

Solid-State Synthesis of Cobalt/NCS Electrocatalyst for Oxygen Reduction Reaction in Dual Chamber Microbial Fuel Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
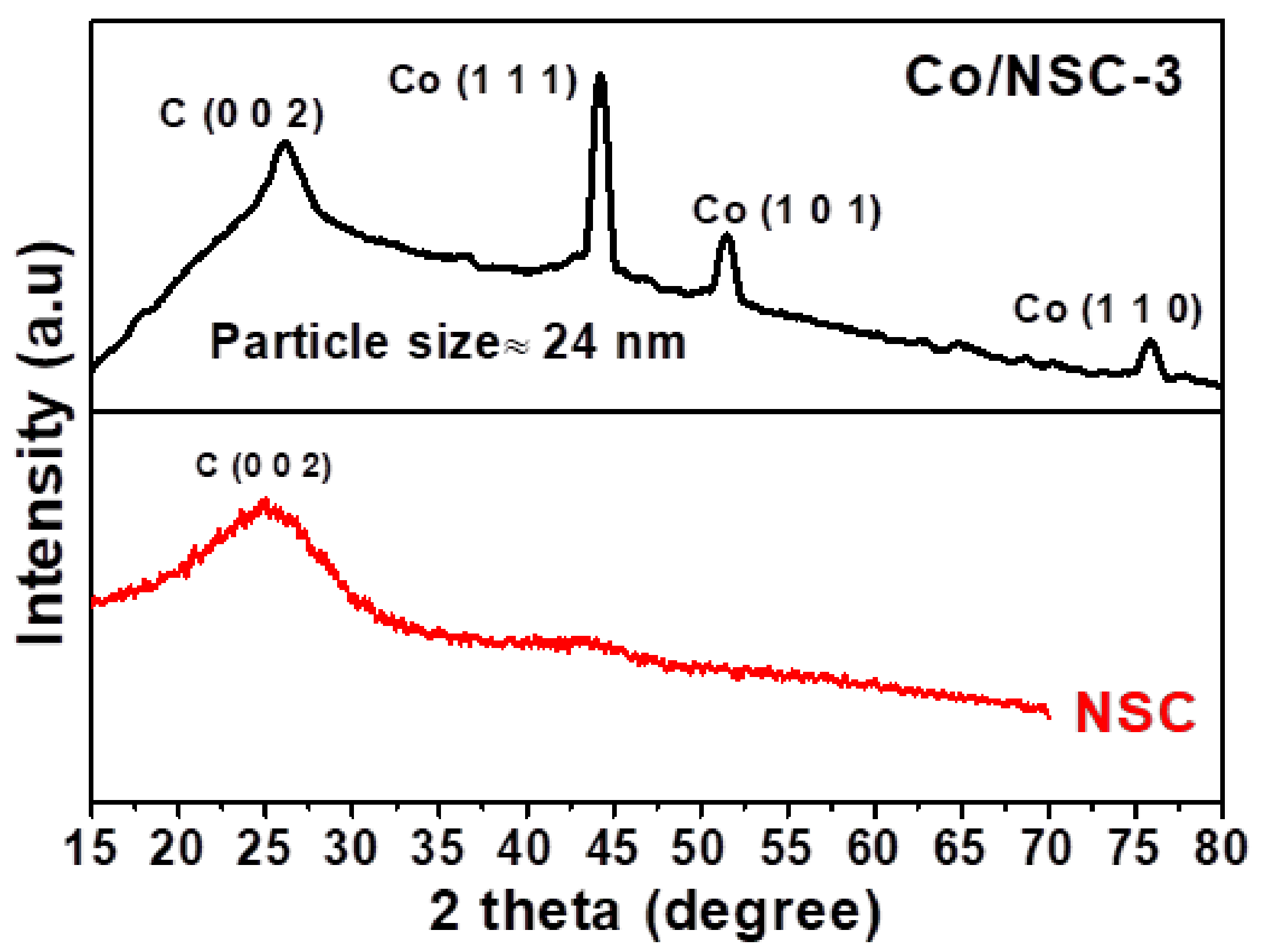
{kind=link}
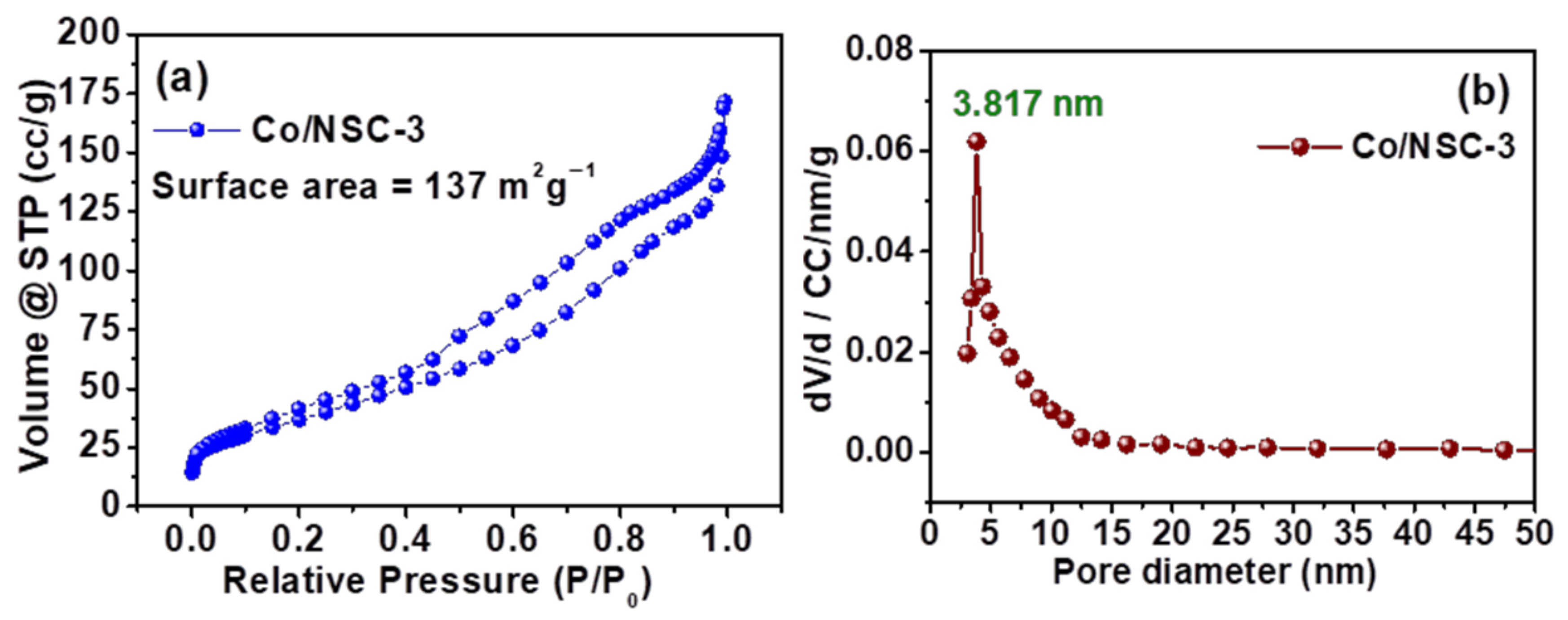
{kind=link}
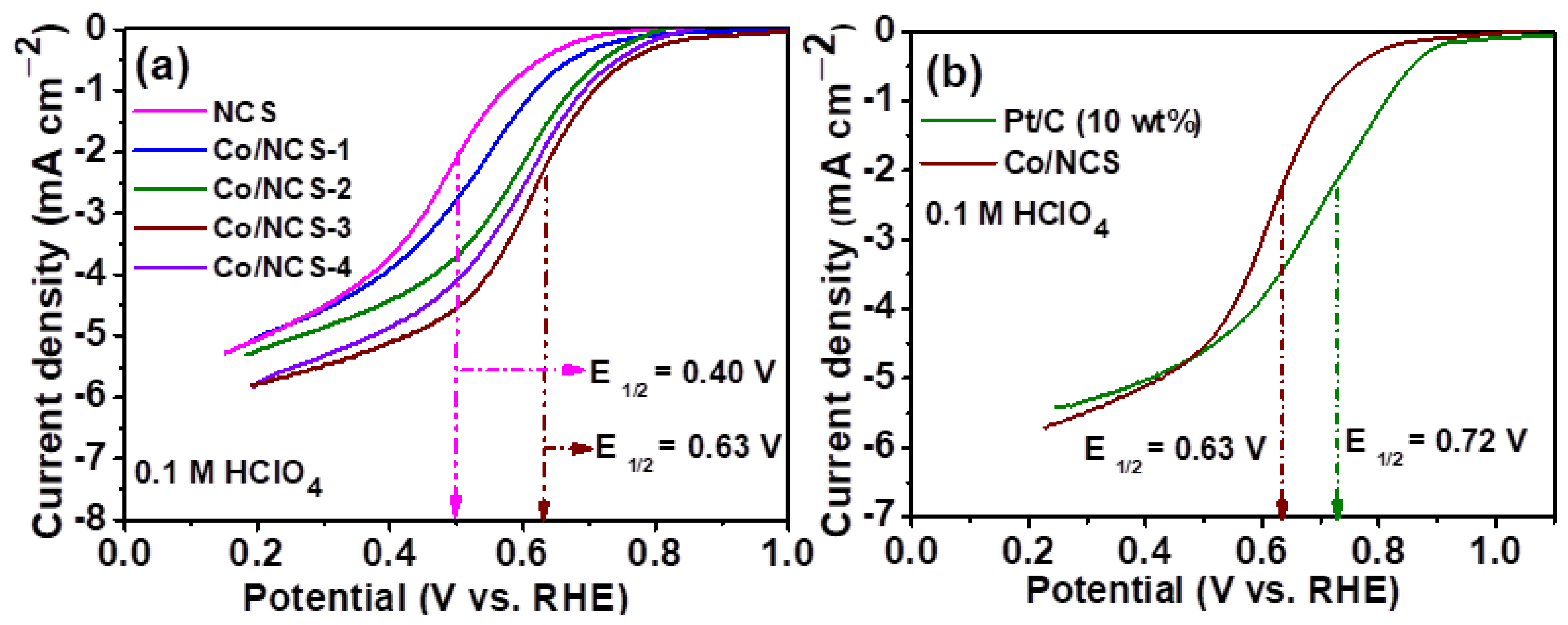
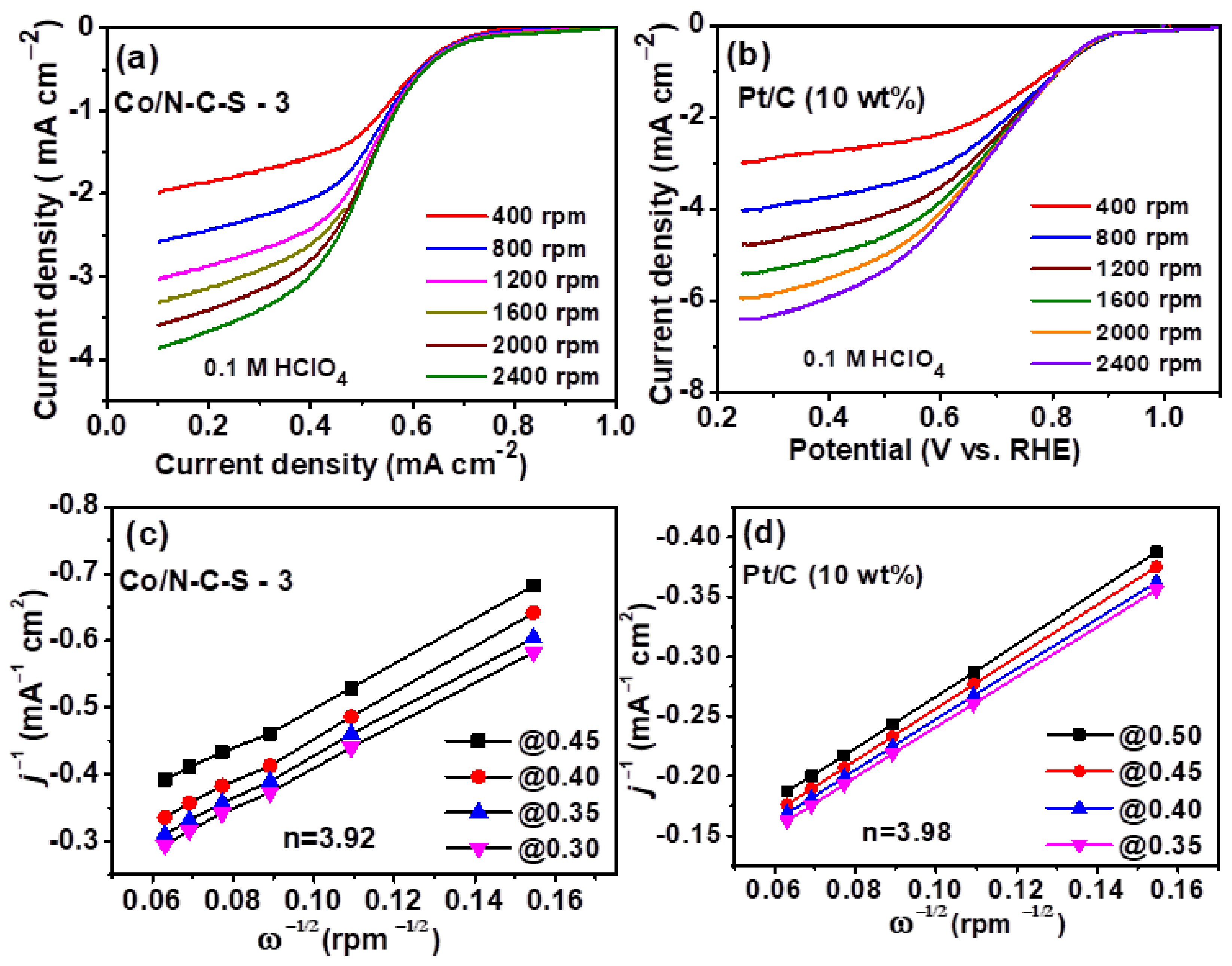{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
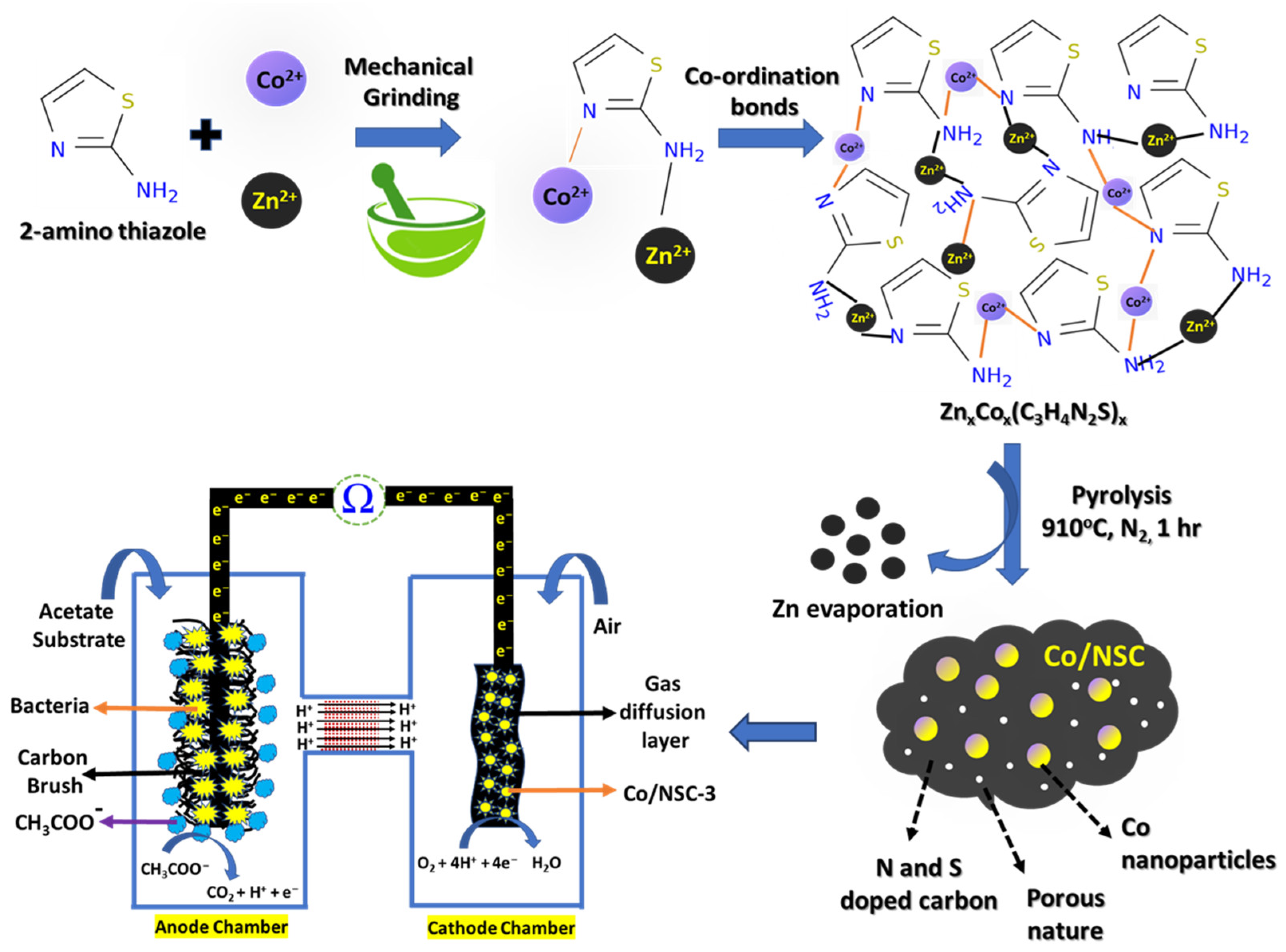
2.1. Synthesis of ZnxCox(C3H4N2S), NCS and Co/NCS Catalyst
2.2. Physical and Electrochemical Characterizations
2.3. Electrochemical Characterizations of the Co/NCS and NCS Catalysts
2.4. Microbial Fuel Cell Operation
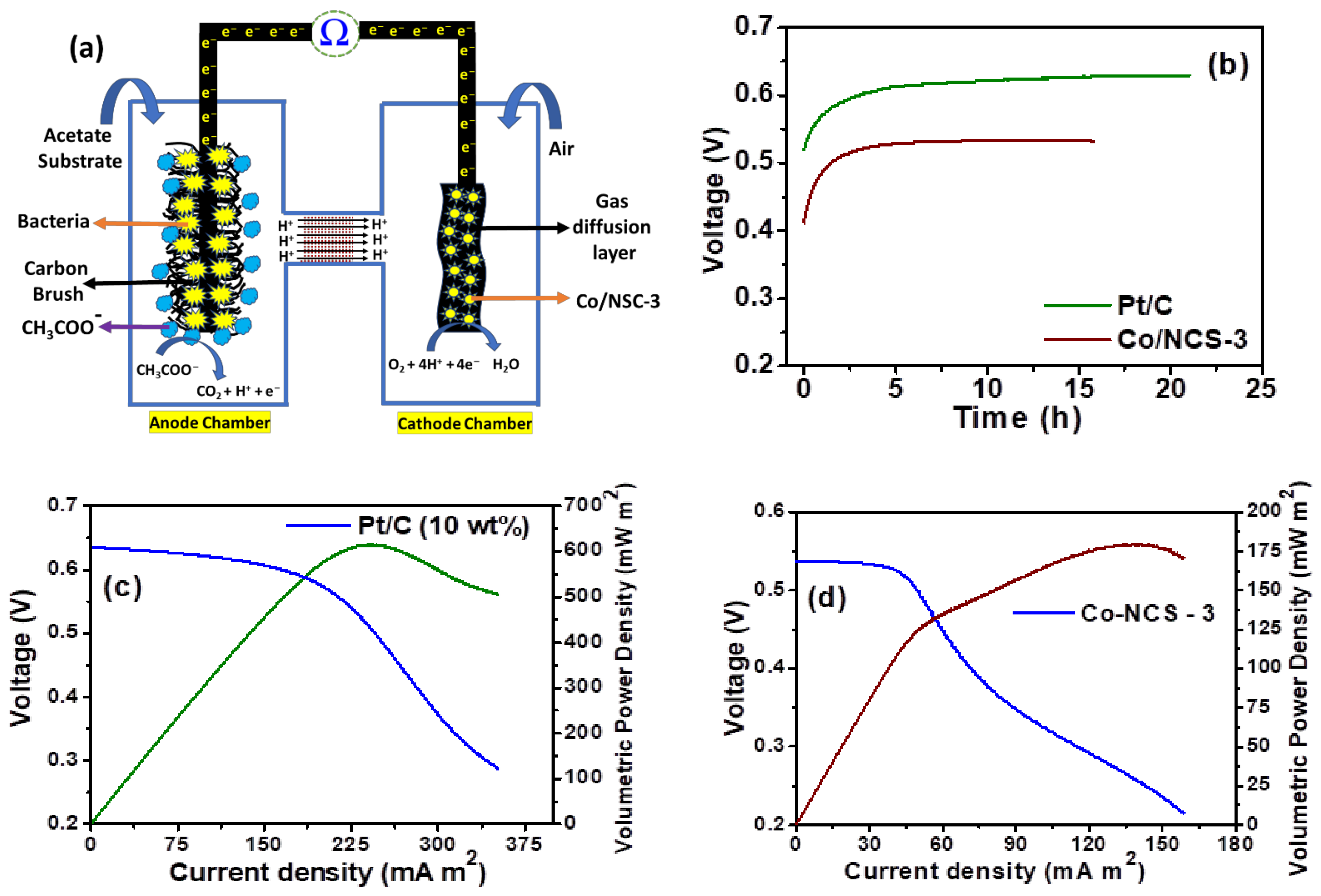
3. Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Slate, A.J.; Whitehead, K.A.; Brownson, D.A.C.; Banks, C.E. Microbial Fuel Cells: An Overview of Current Technology. Renew. Sustain. Energy Rev. 2019, 101, 60–81. [Google Scholar] [CrossRef]
- Zheng, G.; Zhen, H. Long-term performance of a 200-liter modularized microbial fuel cell system treating municipal wastewater: Treatment, energy, and cost. Environ. Sci. Water Res. Technol. 2016, 2, 274–281. [Google Scholar]
- Thiong’o, M.; Osano, A.; Bakari, C. Investigating the ohmic behavior of mediator-less microbial fuel cells using sewerage water as the bio-anode. Cogent Eng. 2022, 9, 2079222. [Google Scholar] [CrossRef]
- Ucar, D.; Zhang, Y.; Angelidaki, I. An Overview of Electron Acceptors in Microbial Fuel Cells. Front. Microbiol. 2017, 8, 643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aftab, S.; Shah, A.; Nisar, J.; Ashiq, M.N.; Akhter, M.S.; Shah, A.H. Marketability Prospects of Microbial Fuel Cells for Sustainable Energy Generation. Energy Fuels 2020, 34, 9108–9136. [Google Scholar] [CrossRef]
