
Molecular Evolution of Metallothioneins of Antarctic Fish: A Physiological Adaptation to Peculiar Seawater Chemical Characteristics
 , ,
, ,  , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Procedures
2.2. Animal Sampling
2.3. cDNA Synthesis and Amplification
2.4. Rapid Amplification of cDNA Ends (3′-RACE and 5’-RACE)
2.5. Cloning and Sequencing of PCR-Amplified cDNAs
2.6. Sequence Analysis and Phylogenetic Reconstruction
2.7. Phylogeny-Based Tests of Selection
2.8. Stringent Functionally Constrained Amino Acids Identification
3. Results
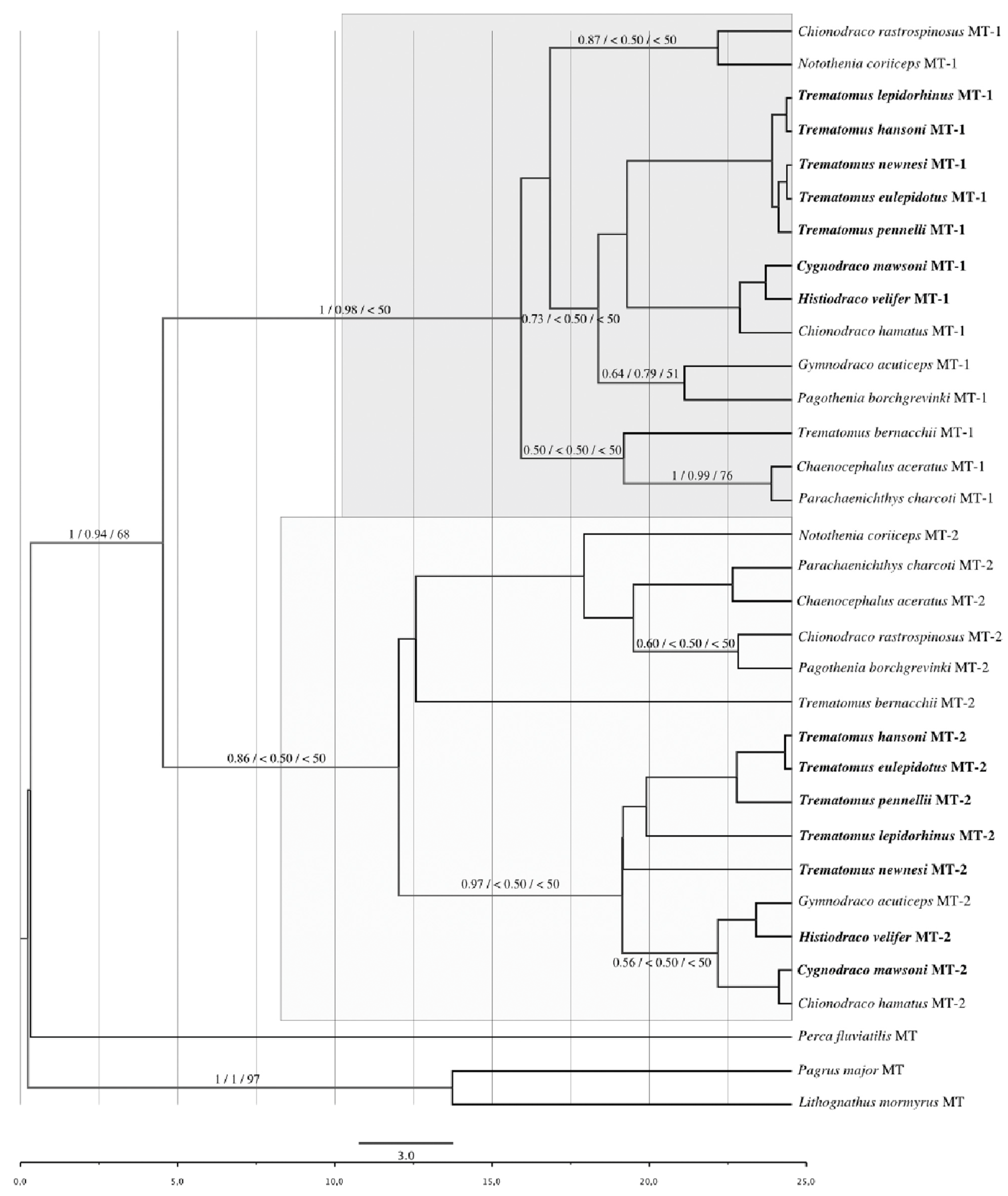
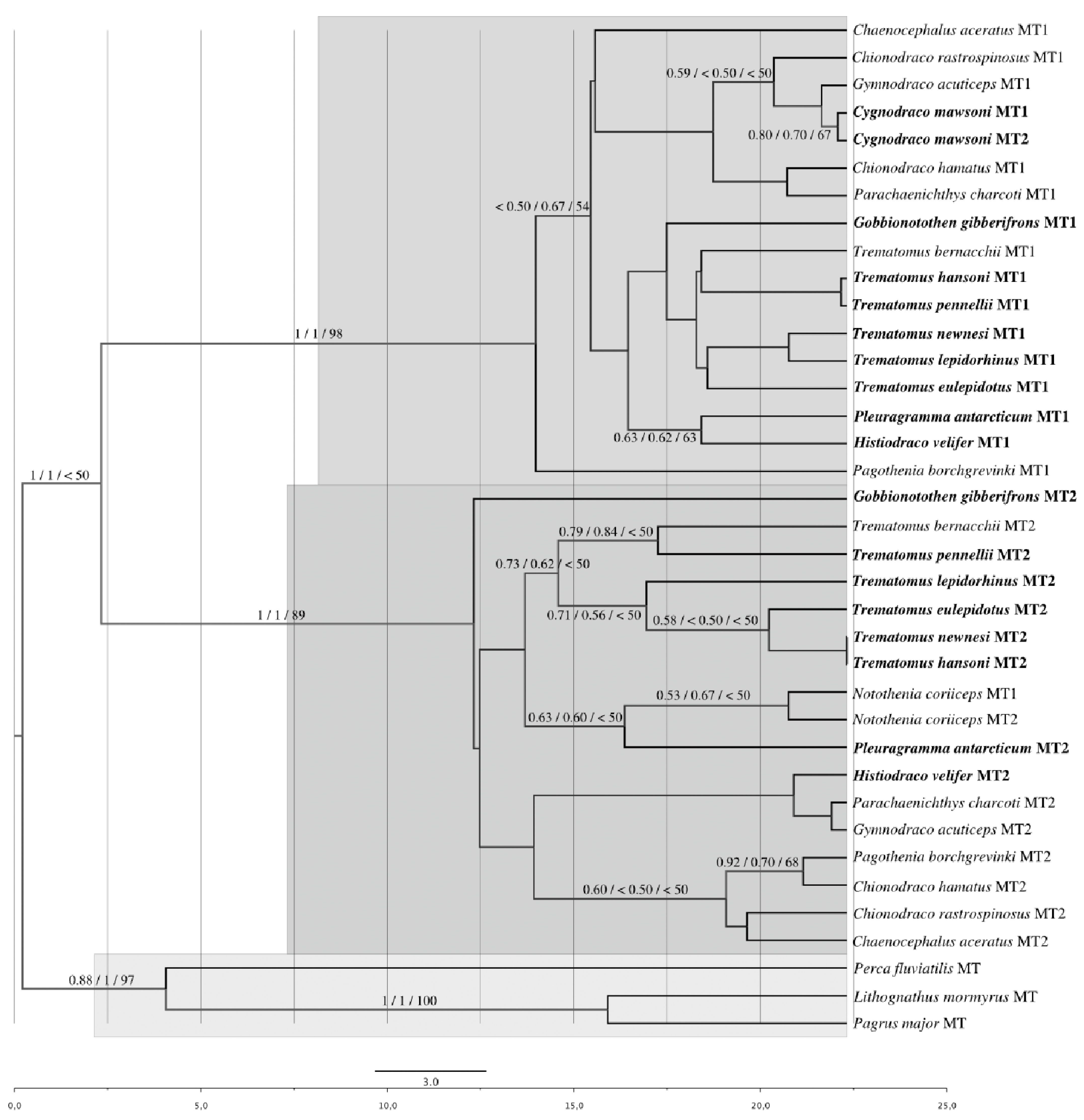
3.1. Phylogenetic Reconstruction Based on Coding Regions
3.2. Positive Selection Analyses on MT Proteins
3.3. Strict Molecular Clock vs. Relaxed Molecular Clock Models
3.4. Positive Selection at the Codon Level
3.5. Stringent Functionally Constrained Amino Acids
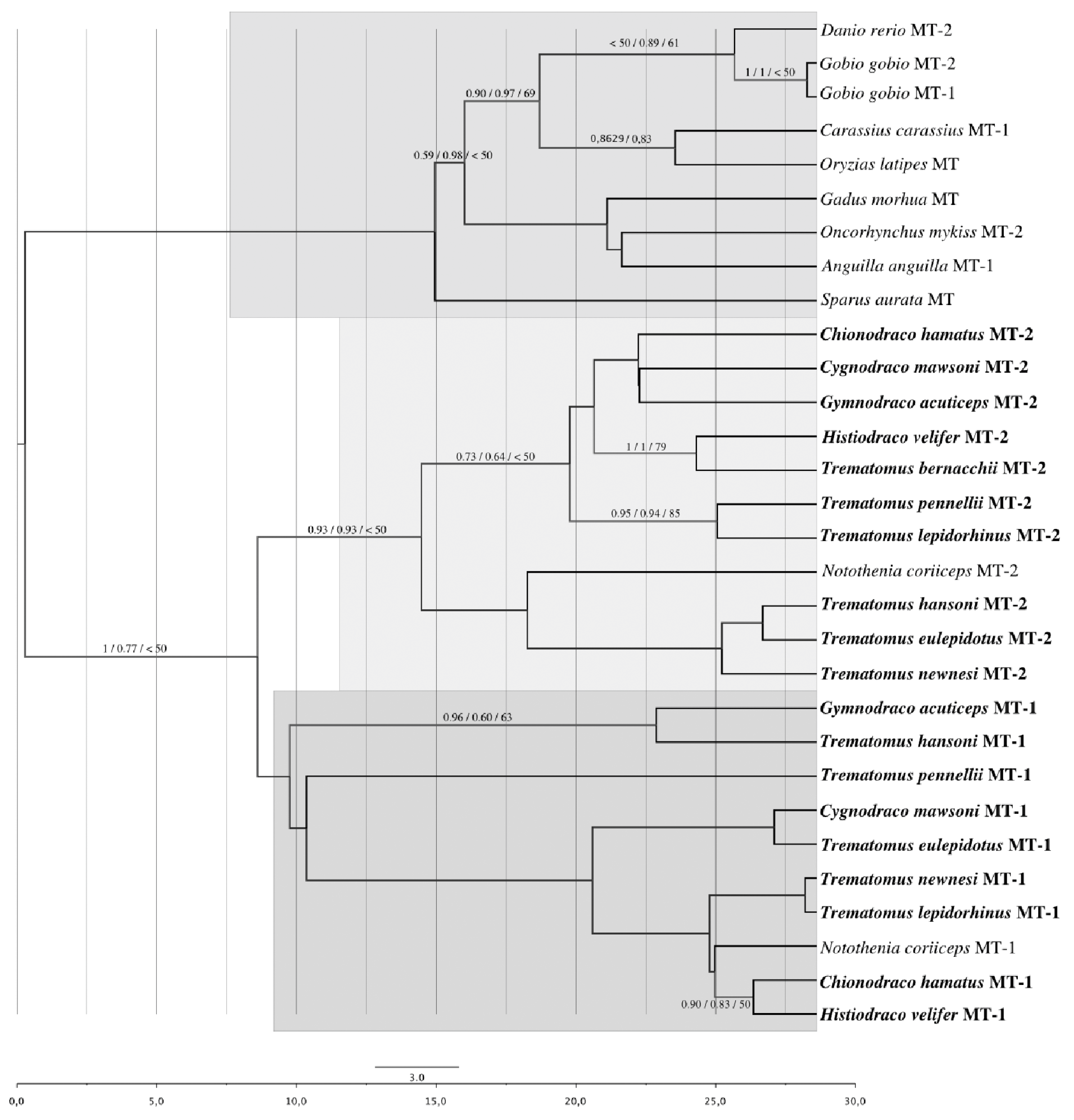
3.6. PhylogeneticAnalysis on UTR Sequences
4. Discussion
4.1. Phylogenetic Analyses of the MT Coding Sequences
4.2. Selection Pressures on MT Paralogs
4.3. Stringent Functionally Constrained Amino Acids
4.4. UTR Regions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lau, Y.-T.; Parker, S.K.; Near, T.J.; Detrich, H.W., 3rd. Evolution and function of the globin intergenic regulatory regions of the antarctic dragonfishes (Notothenioidei: Bathydraconidae). Mol. Biol. Evol. 2012, 29, 1071–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eastman, J.T. The nature of the diversity of Antarctic fishes. Polar Biol. 2005, 28, 93–107. [Google Scholar] [CrossRef]
- Daane, J.M.; Detrich, H.W., 3rd. Adaptations and Diversity of Antarctic Fishes: A Genomic Perspective. Annu. Rev. Anim. Biosci. 2022, 10, 39–62. [Google Scholar] [CrossRef] [PubMed]
- Santovito, G.; Piccinni, E.; Boldrin, F.; Irato, P. Comparative study on metal homeostasis and detoxification in two Antarctic teleosts. Comp. Biochem. Physiol. C 2012, 155, 580–586. [Google Scholar] [CrossRef]
- Welker, A.F.; Moreira, D.C.; Campos, É.G.; Hermes-Lima, M. Role of redox metabolism for adaptation of aquatic animals to drastic changes in oxygen availability. Comp. Biochem. Physiol. A 2013, 165, 384–404. [Google Scholar] [CrossRef]
