

Solid-State Synthesis of Cobalt/NCS Electrocatalyst for Oxygen Reduction Reaction in Dual Chamber Microbial Fuel Cells


Department of Environmental Science, Keimyung University, Daegu 42601, Republic of Korea
*
Authors to whom correspondence should be addressed.
Nanomaterials 2022, 12(24), 4369; https://doi.org/10.3390/nano12244369
Submission received: 8 November 2022
/
Revised: 1 December 2022
/
Accepted: 5 December 2022
/
Published: 7 December 2022
(This article belongs to the Special Issue Editorial Board Members’ Collection Series: New Trends in Inorganic Nanoparticles and Composites from Preparation to Applications)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Due to the high cost of presently utilized Pt/C catalysts, a quick and sustainable synthesis of electrocatalysts made of cost-effective and earth-abundant metals is urgently needed. In this work, we demonstrated a mechanochemically synthesized cobalt nanoparticles supported on N and S doped carbons derived from a solid-state-reaction between zinc acetate and 2-amino thiazole as metal, organic ligand in presence of cobalt (Co) metal ions ZnxCox(C3H4N2S). Pyrolysis of the ZnxCox(C3H4N2S) produced, Co/NSC catalyst in which Co nanoparticles are evenly distributed on the nitrogen and sulfur doped carbon support. The Co/NSC catalyst have been characterized with various physical and electrochemical characterization techniques. The Co content in the ZnxCox(C3H4N2S) is carefully adjusted by varying the Co content and the optimized Co/NSC-3 catalyst is subjected to the oxygen reduction reaction in 0.1 M HClO4 electrolyte. The optimized Co/NSC-3 catalyst reveals acceptable ORR activity with the half-wave potential of ~0.63 V vs. RHE in acidic electrolytes. In addition, the Co/NSC-3 catalyst showed excellent stability with no loss in the ORR activity after 10,000 potential cycles. When applied as cathode catalysts in dual chamber microbial fuel cells, the Co/NCS catalyst delivered satisfactory volumetric power density in comparison with Pt/C.
1. Introduction
Microbial fuel cells (MFC) is growing as a promising technology for simultaneous wastewater treatment and bioelectricity generation [1]. The organic matter in the wastewater is converted into H+ and electrons by the microorganisms found in the anode. Protons migrate from the anode to the cathode via the proton exchange membrane, while electrons move through the external circuit, which creates the electrical current. The MFC efficiency depends on the type, content and growth characteristics of microorganisms, i.e., bacteria. However, keeping in mind, the real situation, such as large-scale wastewater treatment plants, it is almost impossible to control the type and content of bacteria’s, due to the fact that, the wastewater comes from different sources (ex. domestic/industrial/agricultural, etc.) to the treatment facilities. Additionally, therefore, in order to reduce the cost and complexity, in general, the indigenous bacteria present in the wastewater are utilized for the organic matter degradation and simultaneous power production, in practical situations [2,3]. On the cathode, the protons and electrons combine with the electron acceptor, generally O2, to produce water, by oxygen reduction reaction (ORR) [4]. MFC technology is expected to become promising in making wastewater treatment more sustainable and economical in the near future [5]. However, there are a number of problems that severely limit the commercial use of MFC, including effective electron transfer from the bacterial surface to anode electrode, formation of effective biofilm, diffusion of H+ from anode to cathode, faster kinetics of cathodic ORR, design, and integration of MFCs that suits to the practical wastewater treatment applications [6]. Among these factors, the kinetics of ORR on cathode, is found to be a factor that greatly affects the performance of MFC. This is due to the fact that ORR is a multi-step and sluggish, especially in acidic electrolytes. Due to its sluggish nature, the ORR demands highly active catalyst such as Pt, therefore, Pt nanoparticles supported on carbon (Pt/C) is considered as the best ORR catalyst so far [7]. However, Pt based catalysts are not ideal for practical application, due to their high cost, poor durability, and scarcity. Recently, transition metal/nitrogen doped carbon-based materials have been demonstrated as one of the best alternative catalysts to traditional Pt/C catalyst for ORR [8]. A number of transition metals such as Co, Mn, Fe, Ni and perovskite-based nanocomposites have been considered as most efficient catalysts for ORR [9,10]. It is believed that transition metals in combination with N form M-N-C (M=Fe, Co, Mn, Ni etc, N-C=nitrogen doped carbon) type of coordination bonds in the catalyst as ORR active centers [11]. Among these catalysts the Fe-N-C type catalysts are considered as the most attractive catalysts due to their remarkable ORR activity in acidic and alkaline electrolytes. However, Fe-N-C catalysts have some limitations such as their ability to form radicals (OH•) with the H2O2 (generated from the 2-electron reduction in O2). The OH• radicals are known to degrade the nafion membrane and nafion ionomer in the catalyst layer therefore reduces the ionic conductivity and deteriorates the MFC performance [12]. In this regard, the Co based catalysts are promising due to the fact that Co is inactive for Fenton’s reaction [13].
In general, the M-N-C catalyst are synthesized from the mixture of heteroatom containing organic ligands and metal salts, a carbon source mixture which is then pyrolyzed at a definite temperature to produce M-N-C catalysts [14]. A number of M-N-C catalysts have been synthesized through this universal strategy with a moderate to superb ORR activity [15]. It is to be noted that, most of the M-N-C catalysts synthesis processes involves a definite solution chemistry, in which the precursors are dissolved in a variety of solvents such as ethanol, methanol, N,N-dimethylformamide, toluene, acetic acid, dimethylacetamide, etc. [16]. Due to the use of solution synthesis, obviously a lot of solvent waste will be generated during washing and filtering steps after the reaction completion. Unfortunately, most of the laboratories and small-scale industries have no regulations either to treat or recycle this toxic organic waste therefore creating water pollution subsequently threat to the environment [17]. A recent report from the “European Union-Registration, Evaluation, Authorization and Restriction of Chemicals (EU-REACH)” demanding the chemical industries to shift from organic solvents to green solvents or solvent free synthesis processes due to their toxicity on aquatic species [18]. Hence, it is highly desired to develop solvent free synthesis methods to produce active and stable ORR catalysts for MFC cathodes. In this context, solid-state synthesis techniques is emerging as a fantastic tool to synthesize M-N-C catalysts without use of any solvents during the catalyst synthesis process. Due to their distinctive method of quickly stimulating solid phase reactions, mechanochemistry has been developing for millennia and still has a high level of significance [19]. Solid-state synthesis has a number of benefits over traditional solution-based synthesis methods, including scalability, a higher product yield, solvent-free operations, quick reaction times, and simple handling with a potential of mass manufacturing. Therefore, the solid-state synthesis technique has been utilized for the synthesis of various Fe and Co based catalysts [20].
In this work, we synthesized Co/NCS electrocatalysts via a solid-state, solid-state synthesis process by solid-state reaction with Zn, Co as metal atoms, 2-amino thiazole as the organic ligand and carbon, nitrogen and sulphur source. The precursor ZnxCox(C3H4N2S) is subjected to pyrolysis at the temperature of 910 °C to acquire Co/NCS catalyst. The obtained Co/NCS catalyst is characterized by various physicochemical and electrochemical characterizations in 0.1 M HClO4. The Co content in the catalysts is systematically optimized and the optimized catalyst with the ideal Co content has been investigated for the ORR kinetics, stability in acidic electrolytes and in dual chamber microbial fuel cell (DCMFC) operated with acetate substrate and atmospheric air as oxidant. It is found that Co/NSC-3 catalyst showed a half-wave potential of ~0.63 V and excellent stability against potential cycling test for 10,000 cycles, almost no loss in half-wave potential. In addition, the Co/NCS-3 catalyst also delivered a volumetric power density of 180 mW m2 in the DCMFC set up.
2. Materials and Methods
2.1. Synthesis of ZnxCox(C3H4N2S), NCS and Co/NCS Catalyst
The precursor ZnxCox(C3H4N2S) is synthesized by a solid-state synthesis strategy with a laboratory mortar and pestle. The Zn((NO3)2·6H2O, 2-aminothiazole and Co(NO3)2·6H2O are taken in a definite mass ratio (1 g:1 g:0.1/0.150/0.20/0.250 mg, respectively) into a mortar and pestle, and are physically grinded for about 30 min. During the grinding process, the precursors turn into liquid paste and is left at room temperature until it dries to obtain ZnxCo1−x(C3H4N2S) precursor. The silica crucible is then directly placed at the center of the tubular furnace and then subjected to pyrolysis at the temperature of 910 °C with a heating rate of 5 °C/min under continuous N2 atmosphere. Once the desired temperature of 910 °C is reached, the temperature is maintained for 2 h, then the furnace is allowed to cool down naturally to room temperature. A black powder obtained after the pyrolysis are named as Co/NCS-1, Co/NCS-2, Co/NCS-3 and Co/NCS-4, corresponding to the mass ratio of Co(NO3)2 used during the synthesis (0.1/0.150/0.200/0.250 mg). For comparison, a catalyst is also synthesized without Co(NO3)2, and the precursor is pyrolyzed at 910 °C, for 1 hr and the catalyst obtained is named NCS. The Co/NCS and NCS catalysts are directly used for physicochemical and electrochemical characterizations without any further processing.
2.2. Physical and Electrochemical Characterizations
Particle size, graphitic nature of the Co/NCS and NCS catalysts are studied by using powder X-ray diffraction (XRD, Rigaku (D/Max-2500) diffractometer) analysis with Cu-Kα radiation source (λ = 1.5406 Å). The morphology of the Co/NCS and NCS catalysts are studied by scanning electron FESEM (Hitachi SU8220, Chiyoda City, Tokyo) and transmission electron microscope (FETEM, Titan G2 ChemiSTEM Cs Probe (FEI Company, Eindhoven, The Netherlands). The oxidation state of cobalt and identification of different bonding of N and S are examined by X-ray photoelectron spectroscopy (XPS) using a Thermo Scientific K-Alpha X-ray photoelectron spectrometer.
