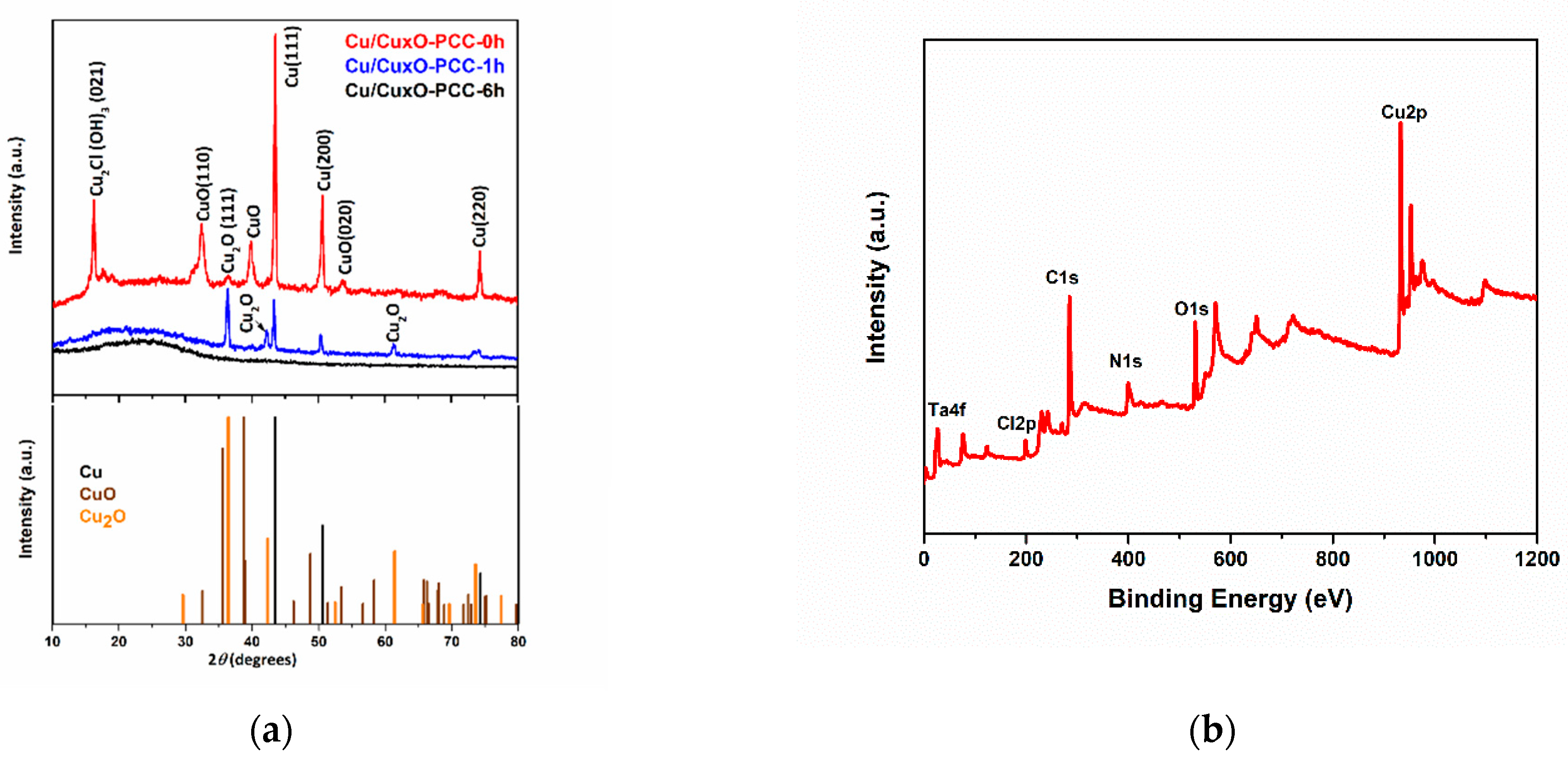
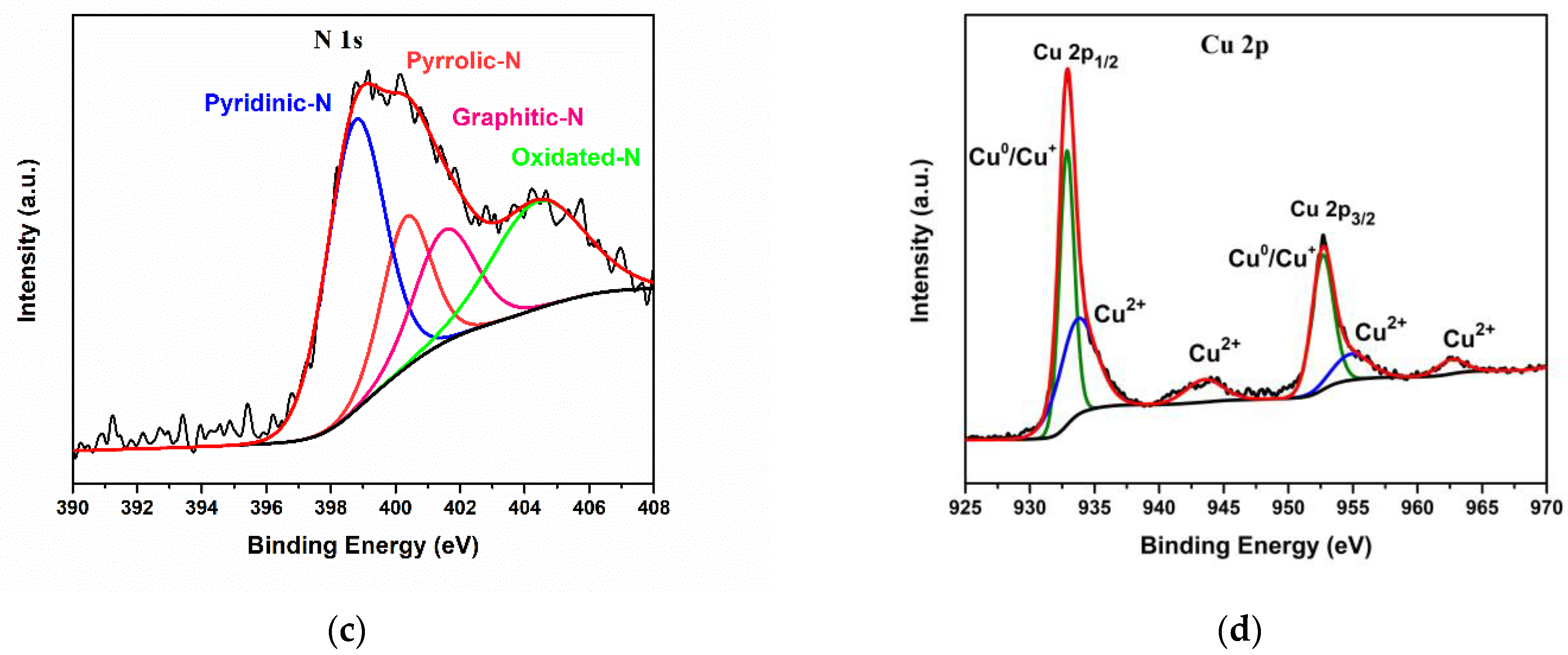
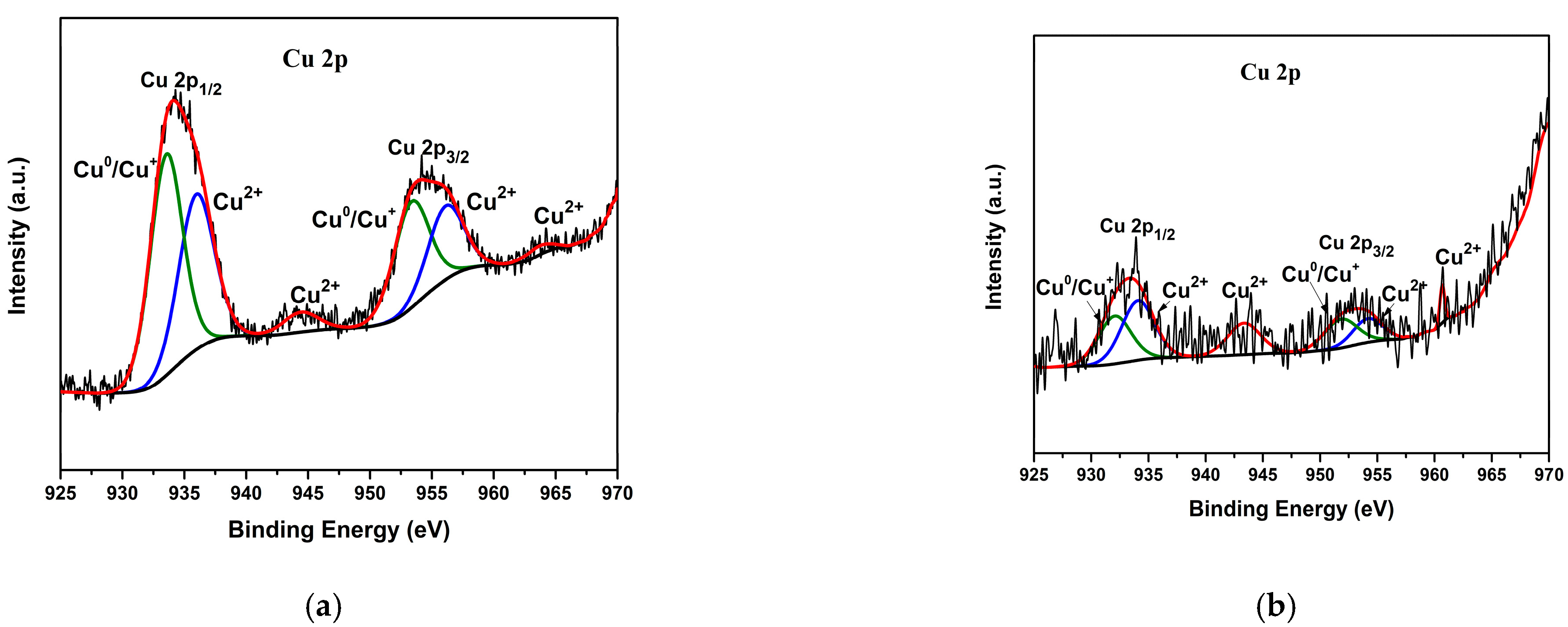
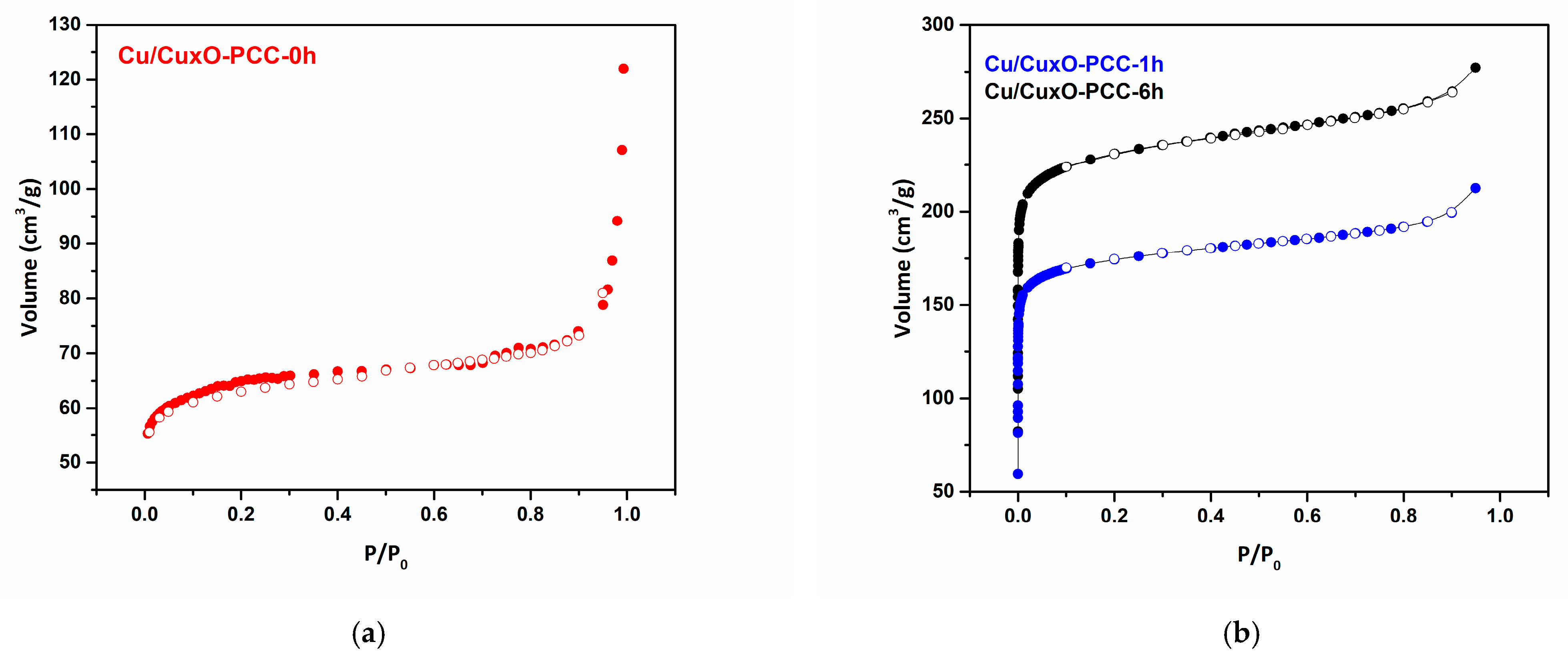
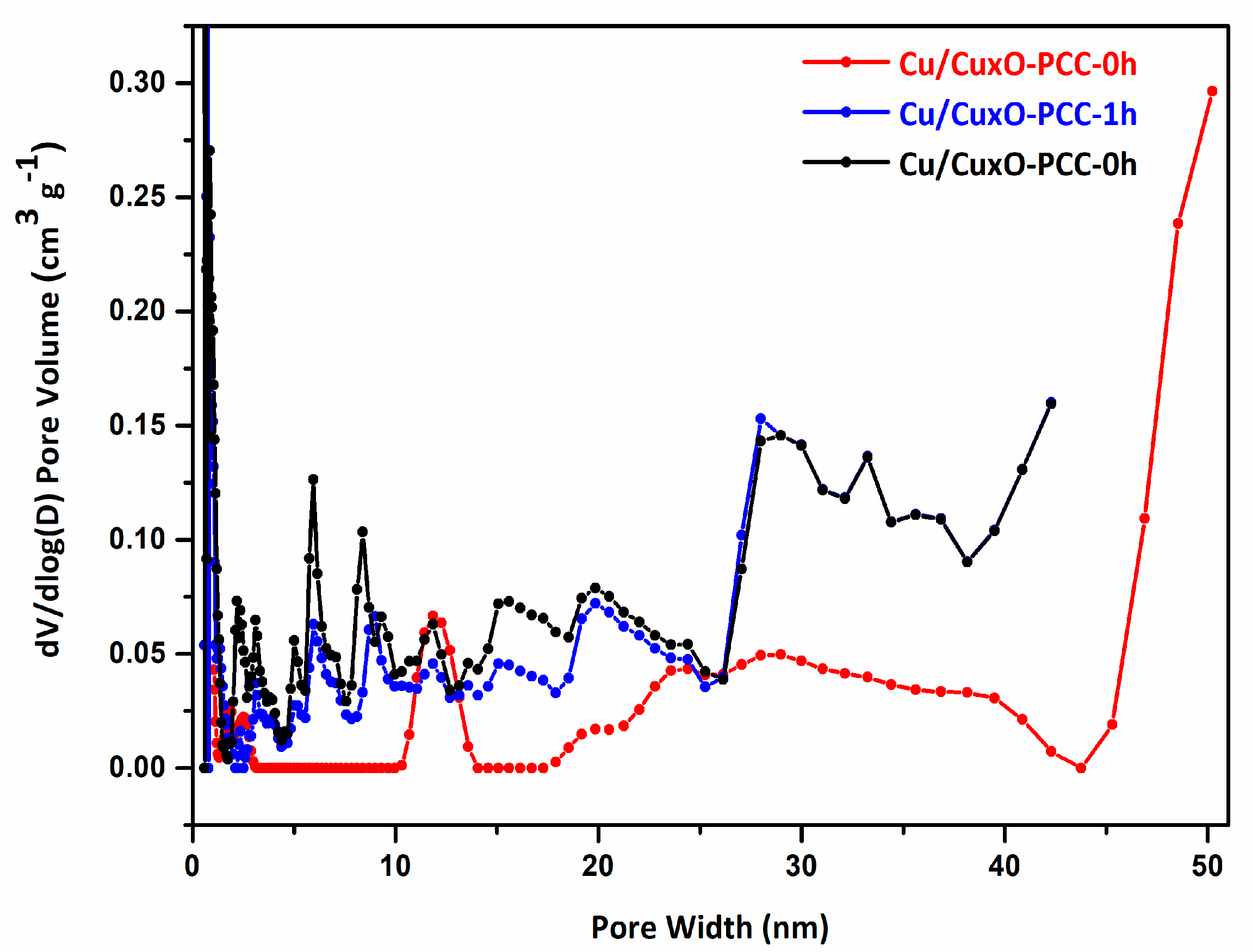
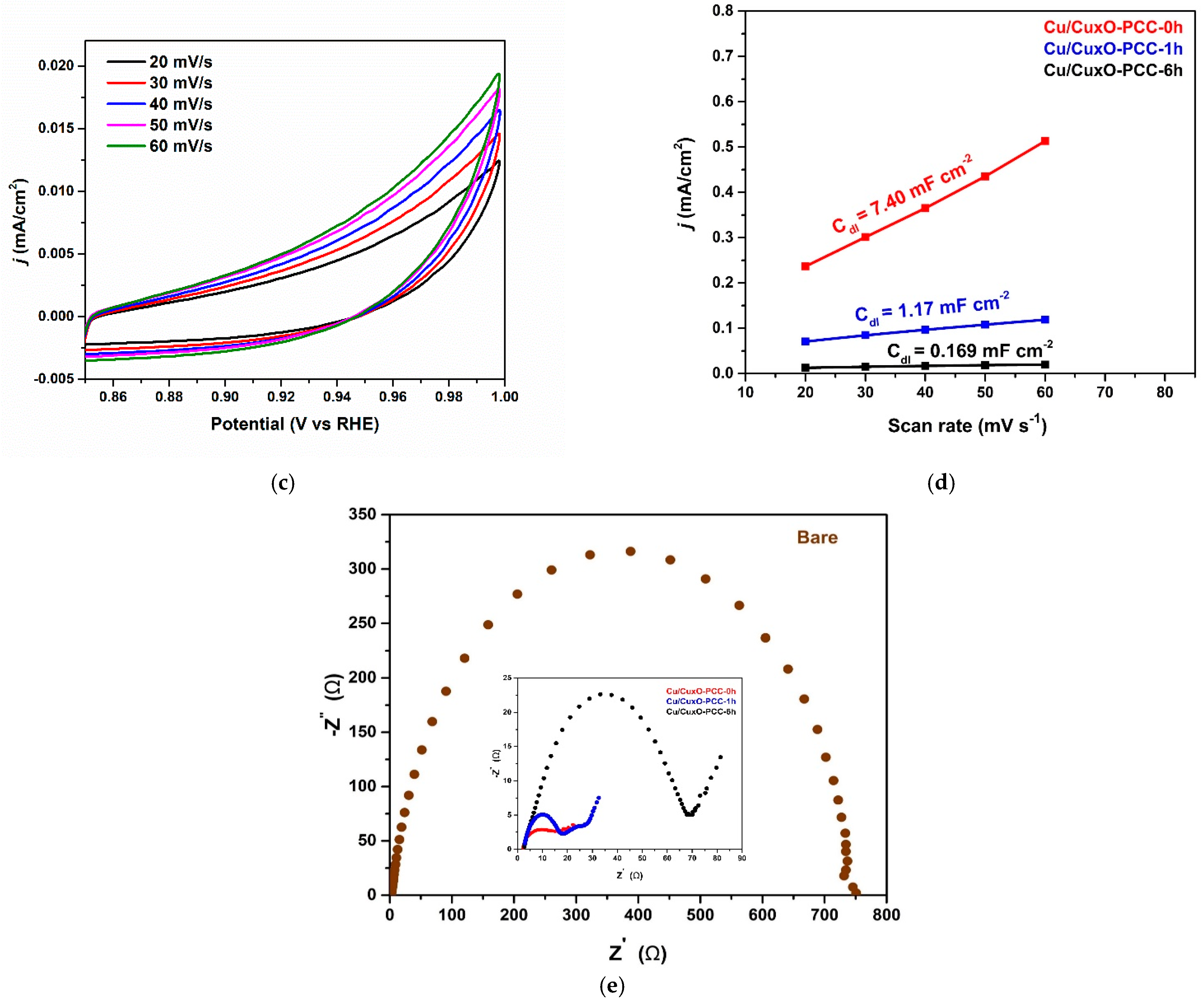
3.1. Morphological and Structural Characterization
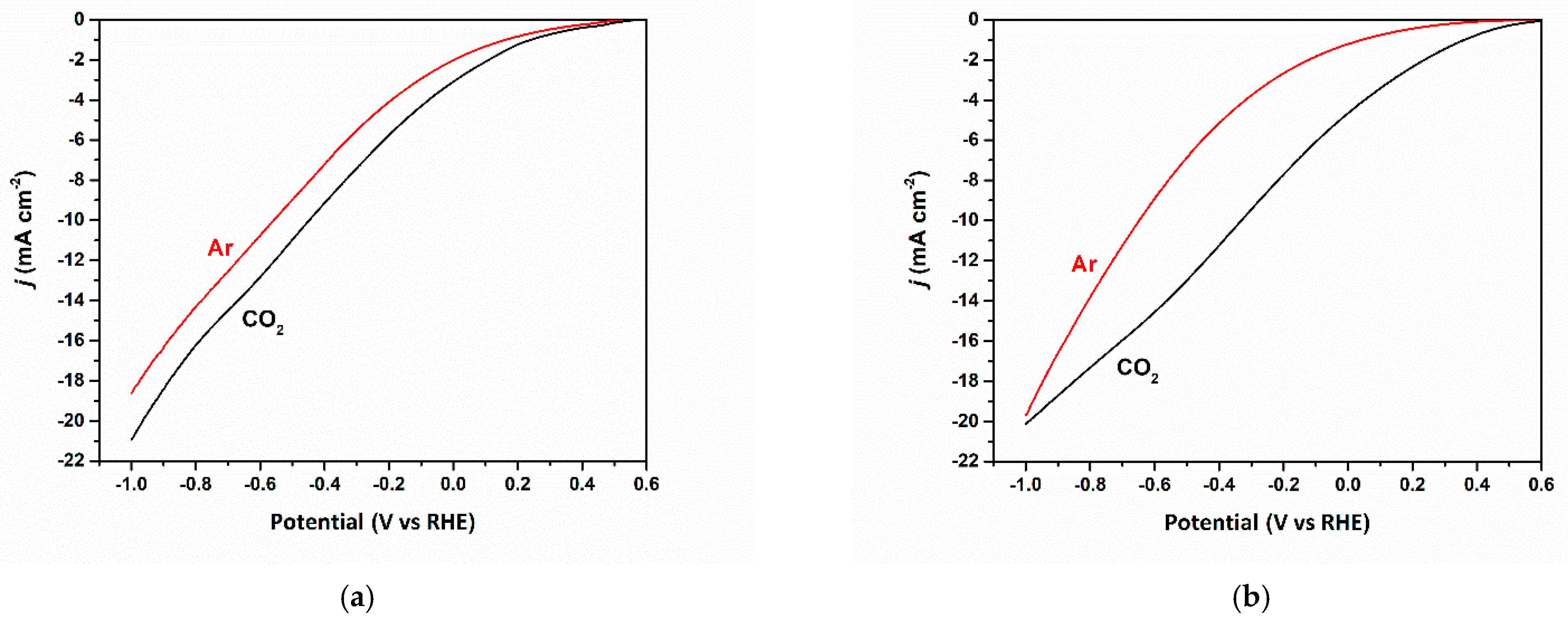
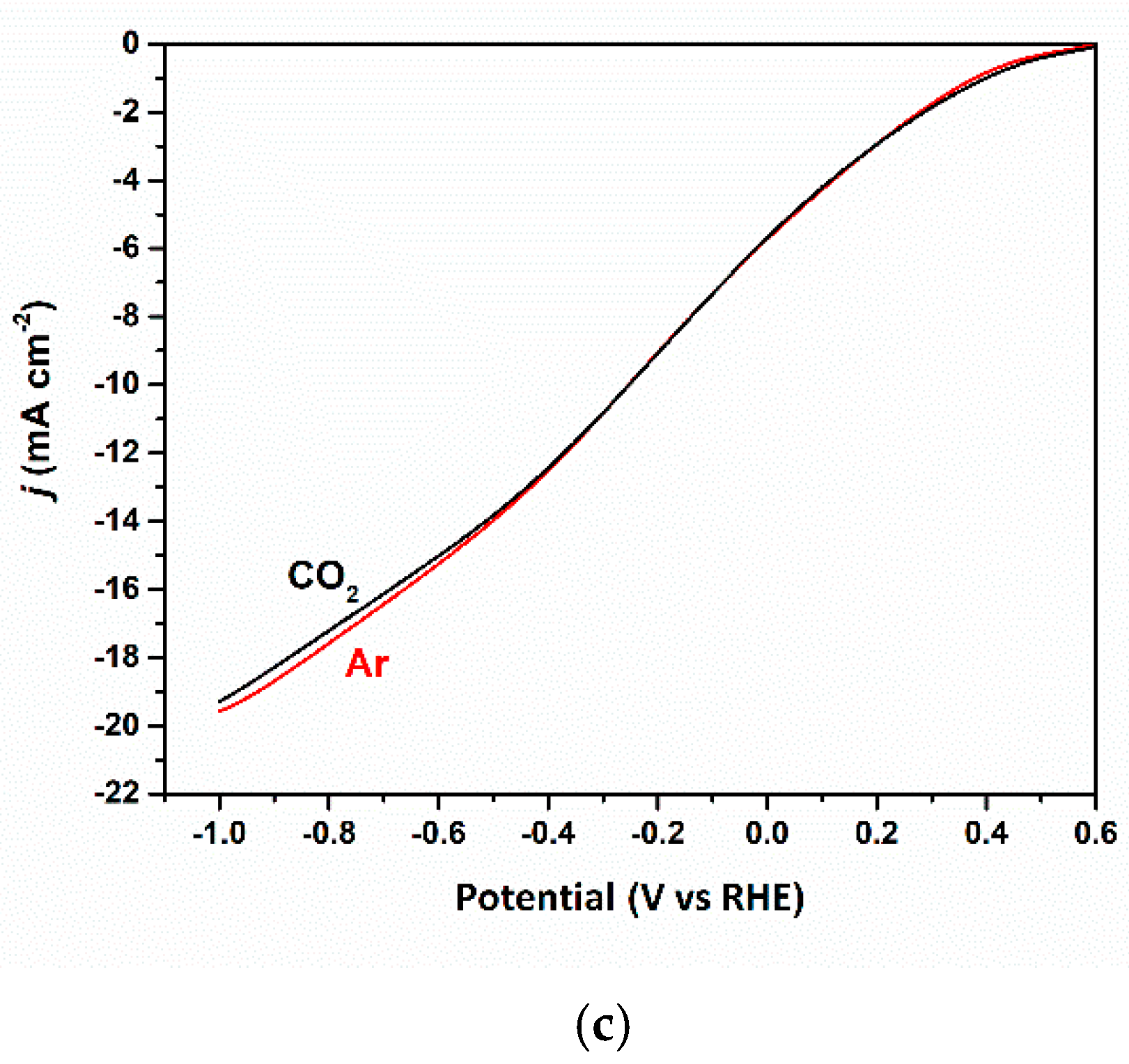
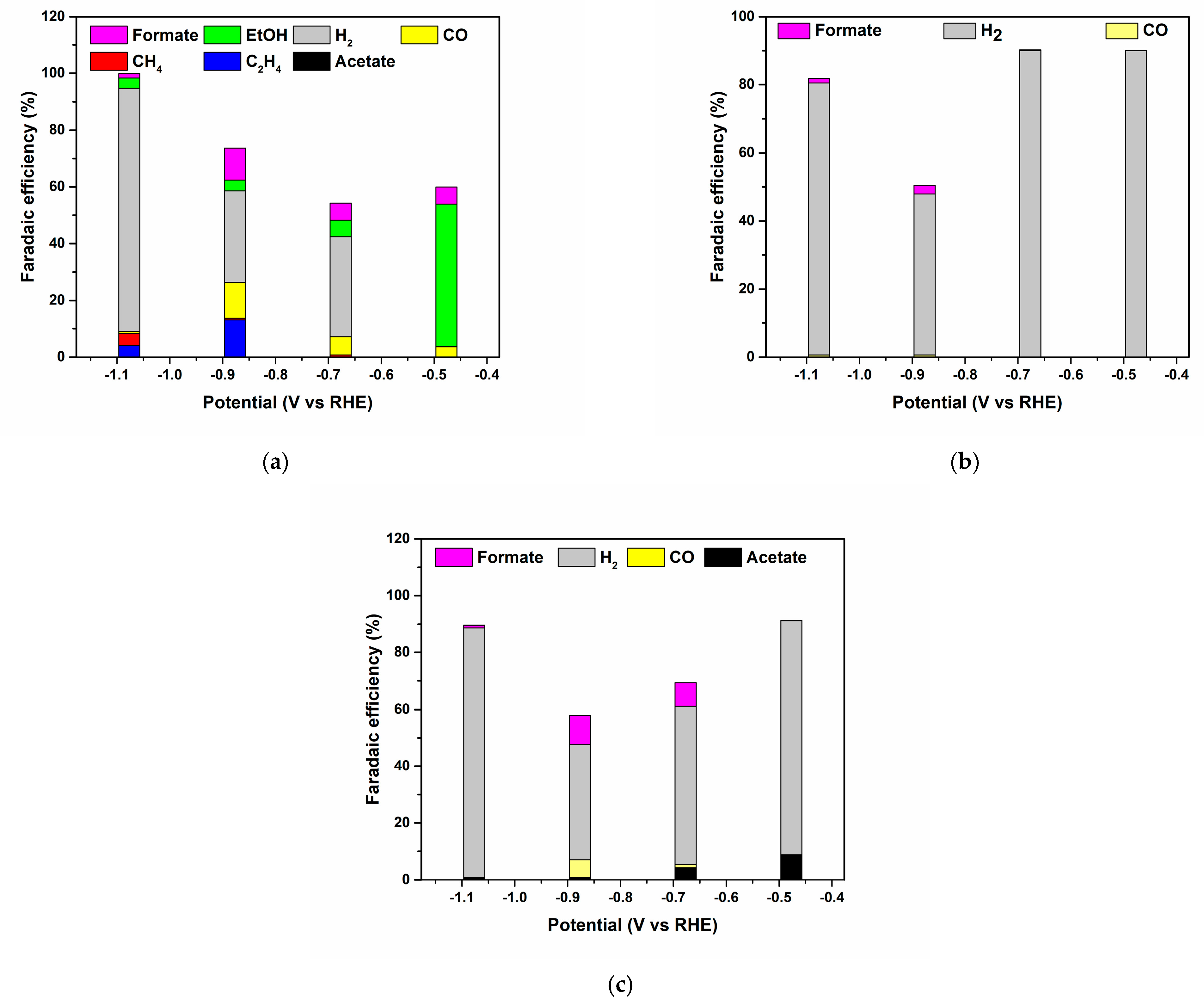
3.2. Electrocatalytic Reduction of CO2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- He, Y.; Manful, D.; Warren, R.; Forstenhäusler, N.; Osborn, T.J.; Price, J.; Jenkins, R.; Wallace, C.; Yamazaki, D. Quantification of Impacts between 1.5 and 4 °C of Global Warming on Flooding Risks in Six Countries. Clim. Chang. 2022, 170, 1–21. [Google Scholar] [CrossRef]
- Matzarakis, A. Communication Aspects about Heat in an Era of Global Warming—The Lessons Learnt by Germany and Beyond. Atmosphere 2022, 13, 226. [Google Scholar] [CrossRef]
- Penuelas, J.; Fernández-Martínez, M.; Vallicrosa, H.; Maspons, J.; Zuccarini, P.; Carnicer, J.; Sanders, T.G.M.; Krüger, I.; Obersteiner, M.; Janssens, I.A.; et al. Increasing Atmospheric CO2 Concentrations Correlate with Declining Nutritional Status of European Forests. Commun. Biol. 2020, 3, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamu, A.; Russo-Abegão, F.; Boodhoo, K. Process Intensification Technologies for CO2 Capture and Conversion—a Review. BMC Chem. Eng. 2020, 2, 1–18. [Google Scholar] [CrossRef]
- Zhu, Q. Developments on CO2-Utilization Technologies. Clean Energy 2019, 3, 85–100. [Google Scholar] [CrossRef] [Green Version]
- Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. A Review of Catalysts for the Electroreduction of Carbon Dioxide to Produce Low-Carbon Fuels. Chem. Soc. Rev. 2014, 43, 631–675. [Google Scholar] [CrossRef]
- Hori, Y. Electrochemical CO2 Reduction on Metal Electrodes. In Modern Aspects of Electrochemistry; Springer: Berlin/Heidelberg, Germany, 2008; pp. 89–189. [Google Scholar]
- Hori, Y.; Murata, A.; Takahashi, R. Formation of Hydrocarbons in the Electrochemical Reduction of Carbon Dioxide at a Copper Electrode in Aqueous Solution. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1989, 85, 2309–2326. [Google Scholar] [CrossRef]
- Hitetomo, N.; IKEDA, S.; Yoshiyuki, O.; Kaname, I. Potential Dependencies of the Products on Electrochemical Reduction of Carbon Dioxide at a Copper Electrode. Chem. Soc. Japan 1989, 18, 289–292. [Google Scholar]