- Do, M.H.; Ngo, H.H.; Guo, W.S.; Liu, Y.; Chang, S.W.; Nguyen, D.D.; Nghiem, L.D.; Ni, B.J. Challenges in the Application of Microbial Fuel Cells to Wastewater Treatment and Energy Production: A Mini Review. Sci. Total Environ. 2018, 639, 910–920. [Google Scholar] [CrossRef]
- Ben Liew, K.; Daud, W.R.W.; Ghasemi, M.; Leong, J.X.; Su Lim, S.; Ismail, M. Non-Pt Catalyst as Oxygen Reduction Reaction in Microbial Fuel Cells: A Review. Int. J. Hydrogen Energy 2014, 39, 4870–4883. [Google Scholar] [CrossRef]
- Peera, S.G.; Maiyalagan, T.; Liu, C.; Ashmath, S.; Lee, T.G.; Jiang, Z.; Mao, S. A Review on Carbon and Non-Precious Metal Based Cathode Catalysts in Microbial Fuel Cells. Int. J. Hydrogen Energy 2021, 46, 3056–3089. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, Y.; Wang, H.; Zhang, J.; Zhao, H.; Chen, L.; Xu, L.; Xie, Y.; Huang, J. MIL-88-Derived N and S Co-Doped Carbon Materials with Supplemental FeSx to Enhance the Oxygen Reduction Reaction Performance. Catalysts 2022, 12, 806. [Google Scholar] [CrossRef]
- Nandikes, G.; Gouse Peera, S.; Singh, L. Perovskite-Based Nanocomposite Electrocatalysts: An Alternative to Platinum ORR Catalyst in Microbial Fuel Cell Cathodes. Energies 2021, 15, 272. [Google Scholar] [CrossRef]
- Xu, H.; Wang, D.; Yang, P.; Liu, A.; Li, R.; Li, Y.; Xiao, L.; Ren, X.; Zhang, J.; An, M. Atomically Dispersed M–N–C Catalysts for the Oxygen Reduction Reaction. J. Mater. Chem. A 2020, 8, 23187–23201. [Google Scholar] [CrossRef]
- Ma, Q.; Jin, H.; Zhu, J.; Li, Z.; Xu, H.; Liu, B.; Zhang, Z.; Ma, J.; Mu, S. Stabilizing Fe–N–C Catalysts as Model for Oxygen Reduction Reaction. Adv. Sci. 2021, 8, 2102209. [Google Scholar] [CrossRef]