2.3. Electrochemical Characterizations of the Co/NCS and NCS Catalysts
Electrochemical studies of the Co/NCS and NCS catalysts are studied in a traditional three electrode system with glassy carbon electrode (effective surface area of 0.1257 cm2) as working electrode, graphite rod and saturated calomel electrode as counter and reference electrodes, respectively. The electrochemical studies were performed by using Biologic instruments SP-150e, potentiostat/galvanostat in 0.1 M HClO4. For rotating disk studies, catalyst (Co/NSC or Pt/C) ink is made by dispersing 5 mg of the catalyst in 1 mL of ethanol: water mixture (1:3) and the solution is ultrasonicated for 30 min, to which a nafion solution (5 wt.%) of 15 µL is added and further ultrasonicated for 30 min. A 14 µL of the resultant catalyst ink is deposited in the glassy carbon electrode and then allowed it to dry at room temperature (catalyst loading: 560 µg cm−2). For comparison, the Pt/C catalyst (10 wt.%) with a catalyst loading of 110 µg cm−2 is also deposited on the GCE.
The ORR electrochemical tests were performed by cyclic voltammetry (CV) and linear sweep voltammetry (LSV). Before the electrochemical tests, the electrolyte 0.1 M HClO4 is saturated with N2/O2 gas. The CV and LSV curves were recorded in the potential range of 0–1.2 V vs. RHE with a scan rate of 50 and 10 mV s−1, respectively. For Koutechy-Levich (K–L) plots, the LSV curves were recorded at different rotations speeds from 800 to 2400 rpm. The number of electrons transferred in ORR process at different electrode potentials are calculated from the slopes of Koutechy-Levich (K-L) plots, i.e., plots of j−1 vs. ω−1/2 using the following questions.
where j is measured current density, jL is diffusion-limiting current density, jk is kinetic current density, ω is angular velocity (rad s−1). B is a parameter calculated from Equation (3); “n” is number of electrons transfer per O2 molecule, F is Faraday constant (96,485 C mol−1), is the concentration of oxygen in electrolyte , is diffusion co-efficient of oxygen in the solution () and υ is kinematic viscosity of 0.1 M aqueous KOH electrolyte (0.01 cm2 s−1). The stability of the Co/NCS and Pt/C catalysts have been carried out by potential cycling of the working electrode by recording repeated cyclic voltammograms between 0 to 1.0 V vs. RHE for 5000 and 10,000 cycles at the scan rate of 50 mV s−1. To assess the degradation of the catalysts, the LSVs were recorded after stability test in O2 saturated 0.1 M HClO4 electrolyte with a scan rate of 10 mV s−1. All the potential was represented on relative hydrogen electrode (RHE) scale for convenience.
2.4. Microbial Fuel Cell Operation
The MFC reactor used was a dual chamber glass reactor with a volume capacity of 250 mL each (with a working volume of 200 mL). The carbon brush was used as an anode. The as received carbon brush was washed with ethanol and water to remove any dust and impurities. The two glass chambers are separated by a proton exchange membrane (Nafion 117). The as received membrane was pre-treated in 30 wt.% H2O2, then in 0.5 M H2SO4 and finally in deionized (DI) water. Each step is performed for 1 h at 80 °C. The pre-treated membrane is then stored in DI until use. The cathode used was a commercial GDL (geometric area = 2.6 × 5.5 cm). The cathode catalyst Co/NSC and Pt/C in the form of catalyst ink is deposited onto the GDL. The cathode catalyst ink is prepared by dispersing definite of the Co/NSC catalyst in isopropyl alcohol and ultrasonicated for 30 min, to which 30 wt.% of Nafion (5 wt.%) solution is added and further ultrasonicated for 30 min to obtain the final catalyst ink. The catalyst ink is then coated onto the GDL by using paint brush layer by layer until a desired catalyst loading 2 mg cm−2 for both Co/NSC catalyst while 0.5 mg cm−2 in case of Pt/C catalyst is achieved. The two MFC chambers were coupled with the Nafion 117 membrane, and two chambers are filled with 200 mL of the buffer solution (pH = 7) on both sides. The anode chamber consists of 20 mM sodium acetate. The cathode is purged continuously with atmospheric air with the help of an air pump at a controlled flow rate (~10 mL min−1). In this work, we have “utilized bacterial seed sample”, from sewage water, collected from the local drainage as the source of bacteria. A sample of 10 mL sewage drainage water sample is added to the anodes of fresh MFC set up, allowing the bacteria to culture naturally in the anode chamber. Sewage water is a good source of mixed bacterial strains and mimics the most realistic wastewater treatment applications. In addition, as a source of carbon, we add 20 mM sodium acetate every alternative day to the anode chamber and allow the bacteria to grow. Over the course of time, bacteria attaches the carbon brush fibers and grow continuously, forming a biofilm [21,22]. The biofilm formation can be assessed from the fact that the OCV slowly rises and reaches the maximum after a period of time. After reaching a steady and stable OCV, then the polarization curves were recorded. The anode and cathodes are connected to the potentiostat to monitor the open circuit voltage. The polarization curves were recorded by linear sweep voltammetry at a scan rate of 1 mV s−1. The current density and power densities were normalized according to the cathode geometric area and volume of the anolyte.
3. Results
A simple one step solid-state synthesis technique was adopted to synthesize Co/N-C-S catalyst. Conventionally, M-N-C catalysts are synthesized by the traditional co-ordination reaction between 2-methyl imidazole, as organic ligand with a zinc/cobalt as metal nodes in a methanolic solution, which yields a precipitate over a reaction time and the precipitate needs to be washed and centrifuged [23]. In this study, a simple, quick, one pot synthesis strategy is developed in which the precursors are simply grinded in the mortar and pestle. During the grinding processes, mechanical energy favors the formation of co-ordination bonds to build the metal organic framework (MOF) like structures just as a methanolic solution reaction does [24,25,26,27]. There are a number of researchers who explored solid-state techniques to synthesize MOFs as can be seen from the literatures [28,29,30]. Even though it is well known that MOFs with N-heterocyclic organic ligands are often employed, there are not many investigations on MOFs with more than one heteroatom in the heterocyclic ring. For instance, the most popular ligand for organic compounds containing N is 2-methyl imidazole [31,32]. In addition to altering the electrical distribution, the presence of many heteroatoms in a single organic linker also affords efficient metal coordination sites during the synthesis of MOFs [33]. When creating MOFs, the presence of several heteroatoms in organic linkers helps to improve bond polarization which further helps in boding with metal ions. Electron-rich atoms, such as nitrogen and sulfur, further assist in unique interactions that occur during the solid-state synthesize of MOFs. These interactions include hydrogen bonding and π-π stacking, both of which contribute to the construction of stable, fascinating supramolecular architectures [33]. Thiazoles and thiazolidines, which promote coordination with both soft and hard metals from the periodic table, are especially attractive N, S-containing heterocyclic compounds for the aim of developing MOFs since they provide a wonderful opportunity to synthesis a range of MOFs [34,35].
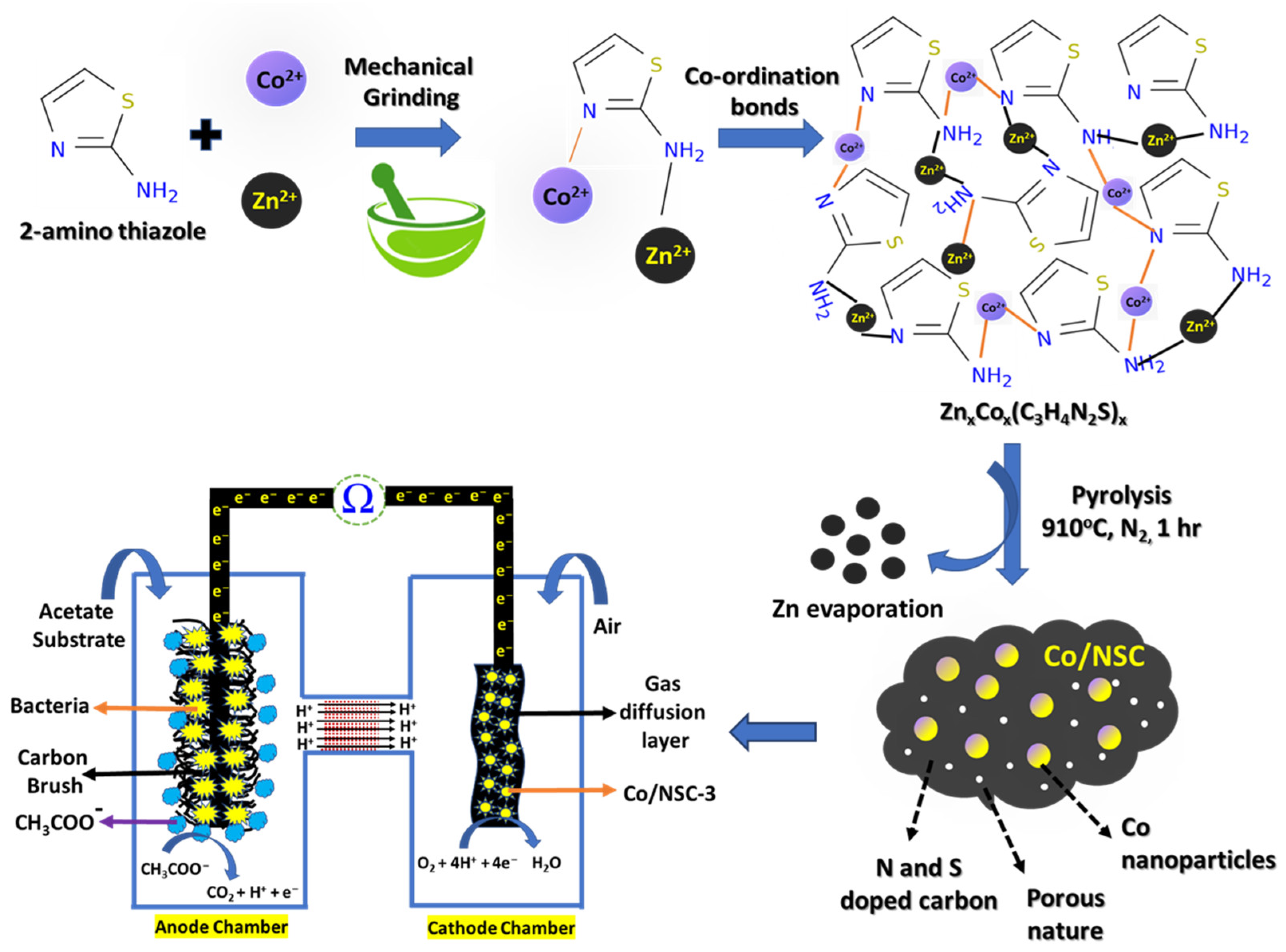
In this work, we used a solid-state synthesis method to create MOFs employing a thiazole organic ligand, namely 2-aminothiazole, with Zn2+ ions and Co2+ ions. This study was inspired by the assertions that were made above. In brief, calculated amount of zinc acetate, 2-aminothiazole with varying amount of cobalt nitrate are taken in a mortar and ground for around thirty minutes. This produces a thick pink color paste that, once dried, which results in the formation of possible intermediate precursor, ZnxCox(C3H4N2S), analogues to Co-MOF. The unique property of 2-aminothiazole is that it reacts in its neutral state, with the nitrogen atom in the ring acting as the more reactive center. The ring N and amino N, both involve in the resonance to form co-ordination bonds with various electrophiles such as transition metal atoms such as Co2+/Zn2+ ions to form intermediate ZnxCox(C3H4N2S) [36]. During the pyrolysis of intermediate precursor, ZnxCox(C3H4N2S) in the presence of a N2 atmosphere, the 2-aminothiazole ligand decomposes, graphitizes, and eventually converts into N, S-doped carbon. This occurs simultaneously with the carbo-reduction in the Co2+ ions into Co metal and Co-N-C active sites, as well as the loss of Zn to ultimately result in the desired Co/NCS catalyst, in which the cobalt nanoparticles are evenly distributed throughout the N and S-doped carbon support [Scheme 1]. The precursor ZnxCox(C3H4N2S) is synthesized by varying composition of Co2+ precursors (0.1/0.150/0.20/0.250 mg) and are pyrolyzed at 900 °C, under N2 atmosphere to obtain the Co/NCS–1, 2, 3 and 4, respectively. The obtained Co/NCS catalysts are characterized by various physical and electrochemical techniques as explained below.
