
Supramolecular Self-Assembly of Dipalmitoylphosphatidylcholine and Carbon Nanotubes: A Dissipative Particle Dynamics Simulation Study


Abstract
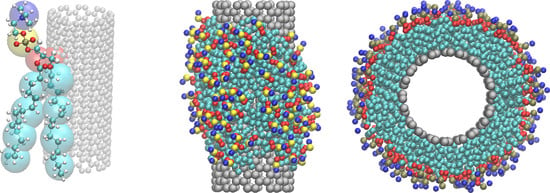
:
1. Introduction
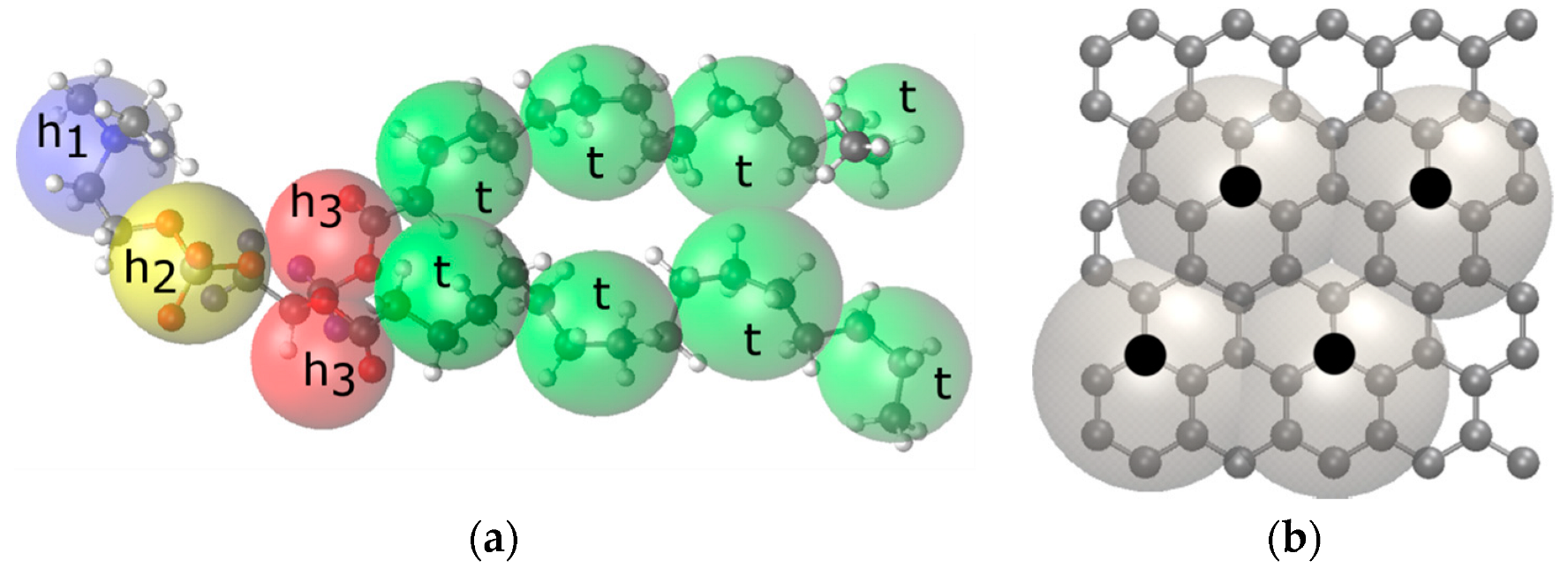
2. Model and Simulation Details
3. Results and Discussion
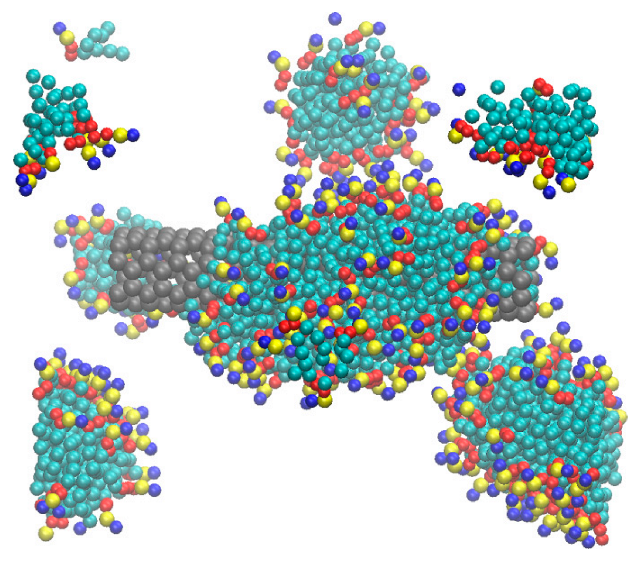
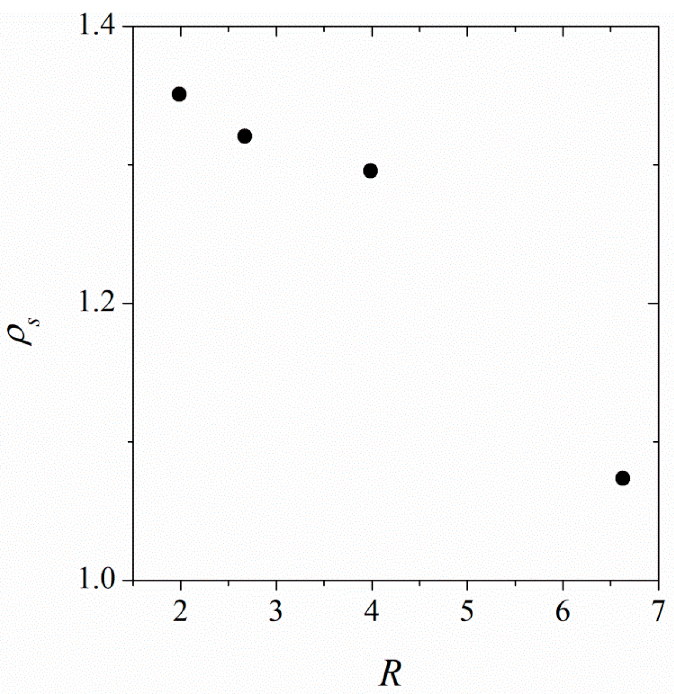
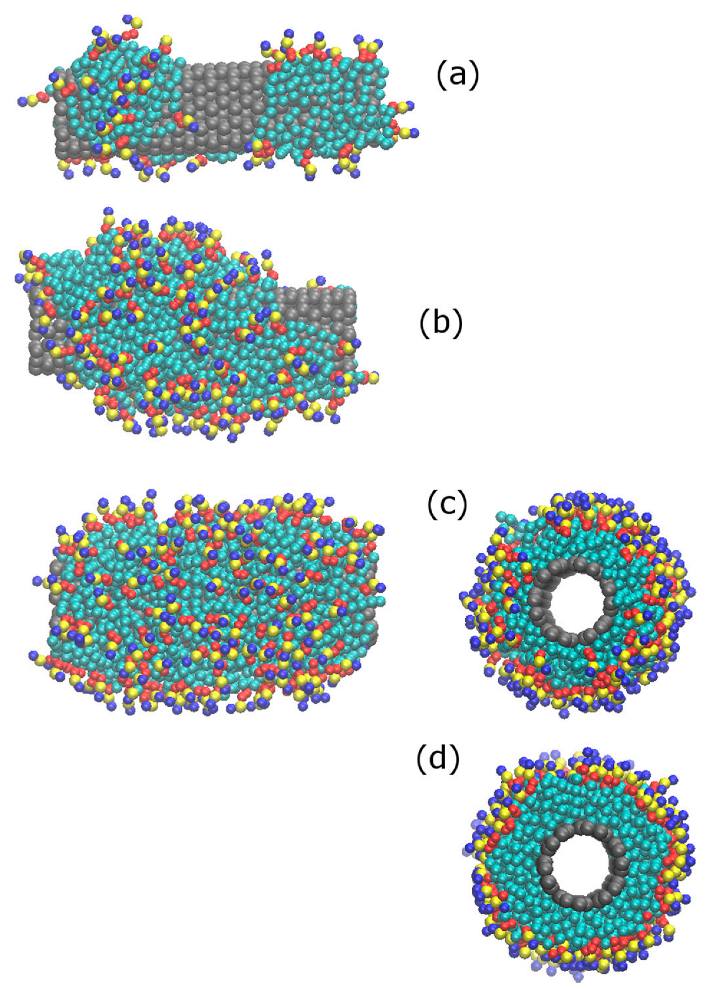
3.1. Clustering and Surface Adsorption of DDPC
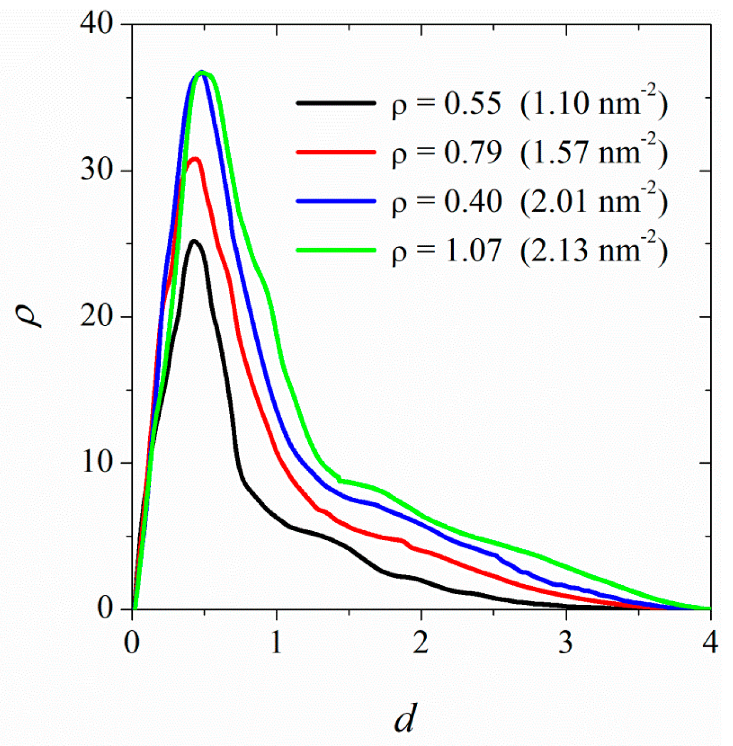
3.2. Surface Saturation of CNTs with DPPC
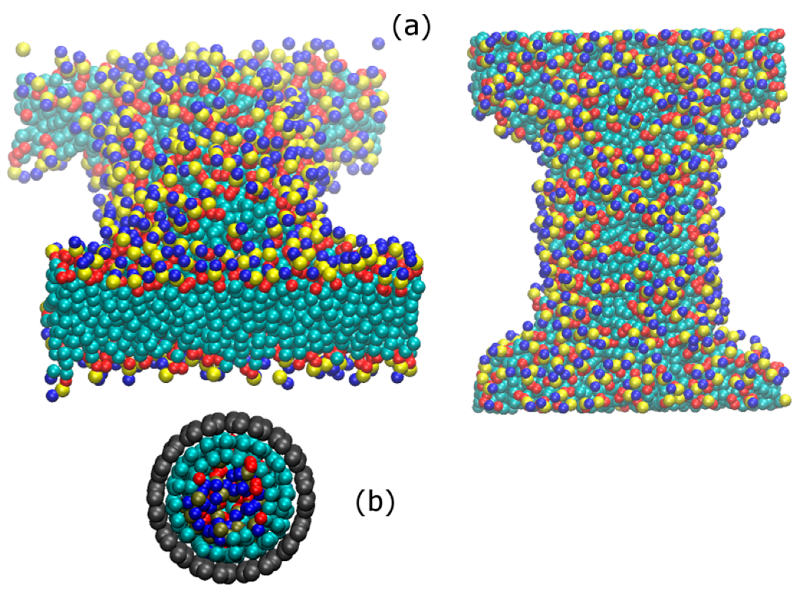
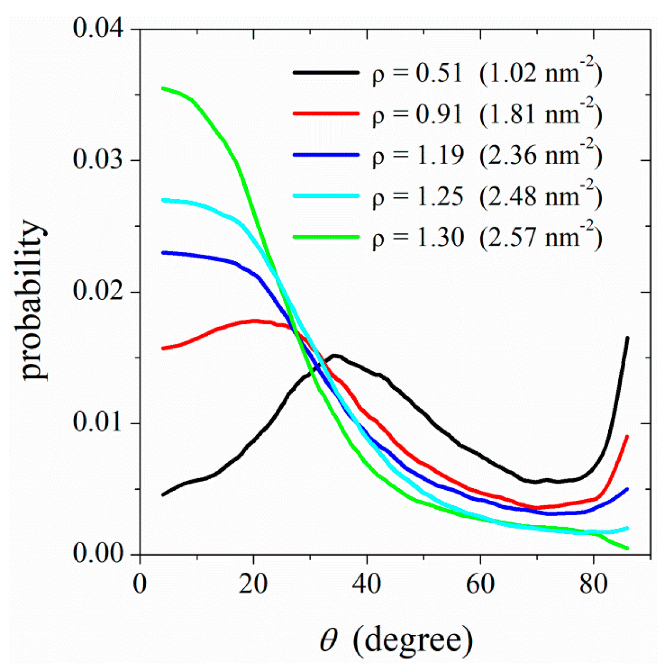
3.3. Ordering of DPPC Molecules on the CNT Surface
3.4. Mechanism of Lipid Ordering on the CNT Surface
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kundu, N.; Banik, D.; Sarkar, N. Self-Assembly of Amphiphiles into Vesicles and Fibrils: Investigation of Structure and Dynamics Using Spectroscopy and Microscopy Techniques. Langmuir 2018, 34, 11637–11654. [Google Scholar] [CrossRef] [PubMed]
- Vauthey, S.; Santoso, S.; Gong, H.; Watson, N.; Zhang, S. Molecular Self-Assembly of Surfactant-Like Peptides to form Nanotubes and Nanovesicles. Proc. Natl. Acad. Sci. USA 2002, 99, 5355–5360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, M.K.; Vermerris, W. Recent Advances in Nanomaterials for Gene Delivery−A Review. Nanomaterials 2017, 7, 94. [Google Scholar] [CrossRef] [Green Version]
- Keshtkar, M.; Mehdipour, N.; Eslami, H. Thermal Conductivity of Polyamide-6,6/Carbon Nanotube Composites: Effects of Tube Diameter and Polymer Linkage between Tubes. Polymers 2019, 11, 1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goh, P.S.; Ismail, A.F. Nanocomposites for Environmental and Energy Applications. Nanomaterials 2021, 11, 345. [Google Scholar] [CrossRef]