- Zitolo, A.; Ranjbar-Sahraie, N.; Mineva, T.; Li, J.; Jia, Q.; Stamatin, S.; Harrington, G.F.; Lyth, S.M.; Krtil, P.; Mukerjee, S.; et al. Identification of Catalytic Sites in Cobalt-Nitrogen-Carbon Materials for the Oxygen Reduction Reaction. Nat. Commun. 2017, 8, 957. [Google Scholar] [CrossRef] [Green Version]
- Osmieri, L. Transition Metal–Nitrogen–Carbon (M–N–C) Catalysts for Oxygen Reduction Reaction. Insights on Synthesis and Performance in Polymer Electrolyte Fuel Cells. ChemEngineering 2019, 3, 16. [Google Scholar] [CrossRef] [Green Version]
- Hao, Z.; Ma, Y.; Chen, Y.; Fu, P.; Wang, P. Non-Noble Metal Catalysts in Cathodic Oxygen Reduction Reaction of Proton Exchange Membrane Fuel Cells: Recent Advances. Nanomaterials 2022, 12, 3331. [Google Scholar] [CrossRef]
- Stock, N.; Biswas, S. Synthesis of Metal-Organic Frameworks (MOFs): Routes to Various MOF Topologies, Morphologies, and Composites. Chem. Rev. 2012, 112, 933–969. [Google Scholar] [CrossRef]
- Welton, T. Solvents and Sustainable Chemistry. Proc. R. Soc. A Math. Phys. Eng. Sci. 2015, 471, 20150502. [Google Scholar] [CrossRef] [Green Version]
- A Better Solution for the Environment. Available online: www.chemistryworld.com/news/enhancing-solvents-sustainability/3010810.article (accessed on 7 August 2019).
- Amrute, A.P.; De Bellis, J.; Felderhoff, M.; Schüth, F. Mechanochemical Synthesis of Catalytic Materials. Chem.–A Eur. J. 2021, 27, 6819–6847. [Google Scholar] [CrossRef]
- Peera, S.G.; Liu, C. Unconventional and Scalable Synthesis of Non-Precious Metal Electrocatalysts for Practical Proton Exchange Membrane and Alkaline Fuel Cells: A Solid-State Co-Ordination Synthesis Approach. Coord. Chem. Rev. 2022, 463, 214554. [Google Scholar] [CrossRef]