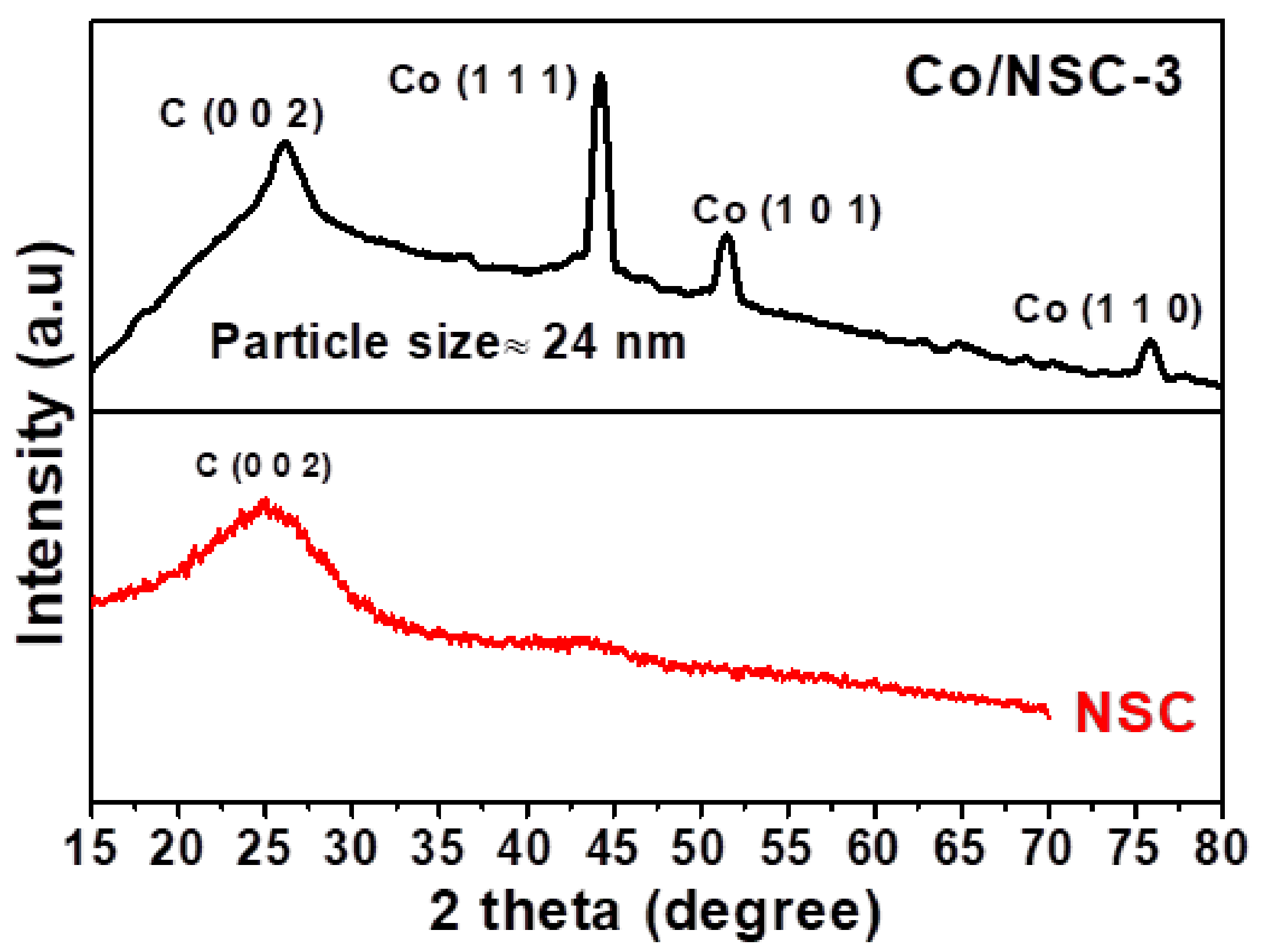
The XRD analysis was performed to identify the phases and crystal size of Co nanoparticles. Figure 1 shows the diffraction pattern of Co/NSC-3 which show diffraction peaks around ~26 (2θ) values corresponding to the carbon (0 0 2) plane, which results from the decomposition and graphitization of the 2-aminothiazole ligand into carbon. At the same time, the Co-NSC-3 sample showed the obvious peaks at the 2θ values of 44°, 51°, and 75° represent the metallic Co (111), Co (101), and Co (110) phases, respectively [37]. The average crystallite size of Co/NCS-3 was calculated using the Scherrer formula, by taking Co (111) phase, the results came out to be ~ 24 nm, respectively, for Co/NCS-3. Field emission scanning electron microscopy (FE-SEM) and transmission electron microscopy (TEM) techniques are used to analyze the morphological observations of the catalysts. Figure 2 shows the SEM images of the pyrolyzed Znx(SN2H4C3) and ZnxCox(C3H4N2S) which results in the formation of NCS and Co/NCS catalysts, respectively. It is observed that NCS catalyst (Figure 2a–c) showed a spherical morphology of the carbon derived from the pyrolysis of Znx(SN2H4C3). In contrast, the ZnxCox(C3H4N2S) derived catalyst, i.e., Co/NSC-3 (Figure 2d–f) showed larger particles of carbon, possibly due to presence of cobalt which induces the graphitization of the carbon during the pyrolysis process which results in increased particle size. Figure 2g–l shows the FE-SEM elemental mapping of the Co-NCS-3 catalyst, which confirm the presence of C, N, O, S and Co in the sample, especially presence of N and S indicates the successful doping of N and S into carbon matrix.
Figure 3 shows the TEM images of the NCS and Co/NCS catalysts. Figure 3a,b shows the TEM images of the NCS catalyst. It is clearly seen that a porous carbon derived from the pyrolysis of ZnxCox(C3H4N2S) catalyst, in which the 2-aminothiazole decomposes into carbon network. During the carbonization process, Zn2+ ions converts into ZnO (<750 °C), At carbonization temperatures > 750 °C, the ZnO clusters were reduced to metallic Zn through carbothermic reduction (ZnO + C → Zn(l) + CO(g)). Subsequently, further increasing the temperature to 910 °C, the Zn metal clusters (boiling point: 908 8C) were vaporized, along with the inert gases, Zn(l) → Zn(g). The pores in the carbon matrix are left by the evaporated gaseous Zn. As a result, Zn works as a porogen (porous inducing agent) in the carbon matrix [38,39]. The carbon’s porosity makes it easier for gaseous O2 to reach the catalytic active site while also facilitating the rapid removal of product water, which lowers the mass transfer limitations during the oxygen reduction reaction. Figure 3c–f shows the TEM images of the Co/NCS-3 catalyst which clearly shows the nanosized metallic Co nanoparticles are evenly distributed on the porous N and S doped carbon support, with the particle size between 8–27 nm, with the average particles are in the range of ~18–20 nm (Figure S1). The Co nanoparticles’ lattice fingers were determined to be Co (1 1 1) planes with an interlayer spacing of 0.209 nm (inset of Figure 3f).
To identify the chemical composition and chemical states of the Co/NCS catalyst, XPS analysis is carried out. The XPS survey spectra (Figure S2a) shows the presence of C, N, S and Co in the catalysts. Further to understand the chemical states, each spectrum is recorded again with a high-resolution scan and deconvoluted. Figure 4a shows the deconvoluted spectra of the C1s which reveals four distinctive peaks at the binding energies of 284.25, 284.9, 286.3, 288.9 eV, corresponding to the bonding of C-C/C=C, C-S, C-N, C=O, respectively [40]. The presence of -C-N- and C-S- bonds strongly suggests that the Co/NSC-3 catalyst possesses nitrogen and sulfur and confirms their successful incorporation into the carbon matrix following the decomposition of 2-aminothiazole. Figure 4b shows the deconvoluted N1s spectra which showed four different deconvoluted peaks at the binding energies of 398.08, 399.08, 400.68 and 401.78 eV corresponding to the N doping in the form of pyridinic-N, pyridinic-N-Cox, pyrrolic-N and graphitic-N, respectively [41].