- Ricci, F.; Lauro, F.M.; Grzymski, J.J.; Read, R.; Bakiu, R.; Santovito, G.; Luporini, P.; Vallesi, A. The Anti-Oxidant Defense System of the Marine Polar Ciliate Euplotes nobilii: Characterization of the MsrB Gene Family. Biology 2017, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Bargagli, R.; Nelli, L.; Ancora, S.; Focardi, S. Elevated cadmium accumulation in marine organisms from Terra Nova Bay (Antarctica). Polar Biol. 1996, 16, 513–520. [Google Scholar] [CrossRef]
- Santovito, G.; Piccinni, E.; Irato, P. An Improved Method for Rapid Determination of the Reduced and Oxidized States of Metallothioneins in Biological Samples; Nova Science Publishers Inc.: New York, NY, USA, 2008; pp. 101–124. [Google Scholar]
- Kägi, J.H. Overview of metallothionein. Methods Enzymol. 1991, 205, 613–626. [Google Scholar] [CrossRef]
- You, C.; Mackay, E.A.; Gehrig, P.M.; Hunziker, P.E.; Kägi, J.H. Purification and characterization of recombinant Caenorhabditis elegans metallothionein. Arch. Biochem. Biophys. 1999, 372, 44–52. [Google Scholar] [CrossRef]
- Formigari, A.; Boldrin, F.; Santovito, G.; Cassidy-Hanley, D.; Clark, T.G.; Piccinni, E. Functional characterization of the 5′-upstream region of MTT5 metallothionein gene from Tetrahymena thermophila. Protist 2010, 161, 71–77. [Google Scholar] [CrossRef]
- Santovito, G.; Trentin, E.; Gobbi, I.; Bisaccia, P.; Tallandini, L.; Irato, P. Non-enzymatic antioxidant responses of Mytilus galloprovincialis: Insights into the physiological role against metal-induced oxidative stress. Comp. Biochem. Physiol. C 2021, 240, 108909. [Google Scholar] [CrossRef] [PubMed]
- Knapen, D.; Redeker, E.S.; Inacio, I.; De Coen, W.; Verheyen, E.; Blust, R. New metallothionein mRNAs in Gobio gobio reveal at least three gene duplication events in cyprinid metallothionein evolution. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2005, 140, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Capasso, C.; Carginale, V.; Crescenzi, O.; Di Maro, D.; Parisi, E.; Spadaccini, R.; Temussi, P.A. Solution structure of MT_nc, a novel metallothionein from the Antarctic fish Notothenia coriiceps. Structure 2003, 11, 435–443. [Google Scholar] [CrossRef] [Green Version]