- Kam, N.W.S.; O’Connell, M.; Wisdom, J.A.; Dai, H. Carbon Nanotubes as Multifunctional Biological Transporters and Near-Infrared Agents for Selective Cancer Cell Destruction. Proc. Natl. Acad. Sci. USA 2005, 102, 11600–11605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metaxa, Z.S.; Boutsioukou, S.; Amenta, M.; Favvas, E.P.; Kourkoulis, S.K.; Alexopoulos, N.D. Dispersion of Multi-Walled Carbon Nanotubes into White Cement Mortars: The Effect of Concentration and Surfactants. Nanomaterials 2022, 12, 1031. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Danné, N.; Godin, A.G.; Lounis, B.; Cognet, L. Evaluation of Different Single-Walled Carbon Nanotube Surface Coatings for Single-Particle Tracking Applications in Biological Environments. Nanomaterials 2017, 7, 393. [Google Scholar] [CrossRef] [Green Version]
- Burdanova, M.G.; Kharlamova, M.V.; Kramberger, C.; Nikitin, M.P. Applications of Pristine and Functionalized Carbon Nanotubes, Graphene, and Graphene Nanoribbons in Biomedicine. Nanomaterials 2021, 11, 3020. [Google Scholar] [CrossRef]
- Richard, C.; Balavoine, F.; Schultz, P.; Ebbesen, T.W.; Mioskowski, C. Supramolecular Self-Assembly of Lipid Derivatives on Carbon Nanotubes. Science 2003, 300, 775–778. [Google Scholar] [CrossRef]
- Wu, Y.; Hudson, J.S.; Lu, Q.; Moore, J.M.; Mount, A.S.; Rao, A.M.; Alexov, E.; Ke, P.C. Coating Single-Walled Carbon Nanotubes with Phospholipids. J. Phys. Chem. B 2006, 110, 2475–2478. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.C. Fiddling the String of Carbon Nanotubes with Amphiphiles. Phys. Chem. Chem. Phys. 2007, 9, 439–447. [Google Scholar] [PubMed]