- Kuhl, K.P.; Cave, E.R.; Abram, D.N.; Jaramillo, T.F. New Insights into the Electrochemical Reduction of Carbon Dioxide on Metallic Copper Surfaces. Energy Environ. Sci. 2012, 5, 7050–7059. [Google Scholar] [CrossRef]
- Nitopi, S.; Bertheussen, E.; Scott, S.B.; Liu, X.; Engstfeld, A.K.; Horch, S.; Seger, B.; Stephens, I.E.L.; Chan, K.; Hahn, C.; et al. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119, 7610–7672. [Google Scholar] [CrossRef] [Green Version]
- Hori, Y.; Konishi, H.; Futamura, T.; Murata, A.; Koga, O.; Sakurai, H.; Oguma, K. “deactivation of Copper Electrode” in Electrochemical Reduction of CO2. Electrochim. Acta 2005, 50, 5354–5369. [Google Scholar] [CrossRef]
- Ren, D.; Deng, Y.; Handoko, A.D.; Chen, C.S.; Malkhandi, S.; Yeo, B.S. Selective Electrochemical Reduction of Carbon Dioxide to Ethylene and Ethanol on Copper(I) Oxide Catalysts. ACS Catal. 2015, 5, 2814–2821. [Google Scholar] [CrossRef]
- Kim, D.; Kley, C.S.; Li, Y.; Yang, P. Copper Nanoparticle Ensembles for Selective Electroreduction of CO2 to C2–C3 Products. Proc. Natl. Acad. Sci. USA 2017, 114, 10560–10565. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.X.; Meng, F.L.; Liu, K.H.; Yi, S.S.; Li, S.J.; Yan, J.M.; Jiang, Q. Amorphizing of Cu Nanoparticles toward Highly Efficient and Robust Electrocatalyst for CO2 Reduction to Liquid Fuels with High Faradaic Efficiencies. Adv. Mater. 2018, 30, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, H.; Yu, J.; Hu, B.; Zhao, H.; Tsiakaras, P.; Song, S. Copper Oxide Derived Nanostructured Self-Supporting Cu Electrodes for Electrochemical Reduction of Carbon Dioxide. Electrochim. Acta 2019, 328, 135083. [Google Scholar] [CrossRef]
- Ke, F.S.; Liu, X.C.; Wu, J.; Sharma, P.P.; Zhou, Z.Y.; Qiao, J.; Zhou, X.D. Selective Formation of C2 Products from the Electrochemical Conversion of CO2 on CuO-Derived Copper Electrodes Comprised of Nanoporous Ribbon Arrays. Catal. Today 2017, 288, 18–23. [Google Scholar] [CrossRef]
- Xie, H.; Wang, T.; Liang, J.; Li, Q.; Sun, S. Cu-Based Nanocatalysts for Electrochemical Reduction of CO2. Nano Today 2018, 21, 41–54. [Google Scholar] [CrossRef]
- Baturina, O.A.; Lu, Q.; Padilla, M.A.; Xin, L.; Li, W.; Serov, A.; Artyushkova, K.; Atanassov, P.; Xu, F.; Epshteyn, A.; et al. CO2 Electroreduction to Hydrocarbons on Carbon-Supported Cu Nanoparticles. ACS Catal. 2014, 4, 3682–3695. [Google Scholar] [CrossRef]
- Ning, H.; Wang, W.; Mao, Q.; Zheng, S.; Yang, Z.; Zhao, Q.; Wu, M. Catalytic Electroreduction of CO2 to C2H4 Using Cu2O Supported on 1-Octyl-3-Methylimidazole Functionalized Graphite Sheets. Wuli Huaxue Xuebao/Acta Phys.-Chim. Sin. 2018, 34, 938–944. [Google Scholar] [CrossRef]