By altering the electroneutrality of the carbon atoms, graphitic-N and pyridinic-N species are known to be the potential active sites for O2 adsorption and its reduction to H2O [42]. However, there exists a lot of controversy whether the pyridinic, graphitic, or both nitrogens’ are active sites for ORR. Some researchers believed that pyridinic N acts as effective active centers to boost electrocatalytic ORR performance [43,44], but others believed that graphitic N, rather than pyridinic N, contributes significantly to ORR performance enhancement [45,46]. Kim et al. proposed a mutual transition between graphitic N and pyridinic N using a cyclic C-N ring opening method to resolve this problem [47]. Due to simultaneous formation of different N species and lack of precisely controlled experimental techniques on type of N species, makes it a difficult task to in elucidating the unique active site. Therefore, it is believed that the synergistic effect of pyridinic-N and graphitic-N are responsible for ORR [48] Additionally, by contributing their electrons to the carbon network and thereby increasing the electron density around Co nanoparticles, pyridinic-N and graphitic-N species can also improve the interaction between O2 and Co nanoparticles in Co/NSC-3. Due to the high electron density around metallic Co catalytic centers, the work function of Co is reduced, which in turn leads to an improvement in the ORR kinetics. In addition to the pyridinic-N, appearance of a distinctive peak ~399.08 eV also suggests that pyridinic-N may also coordinate with Co metallic atoms, therefore, giving rise to a potentially active ORR sites namely Mx-N-C (Cox-N-C) [49]. Numerous theoretical and spectro-analytic studies evidenced that existence of Cox-N-C significantly enhances the ORR by a direct 4 electron transfer [50]. Therefore, presence of pyridinic-N, graphitic-N and Cox-N-C synergistically enhances the ORR activity. Figure 4c shows the deconvoluted S 2p3/2 into three different peaks at the binding energies of 161.68, 162.48 and 163.28 eV. The former two peaks corresponds to the existence of -C-SOx (x = 4/3/2) species and the later peak corresponds to the presence of -C-S-C bonds [51]. The evidence of -C-S-C bonds clearly indicates the effective doping of S atoms into the carbon matrix. It is well established that S atoms increases the spin density of the carbon atoms surrounding the S dopant, which boosts the ORR reactions [52]. The C-SOx-C bonding further shows that S dopant plays an important role in adsorbing O2 atoms during the ORR process. Figure 4d shows the deconvoluted Co2p spectra, which show five peaks at the binding energies of 777.4, 780.1, 782.6 and 796.1 eV, corresponding to metallic Co, Co-N-C/Co-Sx, CoNx, and Co3O4 peaks, respectively [53]. The existence of Co-N-C and Co-Nx species in the Co/NSC catalyst is further confirmed by the occurrence of peaks corresponding to Co-N atoms. Since S atoms are also present in the carbon matrix, it is impossible to rule out the potential of Co species (Co-Sx) forming bonds with S atoms. There is sufficient evidence to suggest that the addition of S atoms to M-N-C active ites further enhance the ORR kinetics by altering the electronic structure of the of M-N-C sites catalytically active centers.
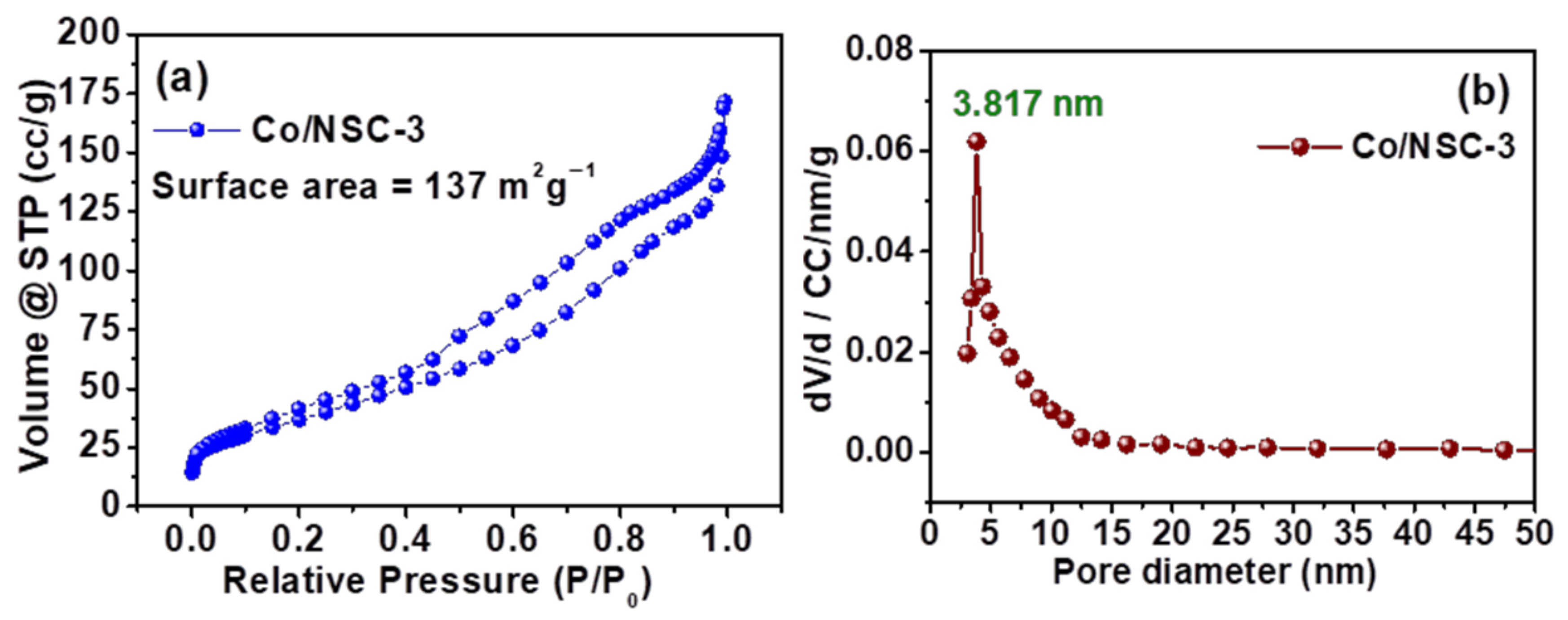
Figure 5 demonstrates the N2 adsorption and desorption isotherms as well as the BJH pore size distribution curves. The existence of mesopores in the catalyst is indicated by the fact that the Co/NCS-3 catalyst displays an isotherm of the type IV kind with a hysteresis loop ranging from a relative pressure of 0.40 to 1.0. The BET surface area that was achieved is 137 m2 g−1, and the mesopores that are present in the Co/NCS-3 catalyst are mostly between 3 and 15 nm in size, with a maximum number of pores of 3.817 nm. It is well knowledge that the existence of mesopores is largely significant for the increased ORR performance because it helps to strike a balance between the mass transfer of reactants towards the catalytic active sites and the removal of product water from the catalyst layer.
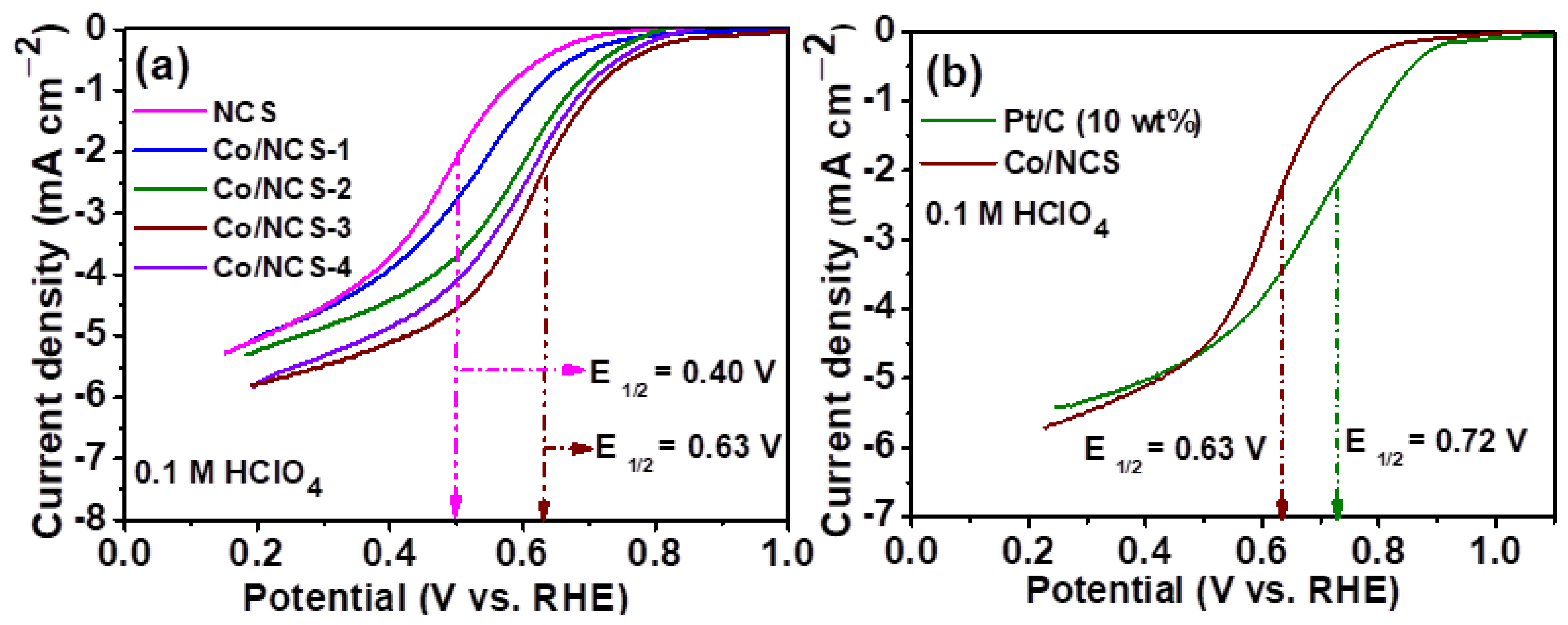
After ascertaining the morphology and chemical nature of the catalysts, the ORR activity of Co/NCS catalysts have been evaluated for ORR in 0.1 M HClO4 electrolytes. Figure 6a shows the ORR curves for Co/NCS catalysts synthesized with different Co contents. The catalyst is also prepared without Co for the ascertaining the role of Co. All the catalysts showed a typical characteristic of an ORR curve, with a well definite kinetic region, mixed kinetic diffusion-controlled region and limiting current regions, indicating that al the catalysts are active for ORR. Half-wave potential (E1/2), i.e a potential at which ORR curve is equal to one half of the limiting current, is calculated to assess the superiority of one catalyst over the other. The catalyst synthesized with 2-amino thiazole (NCS), showed the E1/2 of 0.40 V vs. RHE. The ORR activity of NCS catalyst is due to the presence of various N active sites in the catalyst such as graphitic, pyridinic and pyrrolic-N species, together with spin density induced kinetics by C-S-C active sites. Interestingly, the ORR activity of the NCS catalyst increases significantly when Co is introduced during the synthesis process. This is clearly evidenced from the increased ORR activity of Co/NCS catalysts. The E1/2 of the Co/NCS-1, Co/NCS-2, Co/NCS-3, and Co/NCS-4 catalysts are found to be 0.52, 0.57, 0.63 and 0.59 V vs. RHE, respectively. It is clearly evident that incorporation of Co could enhance the ORR activity by creating Co-N-C/Co-Sx, Co-Nx active sites as ascertained from the XPS analysis. Among the four Co/NCS catalysts, the Co/NCS-3 catalyst showed the optimum ORR activity with the E1/2 of 0.63 V vs. RHE. Therefore, quantitatively, a gain of 223 mV overpotential have been enhanced by Co metal active centers over NCS catalyst. The ORR activity of Co/NCS catalyst is compared with the commercial Pt/C (10 wt.% catalyst as shown in Figure 6b. The Pt/C catalyst exhibits the half wave potential of 0.72 V vs. RHE. Still, Pt/C catalyst show higher activity (~ catalyst loading of 110 µg cm−2) than the Co/NCS catalyst due to the fact that Pt is a noble metal with excellent ORR kinetics. The Co/NCS catalyst E1/2 is 90 mV lower than the Pt/C. Though, the gained ORR activity of Co/NCS is still encouraging due to the fact that Co based catalysts are cheaper than the noble metal catalyst.