- Ferro, D.; Bakiu, R.; De Pittà, C.; Boldrin, F.; Cattalini, F.; Pucciarelli, S.; Miceli, C.; Santovito, G. Cu, Zn superoxide dismutases from Tetrahymena thermophila: Molecular evolution and gene expression of the first line of antioxidant defenses. Protist 2015, 166, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Piva, E.; Schumann, S.; Dotteschini, S.; Brocca, G.; Radaelli, G.; Marion, A.; Irato, P.; Bertotto, D.; Santovito, G. Antioxidant Responses Induced by PFAS Exposure in Freshwater Fish in the Veneto Region. Antioxidants 2022, 11, 1115. [Google Scholar] [CrossRef]
- Rozewicki, J.; Li, S.; Amada, K.M.; Standley, D.M.; Katoh, K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res. 2019, 47, W5–W10. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Wang, J.; Sheng, J.; Gu, Q.; Hong, Y. Molecular characteristics of mitochondrial DNA and phylogenetic analysis of the loach (Misgurnus anguillicaudatus) from the Poyang Lake. Mitochondrial DNA 2012, 23, 187–200. [Google Scholar] [CrossRef]
- Balushkin, A.V. Morphology, classification, and evolution of notothenioid fishes of the Southern Ocean (Notothenioidei, Perciformes). J. Ichthyol. 2000, 40, 74–109. [Google Scholar]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef] [Green Version]
- Bazinet, A.L.; Zwickl, D.J.; Cummings, M.P. A Gateway for phylogenetic analysis powered by grid computing featuring GARLI 2.0. Syst. Biol. 2014, 63, 812–818. [Google Scholar] [CrossRef] [Green Version]
- Rédei, G.P. PAUP (phylogenetic analysis using parsimony). In Encyclopedia of Genetics, Genomics, Proteomics and Informatics; Springer: Dordrecht, The Netherlands, 2008; pp. 23–77. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analyses. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nylander, J.A.; Wilgenbusch, J.C.; Warren, D.L.; Swofford, D.L. AWTY (are we there yet?): A system for graphical exploration of MCMC convergence in Bayesian phylogenetics. Bioinformatics 2008, 24, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Nielsen, R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 2002, 19, 908–917. [Google Scholar] [CrossRef] [Green Version]
- Scheffler, K.; Martin, D.P.; Seoighe, C. Robust inference of positive selection from recombining coding sequences. Bioinformatics 2006, 22, 2493–2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graur, D.; Li, W. Fundamentals of Molecular Evolution, 2nd ed.; Sinauer Ass. Inc.: Sunderland, UK, 2000. [Google Scholar]
- Pond, S.L.; Frost, S.D. Datamonkey: Rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 2005, 21, 2531–2533. [Google Scholar] [CrossRef] [Green Version]