- Attia, D.; Yekymov, E.; Shmidov, Y.; Levi-Kalisman, Y.; Mendelson, O.; Bitton, R.; Yerushalmi-Rozen, R. Surfactant-Mediated Co-Existence of Single-Walled Carbon Nanotube Networks and Cellulose Nanocrystal Mesophases. Nanomaterials 2021, 11, 3059. [Google Scholar] [CrossRef] [PubMed]
- Guzmán-Mendoza, J.J.; Chávez-Flores, D.; Montes-Fonseca, S.L.; González-Horta, C.; Orrantia-Borunda, E.; Sánchez-Ramírez, B. A Novel Method for Carbon Nanotube Functionalization Using Immobilized Candida Antarctica Lipase. Nanomaterials 2022, 12, 1465. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.P.; Qiao, R. Carbon Nanomaterials in Biological Systems. J. Phys. Condens. Matter 2007, 19, 373101. [Google Scholar] [CrossRef]
- Ghellab, S.E.; Han, X. Micrometer-Size Double-Helical Structures from Phospholipid-Modified Carbon Nanotubes. Soft Matter 2022, 18, 2726–2730. [Google Scholar] [CrossRef] [PubMed]
- Bystrov, V.; Likhachev, I.; Sidorova, A.; Filippov, S.; Lutsenko, A.; Shpigun, D.; Belova, E. Molecular Dynamics Simulation Study of the Self-Assembly of Phenylalanine Peptide Nanotubes. Nanomaterials 2022, 12, 861. [Google Scholar] [CrossRef]
- Qiao, R.; Ke, P.C. Lipid-Carbon Nanotube Self-Assembly in Aqueous Solution. J. Am. Chem. Soc. 2006, 128, 13656–13657. [Google Scholar] [CrossRef] [PubMed]
- Obata, S.; Honda, K. Dynamic Behavior of Carbon Nanotube and Bio-Artificial Surfactants Complexes in an Aqueous Environment. J. Phys. Chem. C 2011, 115, 19659–19667. [Google Scholar] [CrossRef]