- Alves, D.C.B.; Silva, R.; Voiry, D.; Asefa, T.; Chhowalla, M. Copper Nanoparticles Stabilized by Reduced Graphene Oxide for CO2 Reduction Reaction. Mater. Renew. Sustain. Energy 2015, 4, 1–7. [Google Scholar] [CrossRef]
- Feng, X.; Jiang, K.; Fan, S.; Kanan, M.W. A Direct Grain-Boundary-Activity Correlation for CO Electroreduction on Cu Nanoparticles. ACS Cent. Sci. 2016, 2, 169–174. [Google Scholar] [CrossRef]
- Kim, B.; Hillman, F.; Ariyoshi, M.; Fujikawa, S.; Kenis, P.J.A. Effects of Composition of the Micro Porous Layer and the Substrate on Performance in the Electrochemical Reduction of CO2 to CO. J. Power Sources 2016, 312, 192–198. [Google Scholar] [CrossRef] [Green Version]
- Daiyan, R.; Saputera, W.H.; Masood, H.; Leverett, J.; Lu, X.; Amal, R. A Disquisition on the Active Sites of Heterogeneous Catalysts for Electrochemical Reduction of CO2 to Value-Added Chemicals and Fuel. Adv. Energy Mater. 2020, 10, 1–36. [Google Scholar] [CrossRef]
- Chai, G.L.; Guo, Z.X. Highly Effective Sites and Selectivity of Nitrogen-Doped Graphene/CNT Catalysts for CO2 Electrochemical Reduction. Chem. Sci. 2016, 7, 1268–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhong, H.R.M.; Tornow, C.E.; Smid, B.; Gewirth, A.A.; Lyth, S.M.; Kenis, P.J.A. A Nitrogen-Doped Carbon Catalyst for Electrochemical CO2 Conversion to CO with High Selectivity and Current Density. ChemSusChem 2017, 10, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Suliman, M.H.; Yamani, Z.H.; Usman, M. Electrochemical Reduction of CO2 to C1 and C2 Liquid Products on Copper-Decorated Nitrogen-Doped Carbon Nanosheets. Nanomaterials 2023, 13, 47. [Google Scholar] [CrossRef]
- Wu, J.; Ma, S.; Sun, J.; Gold, J.I.; Tiwary, C.; Kim, B.; Zhu, L.; Chopra, N.; Odeh, I.N.; Vajtai, R.; et al. A Metal-Free Electrocatalyst for Carbon Dioxide Reduction to Multi-Carbon Hydrocarbons and Oxygenates. Nat. Commun. 2016, 7, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Liu, M.; Sharma, P.P.; Yadav, R.M.; Ma, L.; Yang, Y.; Zou, X.; Zhou, X.D.; Vajtai, R.; Yakobson, B.I.; et al. Incorporation of Nitrogen Defects for Efficient Reduction of CO2 via Two-Electron Pathway on Three-Dimensional Graphene Foam. Nano Lett. 2016, 16, 466–470. [Google Scholar] [CrossRef]
- Liu, X.; Hou, Y.; Tang, M.; Wang, L. Atom Elimination Strategy for MoS2 Nanosheets to Enhance Photocatalytic Hydrogen Evolution. Chinese Chem. Lett. 2022, 34, 107489. [Google Scholar] [CrossRef]