- Dettai, A.; Berkani, M.; Lautredou, A.C.; Couloux, A.; Lecointre, G.; Ozouf-Costaz, C.; Gallut, C. Tracking the elusive monophyly of nototheniid fishes (Teleostei) with multiple mitochondrial and nuclear markers. Mar. Genom. 2012, 8, 49–58. [Google Scholar] [CrossRef]
- Near, T.J.; MacGuigan, D.J.; Parker, E.; Struthers, C.D.; Jones, C.D.; Dornburg, A. Phylogenetic analysis of Antarctic notothenioids illuminates the utility of RADseq for resolving Cenozoic adaptive radiations. Mol. Phylogenet. Evol. 2018, 129, 268–279. [Google Scholar] [CrossRef]
- Bargelloni, L.; Scudiero, R.; Parisi, E.; Carginale, V.; Capasso, C.; Patarnello, T. Metallothioneins in antarctic fish: Evidence for independent duplication and gene conversion. Mol. Biol. Evol. 1999, 16, 885–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatzidimitriou, E.; Bisaccia, P.; Corrà, F.; Bonato, M.; Irato, P.; Manuto, L.; Toppo, S.; Bakiu, R.; Santovito, G. Copper/zinc superoxide dismutase from the crocodile icefish Chionodraco hamatus: Antioxidant defense at constant sub-zero temperature. Antioxidants 2020, 9, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, J.B.; Shipp, S.S.; Lowe, A.L.; Wellner, J.S.; Mosola, A.B. The Antarctic ice sheet during the last glacial maximum and its subsequent retreat history: A review. Quat. Sci. Rev. 2002, 21, 49–70. [Google Scholar] [CrossRef]
- Cheng, C.H.; Chen, L.; Near, T.J.; Jin, Y. Functional antifreeze glycoprotein genes in temperate-water New Zealand nototheniid fish infer an Antarctic evolutionary origin. Mol. Biol. Evol. 2003, 20, 1897–1908. [Google Scholar] [CrossRef] [Green Version]
- Eastman, J.; Eakin, R. Checklist of the species of notothenioid fishes. Antarct. Sci. 2021, 33, 273–280. [Google Scholar] [CrossRef]
- Near, T.; Dornburg, A.; Harrington, R.; Oliveira, C.; Pietsch, T.; Thacker, C.; Satoh, T.; Katayama, E.; Wainwright, P.; Eastman, J.T.; et al. Identification of the notothenioid sister lineage illuminates the biogeographic history of an Antarctic adaptive radiation. BMC Evol. Biol. 2015, 15, 109. [Google Scholar] [CrossRef] [Green Version]
- Pollard, D.A.; Iyer, V.N.; Moses, A.M.; Eisen, M.B. Widespread discordance of gene trees with species tree in Drosophila: Evidence for incomplete lineage sorting. PLoS Genet. 2006, 2, e173. [Google Scholar] [CrossRef] [Green Version]