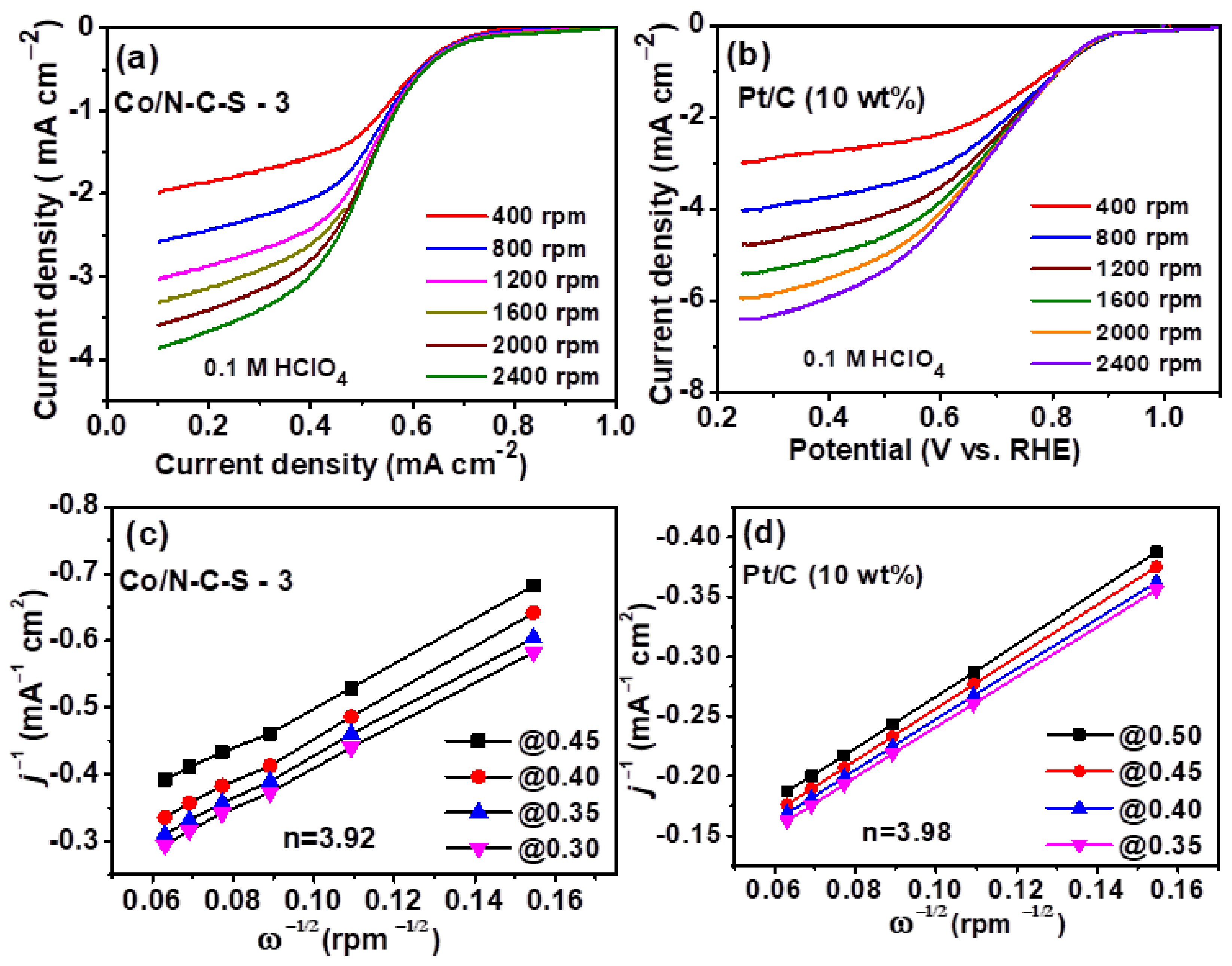
In order to gain knowledge on the ORR pathway, the LSV of Pt/C and Co/NCS catalysts are recorded at different rotations (Figure 7a,b) and K-L plots (Figure 7c,d) are derived. It is well known that ORR precedes either a direct 4 electron reduction process or by a 2 + 2 electron reduction process. The later pathway produces the undesired H2O2. The number of electrons involved in the ORR process is calculated from the slope of K-L plots, i.e., plots of inverse of square root of rotation speed vs. inverse of the current density. With no discernible change in the onset potential, it is shown that both catalysts exhibit increasing ORR limiting currents with regard to an increase in rpm, indicating that the ORR reaction is an O2 diffusion-controlled reaction (Figure 7a,b). Additional linearity in K-L graphs shows that electron transport kinetics at various potentials are virtually identical (Figure 7c,d). The number of electrons (n) were calculated to be 3.98 and 3.92 for Pt/C and Co/NCS catalysts, respectively. The obtained ‘n’ close to 4, clearly suggests that the catalyst perform ORR via a dominant, direct 4 electron pathway albeit a minor proportion of 2 + 2 electron reduction is still possible.
Further, the stability of Co/NCS-3 catalyst is then evaluated by potential cycling test by cyclic voltammetry technique. The catalyst stability is evaluated by taking LSV curve after the potential cycling test (Figure 8a–d). During the stability test analysis, the Pt/C catalyst showed a drastic decrease in the hydrogen adsorption/desorption region as can be seen from Figure 8a. This decrease in the currents of hydrogen adsorption/desorption region represents loss of electrochemical active surface area (ECSA) due to coalescence, Pt nanoparticles agglomeration induced by carbon corrosion [54]. This decrease in the ECSA causes less Pt surface exposure for the ORR catalysts therefore reducing the ORR activity. This was clearly established from the LSV curves recorded after 5000 and 10,000 potentials cycles as shown in Figure 8b. The half-wave potential of the Pt/C decreases by 180 mV after 10,000 potential cycles. Under similar experimental conditions, the Co/NSC-3 catalyst exhibited admirable stability. Figure 8c,d shows the cyclic voltammograms and LSV voltammograms of the Co/NCS-3 catalyst before and after the potential cycles, respectively. It is clearly seen the E1/2 of the Co/NCS-3 catalyst is hardly changed after 10,000 potential cycles, indicating that Co/NCS-3 catalyst have admirable stability. This significant stability of Co/NCS-3 is due to the interaction of Co nanoparticles with the N and S doped carbon support. To verify the stability of CO/NSC-3, we have analyzed the morphology of the Co/NSC-3 catalyst after the stability test, as shown in Figure S3. It is seen that Co nanoparticles seem disordered and along with slightly increased Co particle size after the stability test, due to potential cycling. However, the Co nanoparticles are still seen after the stability test, indicating that Co/NSC-3 catalyst is durable and stable against potential cycling. It is well established that carbon that has been doped with N and S has a high resistance to carbon corrosion during potential cycling. In addition, N-doped carbons offers anchoring effect to the metal nanoparticles, that alleviates the metal leaching in the acidic solution. Further, the stability of the Co/NCS catalyst is also evaluated by the constant voltage @ 0.4 V vs. RHE in 0.1 M HClO4 acidic solution as shown in Figure 8e. After a 17 h of the chronoamperometric measurement, the Co/NCS catalyst maintain ~99% of its initial current. This explains the excellent stability of the Co/NCS catalyst both under potential cycling test and chronoamperometric test.