- Zhou, Z.; Kong, Y.; Tan, H.; Huang, Q.; Wang, C.; Pei, Z.; Wang, H.; Liu, Y.; Wang, Y.; Li, S.; et al. Cation-Vacancy-Enriched Nickel Phosphide for Efficient Electrosynthesis of Hydrogen Peroxides. Adv. Mater. 2022, 34, 2106541. [Google Scholar] [CrossRef]
- Liu, M.; Li, H.; Liu, S.; Wang, L.; Xie, L.; Zhuang, Z.; Sun, C.; Wang, J.; Tang, M.; Sun, S.; et al. Tailoring Activation Sites of Metastable Distorted 1T′-Phase MoS2 by Ni Doping for Enhanced Hydrogen Evolution. Nano Res. 2022, 15, 5946–5952. [Google Scholar] [CrossRef]
- Cheng, X.; Wang, L.; Xie, L.; Sun, C.; Zhao, W.; Liu, X.; Zhuang, Z.; Liu, S.; Zhao, Q. Defect-Driven Selective Oxidation of MoS2 Nanosheets with Photothermal Effect for Photo-Catalytic Hydrogen Evolution Reaction. Chem. Eng. J. 2022, 439, 135757. [Google Scholar] [CrossRef]
- Usman, M.; Humayun, M.; Garba, M.D.; Ullah, L.; Zeb, Z.; Helal, A.; Suliman, M.H.; Alfaifi, B.Y.; Iqbal, N.; Abdinejad, M.; et al. Electrochemical Reduction of CO2: A Review of Cobalt Based Catalysts for Carbon Dioxide Conversion to Fuels. Nanomaterials 2021, 11, 2029. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, K.; Chen, M.; Wang, J.; Liu, J.; Zhang, Y. Cu/Cu2O Nanoparticles Supported on Vertically ZIF-L-Coated Nitrogen-Doped Graphene Nanosheets for Electroreduction of CO2 to Ethanol. ACS Appl. Nano Mater. 2020, 3, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.; Liu, Y.; Quan, X.; Chen, S.; Yu, H. CO2 Electroreduction at Low Overpotential on Oxide-Derived Cu/Carbons Fabricated from Metal Organic Framework. ACS Appl. Mater. Interfaces 2017, 9, 5302–5311. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-J.; Yang, H.; Hong, Y.-H.; Zhang, P.-Y.; Wang, T.; Chen, L.-N.; Zhang, F.-Y.; Wu, Q.-H.; Tian, N.; Zhou, Z.-Y.; et al. Promoting Ethylene Selectivity from CO2 Electroreduction on CuO Supported onto CO2 Capture Materials. ChemSusChem 2018, 11, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Peng, X.; Liu, X.; Li, H.; Luo, J. High Selectivity Toward C2H4 Production over Cu Particles Supported by Butterfly-Wing-Derived Carbon Frameworks. ACS Appl. Mater. Interfaces 2018, 10, 12618–12625. [Google Scholar] [CrossRef]
- Wang, J.; Cheng, T.; Fenwick, A.Q.; Baroud, T.N.; Rosas-Hernández, A.; Ko, J.H.; Gan, Q.; Goddard, W.A.; Grubbs, R.H. Selective CO2 Electrochemical Reduction Enabled by a Tricomponent Copolymer Modifier on a Copper Surface. J. Am. Chem. Soc. 2021, 143, 2857–2865. [Google Scholar] [CrossRef]