- Østbye, T.-K.K.; Wetten, O.F.; Tooming-Klunderud, A.; Jakobsen, K.S.; Yafe, A.; Etzioni, S.; Moen, T.; Andersen, Ø. Myostatin (MSTN) gene duplications in Atlantic salmon (Salmo salar): Evidence for different selective pressure on teleost MSTN-1 and -2. Gene 2007, 403, 159–169. [Google Scholar] [CrossRef]
- Römisch, K.; Matheson, T. Cell biology in the Antarctic: Studying life in the freezer. Nat. Cell Biol. 2003, 5, 3–6. [Google Scholar] [CrossRef]
- Varriale, S.; Ferraresso, S.; Giacomelli, S.; Coscia, M.R.; Bargelloni, L.; Oreste, U. Evolutionary analysis of Antarctic teleost Toll-like receptor 2. Fish Shellfish Immunol. 2012, 33, 1076–1085. [Google Scholar] [CrossRef]
- Santovito, G.; Marino, S.; Sattin, G.; Cappellini, R.; Bubacco, L.; Beltramini, M. Cloning and characterization of cytoplasmic carbonic anhydrase from gills of four Antarctic fish: Insights into the evolution of fish carbonic anhydrase and cold adaptation. Polar Biol. 2012, 35, 1587–1600. [Google Scholar] [CrossRef]
- Sattin, G.; Bakiu, R.; Tolomeo, A.M.; Carraro, A.; Coppola, D.; Ferro, D.; Patarnello, T.; Santovito, G. Characterization and expression of a new cytoplasmic glutathione peroxidase 1 gene in the Antarctic fish Trematomus bernacchii. Hydrobiologia 2015, 761, 363–372. [Google Scholar] [CrossRef]
- Tolomeo, A.M.; Carraro, A.; Bakiu, R.; Toppo, S.; Place, S.P.; Ferro, D.; Santovito, G. Peroxiredoxin 6 from the Antarctic emerald rockcod: Molecular characterization of its response to warming. J. Comp. Physiol. B 2016, 186, 59–71. [Google Scholar] [CrossRef]
- Tolomeo, A.M.; Carraro, A.; Bakiu, R.; Toppo, S.; Garofalo, F.; Pellegrino, D.; Gerdol, M.; Ferro, D.; Place, S.P.; Santovito, G. Molecular Characterization of Novel Mitochondrial Peroxiredoxins from the Antarctic Emerald Rockcod and Their Gene Expression in Response to Environmental Warming. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2019, 225, 108580. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S. Gene duplication and the uniqueness of vertebrate genomes circa 1970–1999. Semin. Cell Dev. Biol. 1999, 10, 517–522. [Google Scholar] [CrossRef]
- Stanley Kim, H.; Yu, Y.; Snesrud, E.C.; Moy, L.P.; Linford, L.D.; Haas, B.J.; Nierman, W.C.; Quackenbush, J. Transcriptional divergence of the duplicated oxidative stress-responsive genes in the Arabidopsis genome. Plant J. 2005, 41, 212–220. [Google Scholar] [CrossRef]