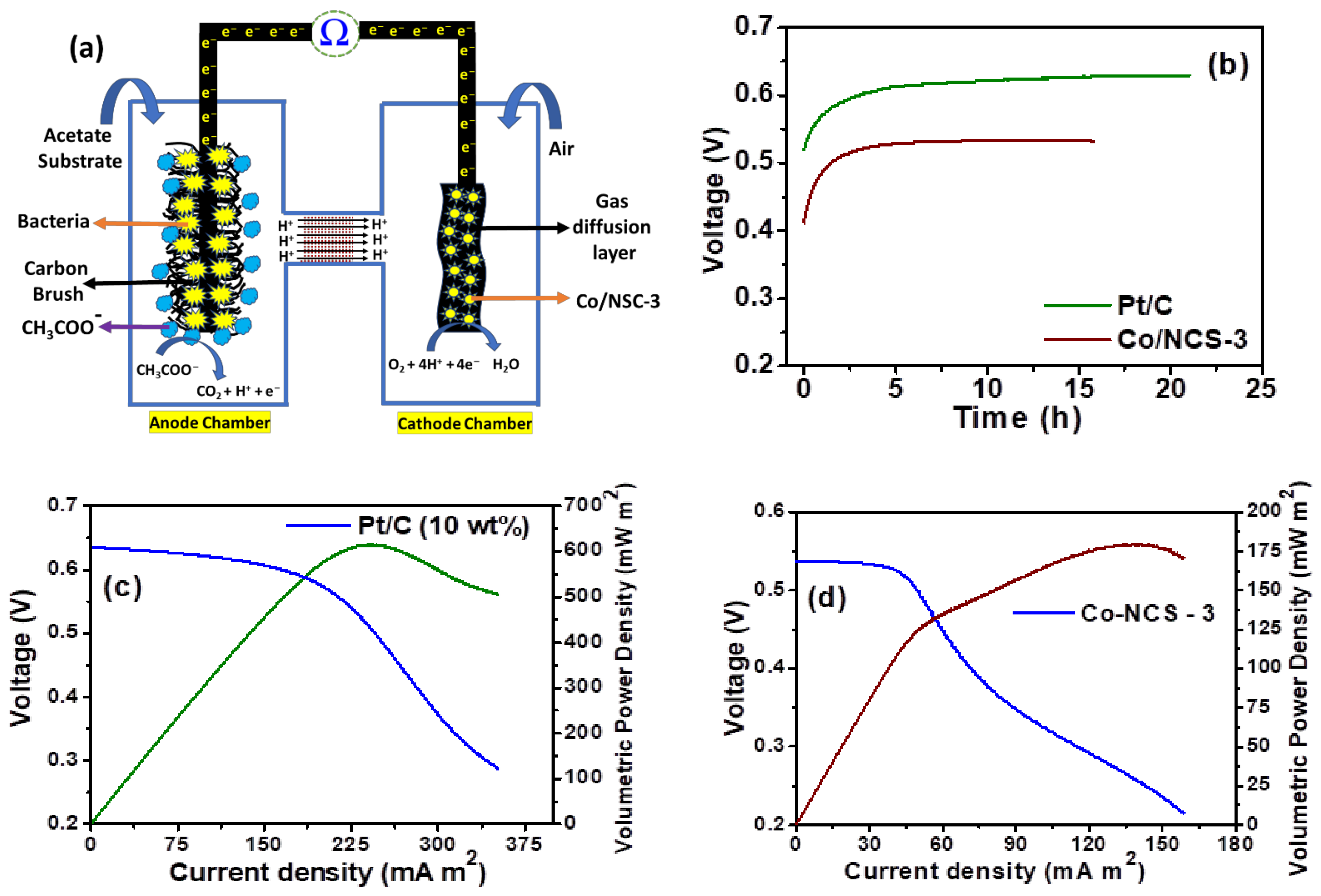
After ascertaining ORR activity of Co/NCS-3 catalyst, it is applied as cathode and its deliverable power density performance was evaluated in a dual chamber MFC set up as shown in in Figure 9a. The Co/NCS-3 catalyst is coated on the commercial gas diffusion layer at the catalyst loading of 2 mg cm−2. At first the open circuit voltage (OCV) was recorded by introducing Co/NSC-3 coated GDL into the cathode chamber of the already activated dual chamber MFC. It can be seen that the starting voltage of 412 mV is seen as soon as the catalyst is introduced into the cathode chamber. The potential slowly rose to a maximum 533 mV after 5 h of time and the voltage stayed relatively constant for about 15 h, indicating that the maximum voltage was achieved. Under similar conditions, Pt/C coated gas diffusion layer is also used as a cathode catalyst, which showed a starting voltage of 523 mV and the potential rose to maximum of 630 mV at the end time of 20 h (Figure 9b).
After achieving a stabilized OCV, the i–v curves are recorded to obtain the achievable power density of Pt/C and Co-NSC-3 catalysts as shown in the (Figure 9b–d). A volumetric power density of about 180 mW m2 was achieved by using Co-NSC-3 catalyst as cathode, catalyst, whereas Pt/C catalyst delivered a volumetric power density of ~615 mW m2. The difference in the obtained power density was reflected from the RDE studies, where Pt/C catalyst still showed better performance than the Co/NCS-3 catalyst. However, the power density of Co/NSC-3 catalyst can be further increased with the increased catalyst loading to match the performance of Pt/C. Increasing the catalyst loading of Co/NSC-3 is still justified, because of the lower cost of cobalt than the Pt/C.
4. Conclusions
Developing cheap, cost-effective catalysts is essential for the successful commercialization of the MFC. We have successfully synthesized the earth’s abundant Co based catalyst by a solid-state synthesis process by utilizing Co nitrate, Zinc nitrate and 2-aminothiazole. Pyrolysis of the (ZnxCo(C3H4N2S)) resulted in Co nanoparticles supported on the N and S doped carbons (Co/NSC). The TEM analysis clearly indicates that Co nanoparticles are evenly distributed on the N and S doped carbon support. The Co/NSC catalyst also showed the porous nature of the N and S doped carbons resulted from the evaporation of metallic Zn during the pyrolysis process. The XPS analysis shows the presence of catalytically actives phases of Co in the form of CoNx/CoSx/metallic Co species. RDE results reveals that Co/NSC-3 catalyst showed optimized ORR activity. The Co/NSC catalyst also showed excellent stability over 10,000 potential cycles and in chronoamperometric studies. In the DCMFC, the Co/NSC catalyst delivered a volumetric power density of ~180 mW m2.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/nano12244369/s1, Details of the commercial GDL used as backing layers for both anodes and cathodes. Figure S1: Particle size distribution of Co nanoparticle in Co-NSC-3 catalysts derived from the TEM images.; Figure S2: (a) XPS survey spectrum for the Co-NSC-3 catalyst (b–d), survey scan of Zn 3p, 3s and 2p spectra, respectively. (e) Zn 2p high resolution scan. Figure S2.T: Elemental composition of Co-NSC-3 catalyst obtained from XPS analysis; Figure S3. TEM images of Co/NSC-3 catalysts after stability test.
Author Contributions
Conceptualization, S.A.; methodology, S.A.; validation, H.-J.K.; formal analysis, S.G.P.; investigation, S.A.; resources, S.G.P. and T.G.L.; data curation, S.A.; writing—original draft preparation, S.A.; writing—review and editing, S.G.P. and T.G.L.; supervision, T.G.L.; project administration, S.G.P.; funding acquisition, S.G.P. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Research Foundation of Korea (NRF) funded by the Korean government (MSIT) (grant no. 2021R1F1A1046648), Republic of Korea.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Bhim Sen Thapa, Department of Biological Sciences, Marquette University, for helping us with the setting up of the MFC set up.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Slate, A.J.; Whitehead, K.A.; Brownson, D.A.C.; Banks, C.E. Microbial Fuel Cells: An Overview of Current Technology. Renew. Sustain. Energy Rev. 2019, 101, 60–81. [Google Scholar] [CrossRef]
- Zheng, G.; Zhen, H. Long-term performance of a 200-liter modularized microbial fuel cell system treating municipal wastewater: Treatment, energy, and cost. Environ. Sci. Water Res. Technol. 2016, 2, 274–281. [Google Scholar]
- Thiong’o, M.; Osano, A.; Bakari, C. Investigating the ohmic behavior of mediator-less microbial fuel cells using sewerage water as the bio-anode. Cogent Eng. 2022, 9, 2079222. [Google Scholar] [CrossRef]
- Ucar, D.; Zhang, Y.; Angelidaki, I. An Overview of Electron Acceptors in Microbial Fuel Cells. Front. Microbiol. 2017, 8, 643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aftab, S.; Shah, A.; Nisar, J.; Ashiq, M.N.; Akhter, M.S.; Shah, A.H. Marketability Prospects of Microbial Fuel Cells for Sustainable Energy Generation. Energy Fuels 2020, 34, 9108–9136. [Google Scholar] [CrossRef]
- Do, M.H.; Ngo, H.H.; Guo, W.S.; Liu, Y.; Chang, S.W.; Nguyen, D.D.; Nghiem, L.D.; Ni, B.J. Challenges in the Application of Microbial Fuel Cells to Wastewater Treatment and Energy Production: A Mini Review. Sci. Total Environ. 2018, 639, 910–920. [Google Scholar] [CrossRef]
- Ben Liew, K.; Daud, W.R.W.; Ghasemi, M.; Leong, J.X.; Su Lim, S.; Ismail, M. Non-Pt Catalyst as Oxygen Reduction Reaction in Microbial Fuel Cells: A Review. Int. J. Hydrogen Energy 2014, 39, 4870–4883. [Google Scholar] [CrossRef]
- Peera, S.G.; Maiyalagan, T.; Liu, C.; Ashmath, S.; Lee, T.G.; Jiang, Z.; Mao, S. A Review on Carbon and Non-Precious Metal Based Cathode Catalysts in Microbial Fuel Cells. Int. J. Hydrogen Energy 2021, 46, 3056–3089. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, Y.; Wang, H.; Zhang, J.; Zhao, H.; Chen, L.; Xu, L.; Xie, Y.; Huang, J. MIL-88-Derived N and S Co-Doped Carbon Materials with Supplemental FeSx to Enhance the Oxygen Reduction Reaction Performance. Catalysts 2022, 12, 806. [Google Scholar] [CrossRef]
- Nandikes, G.; Gouse Peera, S.; Singh, L. Perovskite-Based Nanocomposite Electrocatalysts: An Alternative to Platinum ORR Catalyst in Microbial Fuel Cell Cathodes. Energies 2021, 15, 272. [Google Scholar] [CrossRef]
- Xu, H.; Wang, D.; Yang, P.; Liu, A.; Li, R.; Li, Y.; Xiao, L.; Ren, X.; Zhang, J.; An, M. Atomically Dispersed M–N–C Catalysts for the Oxygen Reduction Reaction. J. Mater. Chem. A 2020, 8, 23187–23201. [Google Scholar] [CrossRef]
- Ma, Q.; Jin, H.; Zhu, J.; Li, Z.; Xu, H.; Liu, B.; Zhang, Z.; Ma, J.; Mu, S. Stabilizing Fe–N–C Catalysts as Model for Oxygen Reduction Reaction. Adv. Sci. 2021, 8, 2102209. [Google Scholar] [CrossRef]
- Zitolo, A.; Ranjbar-Sahraie, N.; Mineva, T.; Li, J.; Jia, Q.; Stamatin, S.; Harrington, G.F.; Lyth, S.M.; Krtil, P.; Mukerjee, S.; et al. Identification of Catalytic Sites in Cobalt-Nitrogen-Carbon Materials for the Oxygen Reduction Reaction. Nat. Commun. 2017, 8, 957. [Google Scholar] [CrossRef] [Green Version]
- Osmieri, L. Transition Metal–Nitrogen–Carbon (M–N–C) Catalysts for Oxygen Reduction Reaction. Insights on Synthesis and Performance in Polymer Electrolyte Fuel Cells. ChemEngineering 2019, 3, 16. [Google Scholar] [CrossRef] [Green Version]
- Hao, Z.; Ma, Y.; Chen, Y.; Fu, P.; Wang, P. Non-Noble Metal Catalysts in Cathodic Oxygen Reduction Reaction of Proton Exchange Membrane Fuel Cells: Recent Advances. Nanomaterials 2022, 12, 3331. [Google Scholar] [CrossRef]
- Stock, N.; Biswas, S. Synthesis of Metal-Organic Frameworks (MOFs): Routes to Various MOF Topologies, Morphologies, and Composites. Chem. Rev. 2012, 112, 933–969. [Google Scholar] [CrossRef]
- Welton, T. Solvents and Sustainable Chemistry. Proc. R. Soc. A Math. Phys. Eng. Sci. 2015, 471, 20150502. [Google Scholar] [CrossRef] [Green Version]
- A Better Solution for the Environment. Available online: www.chemistryworld.com/news/enhancing-solvents-sustainability/3010810.article (accessed on 7 August 2019).