- Rehab, H.M.; Farag, A.S.; Mohamed, K.I.; Gamila, H.A.; Rabeay, Y.A.H. Formation of electroactive biofilms derived by nanostructured anodes surfaces. Bioprocess Biosyst. Eng. 2021, 44, 759–768. [Google Scholar] [CrossRef]
- Numfon, E.; Christina, S.K.; Song, J.R.; Yanasinee, S.; Han, S.K. Enhanced Current Production by Electroactive Biofilm of Sulfate-Reducing Bacteria in the Microbial Fuel Cell. Environ. Eng. Res. 2013, 18, 277–281. [Google Scholar] [CrossRef]
- Yap, M.H.; Fow, K.L.; Chen, G.Z. Synthesis and Applications of MOF-Derived Porous Nanostructures. Green Energy Environ. 2017, 2, 218–245. [Google Scholar] [CrossRef]
- Xiuxiu, L.; Changdong, S.; Changwei, Z.; Meiling, C.; Qi, L.; Guoxiu, W. Cobalt-Based Layered Metal–Organic Framework as an Ultrahigh Capacity Supercapacitor Electrode Material. ACS Appl. Mater. Interfaces 2016, 8, 4585. [Google Scholar] [CrossRef]
- Xiaoshi, H.; Huiping, H.; Chao, L.; Tian, L.; Xiaobing, L.; Qun, C.; Bingwen, H. Cobalt-based metal organic framework with superior lithium anodic performance. J. Solid State Chem. 2016, 242, 71–76. [Google Scholar] [CrossRef]
- Niu, Q.; Guo, J.; Chen, B.; Nie, J.; Guo, X.; Ma, G. Bimetal-organic frameworks/polymer core-shell nanofibers derived heteroatom-doped carbon materials as electrocatalysts for oxygen reduction reaction. Carbon 2017, 114, 250. [Google Scholar] [CrossRef]
- Lu, H.; Zhang, H.; Liu, R.; Zhang, X.; Zhao, H.; Wang, G. Macroscale cobalt-MOFs derived metallic Co nanoparticles embedded in N-doped porous carbon layers as efficient oxygen electrocatalysts. Appl. Surf. Sci. 2017, 392, 402. [Google Scholar] [CrossRef] [Green Version]
- Song, D.; Hu, C.; Gao, Z.; Yang, B.; Li, Q.; Zhan, X.; Tong, X.; Tian, J. Metal–Organic Frameworks (MOFs) Derived Materials Used in Zn–Air Battery. Materials 2022, 15, 5837. [Google Scholar] [CrossRef]