- Maatta, J.; Vierros, S.; Sammalkorpi, M. Controlling Carbon-Nanotube Phospholipid Solubility by Curvature-Dependent Self-Assembly. J. Phys. Chem. B 2015, 119, 4020–4032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patra, N.; Kr, P. Controlled Self-Assembly of Filled Micelles on Nanotubes. J. Am. Chem. Soc. 2011, 133, 6146–6149. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, H. Self-Assembly of Lipids and Single-Walled Carbon Nanotubes: Effects of Lipid Structure and PEGylation. J. Phys. Chem. C 2012, 116, 9327–9333. [Google Scholar] [CrossRef]
- Eslami, H.; Das, S.; Zhou, T.; Müller-Plathe, F. How Alcoholic Disinfectants Affect Coronavirus Model Membranes: Membrane Fluidity, Permeability, and Disintegration. J. Phys. Chem. B 2020, 124, 10374–10385. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Wu, Z.; Das, S.; Eslami, H.; Müller-Plathe, F. How Ethanolic Disinfectants Disintegrate Coronavirus Model Membranes: A Dissipative Particle Dynamics Simulation Study. J. Chem. Theory Comput. 2022, 18, 2597–2615. [Google Scholar] [CrossRef] [PubMed]
- Groot, R.D.; Warren, P.B. Dissipative Particle Dynamics: Bridging the Gap Between Atomistic and Mesoscopic Simulation. J. Chem. Phys. 1997, 107, 4423–4435. [Google Scholar] [CrossRef]
- Atashafrooz, M.; Mehdipour, N. Many-Body Dissipative Particle Dynamics Simulation of Liquid−Vapor Coexisting Curve in Sodium. J. Chem. Eng. Data 2016, 61, 3659–3664. [Google Scholar] [CrossRef]
- Eslami, H.; Bahri, K.; Müller-Plathe, F. Solid−Liquid and Solid−Solid Phase Diagrams of Self-Assembled Triblock Janus Nanoparticles from Solution. J. Phys. Chem. C 2018, 122, 9235–9244. [Google Scholar] [CrossRef]