- Carnera, M.; Schumann, S.; Irato, P.; Santovito, G. Metallothionein gene expression in Trematomus eulepidotus as a response to environmental variation of metal ion con-centrations. ISJ–Invertebr. Surviv. J. 2021, 19, 81. [Google Scholar]
- Cheng, C.H.; Detrich, H.W., 3rd. Molecular ecophysiology of Antarctic notothenioid fishes. Philos. Trans. R Soc. Lond. B Biol. Sci. 2007, 362, 2215–2232. [Google Scholar] [CrossRef] [Green Version]
- Bell, S.G.; Vallee, B.L. The metallothionein/thionein system: An oxidoreductive metabolic zinc link. ChemBioChem 2009, 10, 55–62. [Google Scholar] [CrossRef]
- Kornegay, J.R.; Schilling, J.W.; Wilson, A.C. Molecular adaptation of a leaf-eating bird: Stomach lysozyme of the hoatzin. Mol. Biol. Evol. 1994, 11, 921–928. [Google Scholar] [CrossRef] [Green Version]
- Sidell, B.D. Life at body temperatures below 0 degrees C: The physiology and biochemistry of Antarctic fishes. Gravit. Space Biol. Bull. 2000, 13, 25–34. [Google Scholar] [PubMed]
- Ferro, D.; Franchi, N.; Bakiu, R.; Ballarin, L.; Santovito, G. Molecular characterization and metal induced gene expression of the novel glutathione peroxidase 7 from the chordate invertebrate Ciona robusta. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2018, 205, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Irato, P.; Piccinni, E.; Cassini, A.; Santovito, G. Antioxidant responses to variations in dissolved oxygen of Scapharca inaequivalvis and Tapes philippinarum, two bivalve species from the lagoon of Venice. Mar. Pollut. Bull. 2007, 54, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Ferro, D.; Franchi, N.; Mangano, V.; Bakiu, R.; Cammarata, M.; Parrinello, N.; Santovito, G.; Ballarin, L. Characterization and metal-induced gene transcription of two new copper zinc superoxide dismutases in the solitary ascidian Ciona intestinalis. Aquat. Toxicol. 2013, 140–141, 369–379. [Google Scholar] [CrossRef]
- Franchi, N.; Piccinni, E.; Ferro, D.; Basso, G.; Spolaore, B.; Santovito, G.; Ballarin, L. Characterization and transcription studies of a phytochelatin synthase gene from the solitary tunicate Ciona intestinalis exposed to cadmium. Aquat. Toxicol. 2014, 152, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Ferro, K.; Ferro, D.; Corrà, F.; Bakiu, R.; Santovito, G.; Kurtz, J. Cu, Zn Superoxide Dismutase Genes in Tribolium castaneum: Evolution, Molecular Characterisation, and Gene Expression during Immune Priming. Front. Immunol. 