- Chen, D.; Zhao, J.; Zhang, P.; Dai, S. Mechanochemical Synthesis of Metal–Organic Frameworks. Polyhedron 2019, 162, 59–64. [Google Scholar] [CrossRef]
- Tao, C.-A.; Wang, J.-F. Synthesis of Metal Organic Frameworks by Ball-Milling. Crystals 2020, 11, 15. [Google Scholar] [CrossRef]
- Yang, C.; Ma, X.; Zhou, J.; Zhao, Y.; Xiang, X.; Shang, H.; Zhang, B. Recent Advances in Metal-Organic Frameworks-Derived Carbon-Based Electrocatalysts for the Oxygen Reduction Reaction. Int. J. Hydrogen Energy 2022, 47, 21634–21661. [Google Scholar] [CrossRef]
- Song, Z.; Zhang, L.; Doyle-Davis, K.; Fu, X.; Luo, J.; Sun, X. Recent Advances in MOF-Derived Single Atom Catalysts for Electrochemical Applications. Adv. Energy Mater. 2020, 10, 2001561. [Google Scholar] [CrossRef]
- Rossin, A.; Giambastiani, G. Structural Features and Applications of Metal–Organic Frameworks Containing Thiazole- and Thiazolidine-Based Spacers. CrystEngComm 2015, 17, 218–228. [Google Scholar] [CrossRef]
- Chong-Hyeak, K.; Inn, H.K. Tetra kis(2-amino thia zole-κN 3)dichloridocadmium(II). Acta Crystallogr. Sect. E Struct. Rep. Online 2010, 66, m13. [Google Scholar]
- Suh, S.W.; Kim, C.-H.; Kim, I.H. Bis(2-amino-benzothia-zole-κN)bis-(thio-cyanato-κN)zinc(II). Acta Crystallogr. Sect. E Struct. Rep. Online 2009, 65, m1054. [Google Scholar] [CrossRef] [Green Version]
- Shaik, G.P.; Kwon, H.-J.; Lee, T.G. Highly Efficient Co@NCS Nanosheet Electrocatalyst for Oxygen Reduction Reaction: An Environment-Friendly, Low-Cost and Sustainable Electrocatalyst. Mater. Res. Bull. 2020, 128, 110873. [Google Scholar] [CrossRef]