- Eslami, H.; Karimi-Varzaneh, H.A.; Müller-Plathe, F. Coarse-Grained Computer Simulation of Nanoconfined Polyamide-6,6. Macromolecules 2011, 44, 3117–3128. [Google Scholar] [CrossRef]
- Eslami, H.; Müller-Plathe, F. How Thick is the Interphase in an Ultrathin Polymer Film? Coarse-Grained Molecular Dynamics Simulations of Polyamide-6,6 on Graphene. J. Phys. Chem. C 2013, 117, 5249–5257. [Google Scholar] [CrossRef]
- Eslami, H.; Khani, M.; Müller-Plathe, F. Gaussian Charge Distributions for Incorporation of Electrostatic Interactions in Dissipative Particle Dynamics: Application to Self-Assembly of Surfactants. J. Chem. Theory Comput. 2019, 15, 4197–4207. [Google Scholar] [CrossRef]
- Müller-Plathe, F. YASP: A Molecular Simulation Package. Comput. Phys. Commun. 1993, 78, 77–94. [Google Scholar] [CrossRef]
- Eslami, H.; Rahimi, M.; Müller-Plathe, F. Molecular Dynamics Simulation of a Silica Nanoparticle in Oligomeric Poly(methyl methacrylate): A Model System for Studying the Interphase Thickness in a Polymer–Nanocomposite via Different Properties. Macromolecules 2013, 46, 8680–8692. [Google Scholar] [CrossRef]
- Eslami, H.; Behrouz, M. Molecular Dynamics Simulation of a Polyamide-66/Carbon Nanotube Nanocomposite. J. Phys. Chem. C 2014, 118, 9841–9851. [Google Scholar] [CrossRef]
- Wallace, E.J.; Sansom, M.S.P. Blocking of Carbon Nanotube Based Nanoinjectures by Lipids: A Simulation Study. Nano Lett. 2008, 8, 2751–2756. [Google Scholar] [CrossRef] [PubMed]
- Eslami, H.; Gharibi, A.; Müller-Plathe, F. Mechanisms of Nucleation and Solid-Solid-Phase Transitions in Triblock Janus Assemblies. J. Chem. Theory Comput. 2021, 17, 1742–1754. [Google Scholar] [CrossRef] [PubMed]