2017, 8, 1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakiu, R.; Pacchini, S.; Piva, E.; Schumann, S.; Tolomeo, A.M.; Ferro, D.; Irato, P.; Santovito, G. Metallothionein expression as a physiological response against metal toxicity in the striped rockcod Trematomus hansoni. Int. J. Mol. Sci. 2022, 23, 12799. [Google Scholar] [CrossRef]
- Barreau, C.; Paillard, L.; Osborne, H.B. AU-rich elements and associated factors: Are there unifying principles? Nucleic Acids Res. 2006, 33, 7138–7150. [Google Scholar] [CrossRef] [Green Version]
- Boldrin, F.; Santovito, G.; Formigari, A.; Bisharyan, Y.; Cassidy-Hanley, D.; Clark, T.G.; Piccinni, E. MTT2, a copper inducible metallothionein gene from Tetrahymena thermophila. Comp. Biochem. Physiol. C 2008, 147, 232–240. [Google Scholar] [CrossRef]
- Franchi, N.; Ferro, D.; Ballarin, L.; Santovito, G. Transcription of genes involved in glutathione biosynthesis in the solitary tunicate Ciona intestinalis exposed to metals. Aquat. Toxicol. 2012, 114–115, 14–22. [Google Scholar] [CrossRef]
- Santovito, G.; Boldrin, F.; Irato, P. Metal and metallothionein distribution in different tissues of the Mediterranean clam Venerupis philippinarum during copper treatment and detoxification. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2015, 174–175, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Buchmann, K.; Pedersen, K. A study on teleost phylogeny using specific antisera. J. Fish Biol. 1994, 45, 901–903. [Google Scholar] [CrossRef]
- Arnold, M.L.; Fogarty, N.D. Reticulate Evolution and Marine Organisms: The Final Frontier? Int. J. Mol. Sci. 2009, 10, 3836–3860. [Google Scholar] [CrossRef] [PubMed]
- van Herwerden, L.; Choat, J.H.; Dudgeon, C.L.; Carlos, G.; Newman, S.J.; Frisch, A.; van Oppen, M. Contrasting patterns of genetic structure in two species of the coral trout Plectropomus (Serranidae) from east and west Australia: Introgressive hybridisation or ancestral polymorphisms. Mol. Phylogenet. Evol. 2006, 41, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Marie, A.D.; Van Herwerden, L.; Choat, J.H.; Hobbs, J.P. Hybridization of Reef Fishes at the Indo-Pacific Biogeographic Barrier: A Case Study. Coral Reefs 2007, 26, 841–850. [Google Scholar] [CrossRef]