- Thomou, E.; Basina, G.; Spyrou, K.; Al Wahedi, Y.; Rudolf, P.; Gournis, D. H2S Removal by Copper Enriched Porous Carbon Cuboids. Carbon Trends 2022, 7, 100145. [Google Scholar] [CrossRef]
- Hao, G.P.; Mondin, G.; Zheng, Z.; Biemelt, T.; Klosz, S.; Schubel, R.; Eychmüller, A.; Kaskel, S. Unusual Ultra-Hydrophilic, Porous Carbon Cuboids for Atmospheric-Water Capture. Angew. Chemie -Int. Ed. 2015, 54, 1941–1945. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, B.; Zhong, J.; Cheng, Z. Selective Electrochemical CO2 Reduction over Highly Porous Gold Films. J. Mater. Chem. A 2017, 5, 21955–21964. [Google Scholar] [CrossRef]
- Yang, F.; Elnabawy, A.O.; Schimmenti, R.; Song, P.; Wang, J.; Peng, Z.; Yao, S.; Deng, R.; Song, S.; Lin, Y.; et al. Bismuthene for Highly Efficient Carbon Dioxide Electroreduction Reaction. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Xue, S.; Barber, J.; Zhou, Y.; Meng, J.; Ke, X. An Overview of Cu-Based Heterogeneous Electrocatalysts for CO2 Reduction. J. Mater. Chem. A 2020, 8, 4700–4734. [Google Scholar] [CrossRef]
- Theivasanthi, T. X-ray Diffraction Studies of Copper Nanopowder. arXiv Prepr. arXiv 1936, 101, 593–603. [Google Scholar] [CrossRef]
- Ahmad, T. Ascorbic Acid Assisted Synthesis, Characterization and Catalytic Application of Copper Nanoparticles. Mater. Sci. Eng. Int. J. 2018, 2, 90–94. [Google Scholar] [CrossRef]
- Sudha, V.; Murugadoss, G.; Thangamuthu, R. Structural and Morphological Tuning of Cu-Based Metal Oxide Nanoparticles by a Facile Chemical Method and Highly Electrochemical Sensing of Sulphite. Sci. Rep. 2021, 11, 3413. [Google Scholar] [CrossRef] [PubMed]
- Chmielová, M.; Seidlerová, J.; Weiss, Z. X-ray Diffraction Phase Analysis of Crystalline Copper Corrosion Products after Treatment in Different Chloride Solutions. Corros. Sci. 2003, 45, 883–889. [Google Scholar] [CrossRef]
- Wang, Y.; Lü, Y.; Zhan, W.; Xie, Z.; Kuang, Q.; Zheng, L. Synthesis of Porous Cu2O/CuO Cages Using Cu-Based Metal-Organic Frameworks as Templates and Their Gas-Sensing Properties. J. Mater. Chem. A 2015, 3, 12796–12803. [Google Scholar] [CrossRef]
- Chen, R.X.; Zhu, S.L.; Mao, J.; Cui, Z.D.; Yang, X.J.; Liang, Y.Q.; Li, Z.Y. Synthesis of CuO/Co3O4 Coaxial Heterostructures for Efficient and Recycling Photodegradation. Int. J. Photoenergy 2015, 2015, 183468. [Google Scholar] [CrossRef] [Green Version]