- Bahri, K.; Eslami, H.; Müller-Plathe, F. Self-Assembly of Model Triblock Janus Colloidal Particles in Two Dimensions. J. Chem. Theory Comput. 2022, 18, 1870–1882. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (a) Bonded potential | ||||||
| bond type i-j | () | () | ||||
| h1–h2 | 512 | 0.47 | ||||
| h2–h3 | 512 | 0.47 | ||||
| h3–h3 | 512 | 0.31 | ||||
| h3–t | 512 | 0.59 | ||||
| t–t | 512 | 0.59 | ||||
| (b) Angle-bending potential | ||||||
| angle type i-j-k | () | (degree) | ||||
| h2–h3–h3 | 6 | 120.0 | ||||
| h2–h3–t | 6 | 180.0 | ||||
| h3–t–t | 6 | 180.0 | ||||
| t–t–t | 6 | 180.0 | ||||
| (c) Repulsion parameters, , for all bead types | ||||||
| h1 | h2 | h3 | t | w | CNT | |
| h1 | 110 | 100 | 102 | 130 | 98 | 130 |
| h2 | 100 | 110 | 102 | 130 | 98 | 130 |
| h3 | 102 | 102 | 100 | 110 | 102 | 110 |
| t | 130 | 130 | 110 | 100 | 130 | 100 |
| w | 98 | 98 | 102 | 130 | 100 | 130 |
| CNT | 130 | 130 | 110 | 100 | 130 | 100 |
| Substrate | Number of DPPC Lipid Molecules | Equilibrium Box Size |
|---|---|---|
| CNT (10, 10) | 10 | 17.56 × 22.53 × 17.39 |
| 50 | 17.66 × 22.55 × 17.68 | |
| 100 | 17.89 × 22.52 × 17.93 | |
| 150 | 18.20 × 22.53 × 18.10 | |
| 200 | 18.38 × 22.53 × 18.39 | |
| 250 | 18.66 × 22.55 × 18.56 | |
| 300 | 18.95 × 22.55 × 18.73 | |
| 350 | 18.51 × 22.56 × 19.62 | |
| 400 | 18.99 × 22.41 × 19.72 | |
| 450 | 18.91 × 22.48 × 20.18 | |
| 500 | 19.03 × 22.56 × 20.44 | |
| CNT (14, 14) | 10 | 17.49 × 22.55 × 17.52 |
| 50 | 17.72 × 22.55 × 17.69 | |
| 100 | 17.91 × 22.53 × 17.97 | |
| 150 | 18.17 × 22.53 × 18.18 | |
| 200 | 18.41 × 22.52 × 18.42 | |
| 250 | 18.65 × 22.55 × 18.66 | |
| 300 | 18.93 × 22.45 × 18.88 | |
| 350 | 19.08 × 22.58 × 19.07 | |
| 400 | 19.27 × 22.65 × 19.28 | |