- Peera, S.G.; Balamurugan, J.; Kim, N.H.; Lee, J.H. Sustainable Synthesis of Co@NC Core Shell Nanostructures from Metal Organic Frameworks via Mechanochemical Coordination Self-Assembly: An Efficient Electrocatalyst for Oxygen Reduction Reaction. Small 2018, 14, 1800441. [Google Scholar] [CrossRef]
- Dan, Q.; Cheng, L.; En-Min, W.; Wen-Cui, L.; An-Hui, L. A Method for Creating Microporous Carbon Materials with Excellent CO2-Adsorption Capacity and Selectivity. ChemSusChem 2014, 7, 291–298. [Google Scholar] [CrossRef]
- Bing, Y.; Jiaojiao, Z.; Feng, W.; Luying, Z.; Qian, Z.; Wenhui, X.; Shuijian, H. Review on porous carbon materials engineered by ZnO templates: Design, synthesis and capacitance performance. Mater. Des. 2021, 201, 109518. [Google Scholar] [CrossRef]
- Chao, H.; Chang, Y.; Mingyu, L.; Xiuna, W.; Qiang, D.; Gang, W.; Jieshan, Q. Nitrogen-doped carbon dots decorated on graphene: A novel all-carbon hybrid electrocatalyst for enhanced oxygen reduction reaction. Chem. Commun 2015, 51, 3419–3422. [Google Scholar] [CrossRef]
- Zhang, X.; Fan, Q.; Qu, N.; Yang, H.; Wang, M.; Liu, A.; Yang, J. Ultrathin 2D Nitrogen-Doped Carbon Nanosheets for High Performance Supercapacitors: Insight into the Effects of Graphene Oxides. Nanoscale 2019, 11, 8588–8596. [Google Scholar] [CrossRef]
- James, A.B.; Eric, M.T.; Serban, N.S.; Carlota, D.; Alessandro, I.; Karsten, F.; Khairul, H.; Tatiana, S.P.; Max, G.-M.; Paula, E.C. Untangling Cooperative Effects of Pyridinic and Graphitic Nitrogen Sites at Metal-Free N-Doped Carbon Electrocatalysts for the Oxygen Reduction Reaction. Small 2019, 15, 1902081. [Google Scholar] [CrossRef]