- Zorn, G.; Liu, L.H.; Árnadóttir, L.; Wang, H.; Gamble, L.J.; Castner, D.G.; Yan, M. X-ray Photoelectron Spectroscopy Investigation of the Nitrogen Species in Photoactive Perfluorophenylazide-Modified Surfaces. J. Phys. Chem. C 2014, 118, 376–383. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Maiyalagan, T.; Wang, X. Review on Recent Progress in Nitrogen-Doped Graphene: Synthesis, Characterization, and Its Potential Applications. ACS Catal. 2012, 2, 781–794. [Google Scholar] [CrossRef]
- Tang, W.; Peterson, A.A.; Varela, A.S.; Jovanov, Z.P.; Bech, L.; Durand, W.J.; Dahl, S.; Nørskov, J.K.; Chorkendorff, I. The Importance of Surface Morphology in Controlling the Selectivity of Polycrystalline Copper for CO2 Electroreduction. Phys. Chem. Chem. Phys. 2012, 14, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Palmore, G.T.R.; Sun, S. Electrochemical Reduction of CO2 Catalyzed by Metal Nanocatalysts. Trends Chem. 2019, 1, 739–750. [Google Scholar] [CrossRef]
- Sen, S.; Liu, D.; Palmore, G.T.R. Electrochemical Reduction of CO2 at Copper Nanofoams. ACS Catal. 2014, 4, 3091–3095. [Google Scholar] [CrossRef]
- Du, J.; Li, S.; Liu, S.; Xin, Y.; Chen, B.; Liu, H.; Han, B. Selective Electrochemical Reduction of Carbon Dioxide to Ethanol via a Relay Catalytic Platform. Chem. Sci. 2020, 11, 5098–5104. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Yang, M.P.; Zhi, W.Y.; Wang, H.; Wang, H.; Lu, J.X. Efficient Electrochemical Reduction of CO2 to Ethanol on Cu Nanoparticles Decorated on N-Doped Graphene Oxide Catalysts. J. CO2 Util. 2019, 33, 452–460. [Google Scholar] [CrossRef]
- Song, Y.; Peng, R.; Hensley, D.K.; Bonnesen, P.V.; Liang, L.; Wu, Z.; Meyer, H.M.; Chi, M.; Ma, C.; Sumpter, B.G.; et al. High-Selectivity Electrochemical Conversion of CO2 to Ethanol Using a Copper Nanoparticle/N-Doped Graphene Electrode. ChemistrySelect 2016, 1, 6055–6061. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Tzeng, Y.K.; Ji, Y.; Li, Y.; Li, J.; Zheng, X.; Yang, A.; Liu, Y.; Gong, Y.; Cai, L.; et al. Synergistic Enhancement of Electrocatalytic CO2 Reduction to C2 Oxygenates at Nitrogen-Doped Nanodiamonds/Cu Interface. Nat. Nanotechnol. 2020, 15, 131–137. [Google Scholar] [CrossRef]
- Xu, H.; Rebollar, D.; He, H.; Chong, L.; Liu, Y.; Liu, C.; Sun, C.J.; Li, T.; Muntean, J.V.; Winans, R.E.; et al. Highly Selective Electrocatalytic CO2 Reduction to Ethanol by Metallic Clusters Dynamically Formed from Atomically Dispersed Copper. Nat. Energy 2020, 5, 623–632. [Google Scholar] [CrossRef]
- Song, Y.; Chen, W.; Zhao, C.; Li, S.; Wei, W.; Sun, Y. Metal-Free Nitrogen-Doped Mesoporous Carbon for Electroreduction of CO2 to Ethanol. Angew. Chemie-Int. Ed. 2017, 56, 10840–10844. [Google Scholar] [CrossRef]