- Amrute, A.P.; De Bellis, J.; Felderhoff, M.; Schüth, F. Mechanochemical Synthesis of Catalytic Materials. Chem.–A Eur. J. 2021, 27, 6819–6847. [Google Scholar] [CrossRef]
- Peera, S.G.; Liu, C. Unconventional and Scalable Synthesis of Non-Precious Metal Electrocatalysts for Practical Proton Exchange Membrane and Alkaline Fuel Cells: A Solid-State Co-Ordination Synthesis Approach. Coord. Chem. Rev. 2022, 463, 214554. [Google Scholar] [CrossRef]
- Rehab, H.M.; Farag, A.S.; Mohamed, K.I.; Gamila, H.A.; Rabeay, Y.A.H. Formation of electroactive biofilms derived by nanostructured anodes surfaces. Bioprocess Biosyst. Eng. 2021, 44, 759–768. [Google Scholar] [CrossRef]
- Numfon, E.; Christina, S.K.; Song, J.R.; Yanasinee, S.; Han, S.K. Enhanced Current Production by Electroactive Biofilm of Sulfate-Reducing Bacteria in the Microbial Fuel Cell. Environ. Eng. Res. 2013, 18, 277–281. [Google Scholar] [CrossRef]
- Yap, M.H.; Fow, K.L.; Chen, G.Z. Synthesis and Applications of MOF-Derived Porous Nanostructures. Green Energy Environ. 2017, 2, 218–245. [Google Scholar] [CrossRef]
- Xiuxiu, L.; Changdong, S.; Changwei, Z.; Meiling, C.; Qi, L.; Guoxiu, W. Cobalt-Based Layered Metal–Organic Framework as an Ultrahigh Capacity Supercapacitor Electrode Material. ACS Appl. Mater. Interfaces 2016, 8, 4585. [Google Scholar] [CrossRef]
- Xiaoshi, H.; Huiping, H.; Chao, L.; Tian, L.; Xiaobing, L.; Qun, C.; Bingwen, H. Cobalt-based metal organic framework with superior lithium anodic performance. J. Solid State Chem. 2016, 242, 71–76. [Google Scholar] [CrossRef]
- Niu, Q.; Guo, J.; Chen, B.; Nie, J.; Guo, X.; Ma, G. Bimetal-organic frameworks/polymer core-shell nanofibers derived heteroatom-doped carbon materials as electrocatalysts for oxygen reduction reaction. Carbon 2017, 114, 250. [Google Scholar] [CrossRef]
- Lu, H.; Zhang, H.; Liu, R.; Zhang, X.; Zhao, H.; Wang, G. Macroscale cobalt-MOFs derived metallic Co nanoparticles embedded in N-doped porous carbon layers as efficient oxygen electrocatalysts. Appl. Surf. Sci. 2017, 392, 402. [Google Scholar] [CrossRef] [Green Version]
- Song, D.; Hu, C.; Gao, Z.; Yang, B.; Li, Q.; Zhan, X.; Tong, X.; Tian, J. Metal–Organic Frameworks (MOFs) Derived Materials Used in Zn–Air Battery. Materials 2022, 15, 5837. [Google Scholar] [CrossRef]
- Chen, D.; Zhao, J.; Zhang, P.; Dai, S. Mechanochemical Synthesis of Metal–Organic Frameworks. Polyhedron 2019, 162, 59–64. [Google Scholar] [CrossRef]
- Tao, C.-A.; Wang, J.-F. Synthesis of Metal Organic Frameworks by Ball-Milling. Crystals 2020, 11, 15. [Google Scholar] [CrossRef]
- Yang, C.; Ma, X.; Zhou, J.; Zhao, Y.; Xiang, X.; Shang, H.; Zhang, B. Recent Advances in Metal-Organic Frameworks-Derived Carbon-Based Electrocatalysts for the Oxygen Reduction Reaction. Int. J. Hydrogen Energy 2022, 47, 21634–21661. [Google Scholar] [CrossRef]
- Song, Z.; Zhang, L.; Doyle-Davis, K.; Fu, X.; Luo, J.; Sun, X. Recent Advances in MOF-Derived Single Atom Catalysts for Electrochemical Applications. Adv. Energy Mater. 2020, 10, 2001561. [Google Scholar] [CrossRef]
- Rossin, A.; Giambastiani, G. Structural Features and Applications of Metal–Organic Frameworks Containing Thiazole- and Thiazolidine-Based Spacers. CrystEngComm 2015, 17, 218–228. [Google Scholar] [CrossRef]
- Chong-Hyeak, K.; Inn, H.K. Tetra kis(2-amino thia zole-κN 3)dichloridocadmium(II). Acta Crystallogr. Sect. E Struct. Rep. Online 2010, 66, m13. [Google Scholar]
- Suh, S.W.; Kim, C.-H.; Kim, I.H. Bis(2-amino-benzothia-zole-κN)bis-(thio-cyanato-κN)zinc(II). Acta Crystallogr. Sect. E Struct. Rep. Online 2009, 65, m1054. [Google Scholar] [CrossRef] [Green Version]
- Shaik, G.P.; Kwon, H.-J.; Lee, T.G. Highly Efficient Co@NCS Nanosheet Electrocatalyst for Oxygen Reduction Reaction: An Environment-Friendly, Low-Cost and Sustainable Electrocatalyst. Mater. Res. Bull. 2020, 128, 110873. [Google Scholar] [CrossRef]
- Peera, S.G.; Balamurugan, J.; Kim, N.H.; Lee, J.H. Sustainable Synthesis of Co@NC Core Shell Nanostructures from Metal Organic Frameworks via Mechanochemical Coordination Self-Assembly: An Efficient Electrocatalyst for Oxygen Reduction Reaction. Small 2018, 14, 1800441. [Google Scholar] [CrossRef]
- Dan, Q.; Cheng, L.; En-Min, W.; Wen-Cui, L.; An-Hui, L. A Method for Creating Microporous Carbon Materials with Excellent CO2-Adsorption Capacity and Selectivity. ChemSusChem 2014, 7, 291–298. [Google Scholar] [CrossRef]
- Bing, Y.; Jiaojiao, Z.; Feng, W.; Luying, Z.; Qian, Z.; Wenhui, X.; Shuijian, H. Review on porous carbon materials engineered by ZnO templates: Design, synthesis and capacitance performance. Mater. Des. 2021, 201, 109518. [Google Scholar] [CrossRef]
- Chao, H.; Chang, Y.; Mingyu, L.; Xiuna, W.; Qiang, D.; Gang, W.; Jieshan, Q. Nitrogen-doped carbon dots decorated on graphene: A novel all-carbon hybrid electrocatalyst for enhanced oxygen reduction reaction. Chem. Commun 2015, 51, 3419–3422. [Google Scholar] [CrossRef]
- Zhang, X.; Fan, Q.; Qu, N.; Yang, H.; Wang, M.; Liu, A.; Yang, J. Ultrathin 2D Nitrogen-Doped Carbon Nanosheets for High Performance Supercapacitors: Insight into the Effects of Graphene Oxides. Nanoscale 2019, 11, 8588–8596. [Google Scholar] [CrossRef]
- James, A.B.; Eric, M.T.; Serban, N.S.; Carlota, D.; Alessandro, I.; Karsten, F.; Khairul, H.; Tatiana, S.P.; Max, G.-M.; Paula, E.C. Untangling Cooperative Effects of Pyridinic and Graphitic Nitrogen Sites at Metal-Free N-Doped Carbon Electrocatalysts for the Oxygen Reduction Reaction. Small 2019, 15, 1902081. [Google Scholar] [CrossRef]
- Qu, L.; Liu, Y.; Baek, J.-B.; Dai, L. Nitrogen-doped graphene as efficient metal-free electrocatalyst for oxygen reduction in fuel cells. ACS Nano 2010, 4, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Chitturi, V.R.; Carlos, R.C.; Yasuyuki, I. In search of the active site in nitrogen-doped carbon nanotube electrodes for the oxygen reduction reaction. J. Phys. Chem. Lett. 2010, 1, 2622–2627. [Google Scholar] [CrossRef]
- Kabir, S.; Artyushkova, K.; Serov, A.; Kiefer, B.; Atanassov, P. Binding Energy Shifts for Nitrogen-containing Graphene-based Electrocatalysts—Experiments and DFT Calculations. Surf. Interface Anal. 2016, 48, 293–300. [Google Scholar] [CrossRef]