- Qu, L.; Liu, Y.; Baek, J.-B.; Dai, L. Nitrogen-doped graphene as efficient metal-free electrocatalyst for oxygen reduction in fuel cells. ACS Nano 2010, 4, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Chitturi, V.R.; Carlos, R.C.; Yasuyuki, I. In search of the active site in nitrogen-doped carbon nanotube electrodes for the oxygen reduction reaction. J. Phys. Chem. Lett. 2010, 1, 2622–2627. [Google Scholar] [CrossRef]
- Kabir, S.; Artyushkova, K.; Serov, A.; Kiefer, B.; Atanassov, P. Binding Energy Shifts for Nitrogen-containing Graphene-based Electrocatalysts—Experiments and DFT Calculations. Surf. Interface Anal. 2016, 48, 293–300. [Google Scholar] [CrossRef]
- Khaled, P.; Shubin, Y.; Yenny, H.; Andreas, W.; Andrey, T.; Xinliang, F.; Klaus, M. Nitrogen-doped graphene and its iron-based composite as efficient electrocatalysts for oxygen reduction reaction. ACS Nano 2012, 6, 9541–9550. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Wu, D.; Feng, X.; Müllen, K. Nitrogen-doped ordered mesoporous graphitic arrays with high electrocatalytic activity for oxygen reduction. Angew. Chem. Int. Ed. 2010, 49, 2565–2569. [Google Scholar] [CrossRef]
- Kim, H.; Lee, K.; Woo, S.I.; Jung, Y. On the mechanism of enhanced oxygen reduction reaction in nitrogen-doped graphene nanoribbons. Phys. Chem. Chem. Phys. 2011, 13, 17505–17510. [Google Scholar] [CrossRef]
- Ning, X.; Li, Y.; Ming, J.; Wang, Q.; Wang, H.; Cao, Y.; Peng, F.; Yang, Y.; Yu, H. Electronic synergism of pyridinic- and graphitic-nitrogen on N-doped carbons for the oxygen reduction reaction. Chem. Sci. 2019, 10, 1589–1596. [Google Scholar] [CrossRef] [Green Version]
- Tylus, U.; Jia, Q.; Strickland, K.; Ramaswamy, N.; Serov, A.; Atanassov, P.; Mukerjee, S. Elucidating Oxygen Reduction Active Sites in Pyrolyzed Metal–Nitrogen Coordinated Non-Precious-Metal Electrocatalyst Systems. J. Phys. Chem. C 2014, 118, 8999–9008. [Google Scholar] [CrossRef]