- Singh, G.; Lee, J.; Karakoti, A.; Bahadur, R.; Yi, J.; Zhao, D.; Albahily, K.; Vinu, A. Emerging Trends in Porous Materials for CO2 capture and Conversion. Chem. Soc. Rev. 2020, 49, 4360–4404. [Google Scholar] [CrossRef] [PubMed]
- Baroud, T.N.; Giannelis, E.P. Role of Mesopore Structure of Hierarchical Porous Carbons on the Electrosorption Performance of Capacitive Deionization Electrodes. ACS Sustain. Chem. Eng. 2019, 7, 7580–7596. [Google Scholar] [CrossRef]
- Ouyang, T.; Ye, Y.Q.; Tan, C.; Guo, S.T.; Huang, S.; Zhao, R.; Zhao, S.; Liu, Z.Q. 1D α-Fe2O3/ZnO Junction Arrays Modified by Bi as Photocathode: High Efficiency in Photoelectrochemical Reduction of CO2 to HCOOH. J. Phys. Chem. Lett. 2022, 13, 6867–6874. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.N.; Guo, J.; Sachindran, A.; Li, F.; Seifitokaldani, A.; Dinh, C.T. Electrochemical CO2 reduction to Ethanol: From Mechanistic Understanding to Catalyst Design. J. Mater. Chem. A 2021, 9, 12474–12494. [Google Scholar] [CrossRef]
- Yan, Z.; Wang, X.; Tan, Y.; Liu, A.; Luo, F.; Zhang, M.; Zeng, L.; Zhang, Y. Thein Situgrowth of Cu2O with a Honeycomb Structure on a Roughed Graphite Paper for the Efficient Electroreduction of CO2 to C2H4. Catal. Sci. Technol. 2021, 11, 6742–6749. [Google Scholar] [CrossRef]
- Kuang, M.; Guan, A.; Gu, Z.; Han, P.; Qian, L.; Zheng, G. Enhanced N-Doping in Mesoporous Carbon for Efficient Electrocatalytic CO2 Conversion. Nano Res. 2019, 12, 2324–2329. [Google Scholar] [CrossRef]
- Zhao, K.; Quan, X. Carbon-Based Materials for Electrochemical Reduction of CO2 to C2+ oxygenates: Recent Progress and Remaining Challenges. ACS Catal. 2021, 11, 2076–2097. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Z.; García de Arquer, F.P.; Dinh, C.T.; Ozden, A.; Li, Y.C.; Nam, D.H.; Li, J.; Liu, Y.S.; Wicks, J.; et al. Efficient Electrically Powered CO2-to-Ethanol via Suppression of Deoxygenation. Nat. Energy 2020, 5, 478–486. [Google Scholar] [CrossRef]
- Marepally, B.C.; Ampelli, C.; Genovese, C.; Tavella, F.; Veyre, L.; Quadrelli, E.A.; Perathoner, S.; Centi, G. Role of Small Cu Nanoparticles in the Behaviour of Nanocarbon-Based Electrodes for the Electrocatalytic Reduction of CO2. J. CO2 Util. 2017, 21, 534–542. [Google Scholar] [CrossRef]
- Lv, K.; Fan, Y.; Zhu, Y.; Yuan, Y.; Wang, J.; Zhu, Y.; Zhang, Q. Elastic Ag-Anchored N-Doped Graphene/Carbon Foam for the Selective Electrochemical Reduction of Carbon Dioxide to Ethanol. J. Mater. Chem. A 2018, 6, 5025–5031. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | BET Surface Area [m2 g−1] | Total Pore Volume [cm3 g−1] | Mesopores Pore Volume [cm3 g−1] | Micropores Pore Volume [cm3 g−1] | Pore Width [nm] |