| 450 | 19.62 × 22.49 × 19.51 | |
| 500 | 19.70 × 22.56 × 19.79 | |
| CNT (20, 20) | 10 | 17.73 × 22.53 × 17.41 |
| 50 | 17.65 × 22.55 × 17.86 | |
| 100 | 17.96 × 22.55 × 18.03 | |
| 150 | 18.32 × 22.59 × 18.14 | |
| 200 | 18.41 × 22.59 × 18.51 | |
| 250 | 18.70 × 22.55 × 18.66 | |
| 300 | 18.92 × 22.45 × 18.99 | |
| 350 | 19.08 × 22.59 × 19.15 | |
| 400 | 19.31 × 22.66 × 19.31 | |
| 450 | 18.44 × 22.51 × 20.83 | |
| 500 | 18.37 × 22.58 × 21.31 | |
| CNT (34, 34) | 10 | 17.76 × 22.55 × 17.56 |
| 50 | 18.15 × 22.55 × 17.56 | |
| 100 | 18.19 × 22.53 × 17.99 | |
| 150 | 18.32 × 22.53 × 18.32 | |
| 200 | 18.58 × 22.52 × 18.52 | |
| 250 | 18.66 × 22.53 × 18.90 | |
| 300 | 18.97 × 22.55 × 19.08 | |
| 350 | 18.90 × 22.59 × 19.49 | |
| 400 | 19.66 × 22.44 × 19.31 | |
| 450 | 19.41 × 22.52 × 19.91 | |
| 500 | 19.80 × 22.59 × 19.89 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keshtkar, M.; Mehdipour, N.; Eslami, H. Supramolecular Self-Assembly of Dipalmitoylphosphatidylcholine and Carbon Nanotubes: A Dissipative Particle Dynamics Simulation Study. Nanomaterials 2022, 12, 2653. https://doi.org/10.3390/nano12152653
Keshtkar M, Mehdipour N, Eslami H. Supramolecular Self-Assembly of Dipalmitoylphosphatidylcholine and Carbon Nanotubes: A Dissipative Particle Dynamics Simulation Study. Nanomaterials. 2022; 12(15):2653. https://doi.org/10.3390/nano12152653
Chicago/Turabian StyleKeshtkar, Mahboube, Nargess Mehdipour, and Hossein Eslami. 2022. "Supramolecular Self-Assembly of Dipalmitoylphosphatidylcholine and Carbon Nanotubes: A Dissipative Particle Dynamics Simulation Study" Nanomaterials 12, no. 15: 2653. https://doi.org/10.3390/nano12152653