- Zhang, C.; An, B.; Yang, L.; Wu, B.; Shi, W.; Wang, Y.-C.; Long, L.-S.; Wang, C.; Lin, W. Sulfur-Doping Achieves Efficient Oxygen Reduction in Pyrolyzed Zeolitic Imidazolate Frameworks. J. Mater. Chem. A 2016, 4, 4457–4463. [Google Scholar] [CrossRef]
- Gouse Peera, S.; Kwon, H.-J.; Lee, T.G.; Hussain, A.M. Heteroatom- and Metalloid-Doped Carbon Catalysts for Oxygen Reduction Reaction: A Mini-Review. Ionics 2020, 26, 1563–1589. [Google Scholar] [CrossRef]
- Wu, W.; Zhang, Q.; Wang, X.; Han, C.; Shao, X.; Wang, Y.; Liu, J.; Li, Z.; Lu, X.; Wu, M. Enhancing Selective Photooxidation through Co–Nx-Doped Carbon Materials as Singlet Oxygen Photosensitizers. ACS Catal. 2017, 7, 7267–7273. [Google Scholar] [CrossRef]
- Peera, S.G.; Koutavarapu, R.; Akula, S.; Asokan, A.; Moni, P.; Selvaraj, M.; Balamurugan, J.; Kim, S.O.; Liu, C.; Sahu, A.K. Carbon Nanofibers as Potential Catalyst Support for Fuel Cell Cathodes: A Review. Energy Fuels 2021, 35, 11761–11799. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashmath, S.; Kwon, H.-J.; Peera, S.G.; Lee, T.G. Solid-State Synthesis of Cobalt/NCS Electrocatalyst for Oxygen Reduction Reaction in Dual Chamber Microbial Fuel Cells. Nanomaterials 2022, 12, 4369. https://doi.org/10.3390/nano12244369
Ashmath S, Kwon H-J, Peera SG, Lee TG. Solid-State Synthesis of Cobalt/NCS Electrocatalyst for Oxygen Reduction Reaction in Dual Chamber Microbial Fuel Cells. Nanomaterials. 2022; 12(24):4369. https://doi.org/10.3390/nano12244369
Chicago/Turabian StyleAshmath, Shaik, Hyuk-Jun Kwon, Shaik Gouse Peera, and Tae Gwan Lee. 2022. "Solid-State Synthesis of Cobalt/NCS Electrocatalyst for Oxygen Reduction Reaction in Dual Chamber Microbial Fuel Cells" Nanomaterials 12, no. 24: 4369. https://doi.org/10.3390/nano12244369