- Khaled, P.; Shubin, Y.; Yenny, H.; Andreas, W.; Andrey, T.; Xinliang, F.; Klaus, M. Nitrogen-doped graphene and its iron-based composite as efficient electrocatalysts for oxygen reduction reaction. ACS Nano 2012, 6, 9541–9550. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Wu, D.; Feng, X.; Müllen, K. Nitrogen-doped ordered mesoporous graphitic arrays with high electrocatalytic activity for oxygen reduction. Angew. Chem. Int. Ed. 2010, 49, 2565–2569. [Google Scholar] [CrossRef]
- Kim, H.; Lee, K.; Woo, S.I.; Jung, Y. On the mechanism of enhanced oxygen reduction reaction in nitrogen-doped graphene nanoribbons. Phys. Chem. Chem. Phys. 2011, 13, 17505–17510. [Google Scholar] [CrossRef]
- Ning, X.; Li, Y.; Ming, J.; Wang, Q.; Wang, H.; Cao, Y.; Peng, F.; Yang, Y.; Yu, H. Electronic synergism of pyridinic- and graphitic-nitrogen on N-doped carbons for the oxygen reduction reaction. Chem. Sci. 2019, 10, 1589–1596. [Google Scholar] [CrossRef] [Green Version]
- Tylus, U.; Jia, Q.; Strickland, K.; Ramaswamy, N.; Serov, A.; Atanassov, P.; Mukerjee, S. Elucidating Oxygen Reduction Active Sites in Pyrolyzed Metal–Nitrogen Coordinated Non-Precious-Metal Electrocatalyst Systems. J. Phys. Chem. C 2014, 118, 8999–9008. [Google Scholar] [CrossRef]
- Zhang, C.; An, B.; Yang, L.; Wu, B.; Shi, W.; Wang, Y.-C.; Long, L.-S.; Wang, C.; Lin, W. Sulfur-Doping Achieves Efficient Oxygen Reduction in Pyrolyzed Zeolitic Imidazolate Frameworks. J. Mater. Chem. A 2016, 4, 4457–4463. [Google Scholar] [CrossRef]
- Gouse Peera, S.; Kwon, H.-J.; Lee, T.G.; Hussain, A.M. Heteroatom- and Metalloid-Doped Carbon Catalysts for Oxygen Reduction Reaction: A Mini-Review. Ionics 2020, 26, 1563–1589. [Google Scholar] [CrossRef]
- Wu, W.; Zhang, Q.; Wang, X.; Han, C.; Shao, X.; Wang, Y.; Liu, J.; Li, Z.; Lu, X.; Wu, M. Enhancing Selective Photooxidation through Co–Nx-Doped Carbon Materials as Singlet Oxygen Photosensitizers. ACS Catal. 2017, 7, 7267–7273. [Google Scholar] [CrossRef]
- Peera, S.G.; Koutavarapu, R.; Akula, S.; Asokan, A.; Moni, P.; Selvaraj, M.; Balamurugan, J.; Kim, S.O.; Liu, C.; Sahu, A.K. Carbon Nanofibers as Potential Catalyst Support for Fuel Cell Cathodes: A Review. Energy Fuels 2021, 35, 11761–11799. [Google Scholar] [CrossRef]
Scheme 1.
Graphical representation of the Co/NSC catalyst synthesis process and its application in DCMFC.
Scheme 1.
Graphical representation of the Co/NSC catalyst synthesis process and its application in DCMFC.

Figure 1.
XRD patterns of NSC and Co/NSC-3 catalysts.

Figure 2.
SEM images of NCS (a–c), Co/NSC-3 (d–f) and SEM elemental mapping for Co/NSC-3 catalyst (g) images area (h) mapping for C (i) N (j) O (k) S (l) Co atoms.
Figure 2.
SEM images of NCS (a–c), Co/NSC-3 (d–f) and SEM elemental mapping for Co/NSC-3 catalyst (g) images area (h) mapping for C (i) N (j) O (k) S (l) Co atoms.

Figure 3.
TEM images of NCS (a,b), Co/NSC-3 (c–f). The yellow box in figure (b) indicates the porous nature of NCS carbon. Inset of (f) shows the lattice fringes with the d = 0.209 nm corresponds to Co (1 1 1).
Figure 3.
TEM images of NCS (a,b), Co/NSC-3 (c–f). The yellow box in figure (b) indicates the porous nature of NCS carbon. Inset of (f) shows the lattice fringes with the d = 0.209 nm corresponds to Co (1 1 1).

Figure 4.
XPS deconvoluted spectra for (a) C 1 s (b) N 1 s (c) S 2p3/2 (d) Co 2p spectra.

Figure 5.
(a) N2 adsorption/desorption isotherm curves of Co/NSC-3 and (b) pore size distribution curves of Co/NSC-3 catalyst.
Figure 5.
(a) N2 adsorption/desorption isotherm curves of Co/NSC-3 and (b) pore size distribution curves of Co/NSC-3 catalyst.

Figure 6.
(a) Linear Sweep Voltametric (LSV) curves of Co/NSC catalysts with different Co contents (b) Comparison of LSV curves of Co@NSC-3 catalyst with commercial Pt/C (10 wt.%). Electrolyte: 0.1 M HClO4, scan rate: 10 mV/s.
Figure 6.
(a) Linear Sweep Voltametric (LSV) curves of Co/NSC catalysts with different Co contents (b) Comparison of LSV curves of Co@NSC-3 catalyst with commercial Pt/C (10 wt.%). Electrolyte: 0.1 M HClO4, scan rate: 10 mV/s.

Figure 7.
LSV curves of (a) Co/NSC-3 (b) Pt/C (10 wt.%) recorded at different rotation speeds and their respective K-L plots (c,d), respectively.
Figure 7.
LSV curves of (a) Co/NSC-3 (b) Pt/C (10 wt.%) recorded at different rotation speeds and their respective K-L plots (c,d), respectively.

Figure 8.
Cyclic voltammograms of (a) Pt/C (10 wt.%) (c) Co/NSC-3 recorded before and after the stability test. LSV curves recorded after 5000 and/or 100,000 potential cycles of cyclic voltammograms for (b) Pt/C (10 wt.%) (d) Co/NSC-3. (e) Chronoamperometric responses of Co/NSC-3 in O2 saturated electrolyte. Electrolyte: 0.1 M HClO4, scan rate: 50 mV/sec (CV) 10 mV/sec (LSV).
Figure 8.
Cyclic voltammograms of (a) Pt/C (10 wt.%) (c) Co/NSC-3 recorded before and after the stability test. LSV curves recorded after 5000 and/or 100,000 potential cycles of cyclic voltammograms for (b) Pt/C (10 wt.%) (d) Co/NSC-3. (e) Chronoamperometric responses of Co/NSC-3 in O2 saturated electrolyte. Electrolyte: 0.1 M HClO4, scan rate: 50 mV/sec (CV) 10 mV/sec (LSV).

Figure 9.
(a) Schematic representation of dual chamber microbial fuel cells (b) recorded OCV values of Co/NSC-3 and Pt/C (10 wt.%). The i-v characteristics of (c) Pt/C (10 wt.%) (d) Co-NSC-3 catalysts in dual chamber microbial fuel cells. Anolyte: 20 mM sodium acetate + pH 7 buffer, catholyte: Air saturated pH 7 buffer.
Figure 9.
(a) Schematic representation of dual chamber microbial fuel cells (b) recorded OCV values of Co/NSC-3 and Pt/C (10 wt.%). The i-v characteristics of (c) Pt/C (10 wt.%) (d) Co-NSC-3 catalysts in dual chamber microbial fuel cells. Anolyte: 20 mM sodium acetate + pH 7 buffer, catholyte: Air saturated pH 7 buffer.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ashmath, S.; Kwon, H.-J.; Peera, S.G.; Lee, T.G. Solid-State Synthesis of Cobalt/NCS Electrocatalyst for Oxygen Reduction Reaction in Dual Chamber Microbial Fuel Cells. Nanomaterials 2022, 12, 4369. https://doi.org/10.3390/nano12244369
AMA Style
Ashmath S, Kwon H-J, Peera SG, Lee TG. Solid-State Synthesis of Cobalt/NCS Electrocatalyst for Oxygen Reduction Reaction in Dual Chamber Microbial Fuel Cells. Nanomaterials. 2022; 12(24):4369. https://doi.org/10.3390/nano12244369
Chicago/Turabian StyleAshmath, Shaik, Hyuk-Jun Kwon, Shaik Gouse Peera, and Tae Gwan Lee. 2022. "Solid-State Synthesis of Cobalt/NCS Electrocatalyst for Oxygen Reduction Reaction in Dual Chamber Microbial Fuel Cells" Nanomaterials 12, no. 24: 4369. https://doi.org/10.3390/nano12244369
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.