|---|---|---|---|---|---|
| Cu/CuxO-PCC-0h | 250.9 | 0.189 | 0.089 | 0.100 | 50.232 |
| Cu/CuxO-PCC-1h | 689.5 | 0.330 | 0.091 | 0.239 | 0.545 |
| Cu/CuxO-PCC-6h | 909.2 | 0.430 | 0.117 | 0.313 | 0.545 |
| Catalyst | Electrolyte | Faradic Efficiency% | Potential [V vs. RHE] | Ref. |
|---|---|---|---|---|
| Cu/CuxO nanoparticles embedded on porous carbon cuboids | 0.5 M KHCO3 | 50 | −0.5 | This work |
| Oxide-derived Cu/C catalysts by facile carbonization of Cu-based MOF | 0.1 M KHCO3 | 35 | −0.5 | [36] |
| Cu/Cu2O nanocomposite loaded on the surface of carbon ZIF-L coated on GO | 0.5 M KHCO3 | 70.52 | −0.87 | [35] |
| Cobalt oxide anchored on N-doped Mesoporous carbon and CNTs | 0.5 M KHCO3 | 60.1 | −0.32 | [56] |
| Cu nanoparticles decorated on pyridoxine modification graphene oxide sheets | 0.1 M KHCO3 | 56.3 | −0.25 | [57] |
| Copper nanoparticle ensembles | 0.1 M KHCO3 | 16.6 | −0.86 | [14] |
| Nanoflowers and nanosheets with Cu foam as a substrate | 1 M KHCO3 | 35.7 | −0.4 | [16] |
| Cu nanoparticles on highly textured nitrogen-doped carbon nanospike | 0.1 M KHCO3 | 63 | −1.2 | [58] |
| N-doped nanodiamonds and Cu nanoparticles | 0.5 M KHCO3 | 28.9 | −0.6 | [59] |
| Carbon-supported Cu catalyst synthesized by an amalgamated Cu–Li method | 0.1 M KHCO3 | 91 | −0.7 | [60] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkoshab, M.Q.; Thomou, E.; Abdulazeez, I.; Suliman, M.H.; Spyrou, K.; Iali, W.; Alhooshani, K.; Baroud, T.N. Low Overpotential Electrochemical Reduction of CO2 to Ethanol Enabled by Cu/CuxO Nanoparticles Embedded in Nitrogen-Doped Carbon Cuboids. Nanomaterials 2023, 13, 230. https://doi.org/10.3390/nano13020230
Alkoshab MQ, Thomou E, Abdulazeez I, Suliman MH, Spyrou K, Iali W, Alhooshani K, Baroud TN. Low Overpotential Electrochemical Reduction of CO2 to Ethanol Enabled by Cu/CuxO Nanoparticles Embedded in Nitrogen-Doped Carbon Cuboids. Nanomaterials. 2023; 13(2):230. https://doi.org/10.3390/nano13020230
Chicago/Turabian StyleAlkoshab, Monther Q., Eleni Thomou, Ismail Abdulazeez, Munzir H. Suliman, Konstantinos Spyrou, Wissam Iali, Khalid Alhooshani, and Turki N. Baroud. 2023. "Low Overpotential Electrochemical Reduction of CO2 to Ethanol Enabled by Cu/CuxO Nanoparticles Embedded in Nitrogen-Doped Carbon Cuboids" Nanomaterials 13, no. 2: 230. https://doi.org/10.3390/nano13020230