- Marrone, A.; La Russa, D.; Brunelli, E.; Santovito, G.; La Russa, M.F.; Barca, D.; Pellegrino, D. Antarctic Fish as a Global Pollution Sensor: Metals Biomonitoring in a Twelve-Year Period. Front. Mol. Biosci. 2021, 8, 794946. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Models | Codon Frequency Models | d.f. | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F3 × 4 | F61 | ||||||||||||||
| ω1 | ω2 | ω3 | ω4 | L | LRT | P | ω1 | ω2 | ω3 | ω4 | L | LRT | P | ||
| H0: ω1 = ω2 = ω3 = ω4 | 0.0972 | 0.0972 | 0.0972 | 0.0972 | 1.4581 | 0.1670 | 0.1670 | 0.1670 | 0.1670 | 1.6008 | |||||
| H1: ω1 = ω2 = ω3 ≠ ω4 | 0.1044 | 0.1044 | 0.1044 | 0.0001 | 1.4615 | 0.2843 | 0.5939 | 0.1811 | 0.1811 | 0.1811 | 0.0001 | 1.5848 | 4.2976 | 0.0382 | 1 |
| H2: ω1 = ω2 ≠ ω3 = ω4 | 0.0953 | 0.0953 | 0.1017 | 0.1017 | 1.4598 | 0.0151 | 0.9022 | 0.1596 | 0.1596 | 0.1860 | 0.1860 | 1.6058 | 1.3137 | 0.2517 | 1 |
| H3: ω1 ≠ ω2 ≠ ω3 = ω4 | 0.0446 | 0.1213 | 0.1274 | 0.1274 | 1.4429 | 2.9085 | 0.0881 | 0.0724 | 0.2150 | 0.2186 | 0.2186 | 1.5889 | 4.3021 | 0.0381 | 1 |
| Models Compared | Codon Frequency Models | d.f. | |||
|---|---|---|---|---|---|
| F3 × 4 | F61 | ||||
| LRT | P | LRT | P | ||
| M0 vs. M3 | 28.1828 | <0.00001 | 17.8792 | 0.0013 | 4 |
| M1 vs. M2 | 3.6376 | 0.1622 | 0.2421 | 0.8860 | 2 |
| M7 vs. M8 | 2.5357 | 0.2814 | 1.3829 | 0.3679 | 2 |
| Amino Acid | Codon | Codon Change | Number of Parallel Changes | Number of Convergent Changes | Maximum Likelihood Method | Clade | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SLAC | FEL | REL | ||||||||||
| Normalized E[dN-dS] | P | Normalized E[dN-dS] | P | Normalized E[dN-dS] | Posterior Probability | Bayes Factor | ||||||
| Asp | 2 | GAT—GAC | 3 | 1 | −16.1540 | 0.0010 | −4.1510 | 0.0030 | −2.3512 | 0.9989 | 221.0540 | O, A |
| Pro | 3 | CCC(G)—CCT | 4 | 1 | −11.9160 | 0.0040 | −7.0040 | <0.0001 | −2.7068 | 1 | 11,742.7000 | O, A1 |
| Lys | 8 | AAA—AAG | 0 | 2 | −14.1570 | 0.0060 | −26.0830 | <0.0001 | −4.5843 | 0.9998 | 1359.5700 | A1 |
| Cys | 47 | TGC—TGT | 5 | 0 | −20.1930 | <0.0001 | −19.9510 | <0.0001 | −4.9403 | 1 | 26,838.5000 | O, A |
| Lys | 52 | AAG—AAA | 2 | 0 | −9.4070 | 0.0330 | −10.4540 | 0.0020 | −3.0828 | 0.9968 | 74.4374 | O, A2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakiu, R.; Boldrin, F.; Pacchini, S.; Schumann, S.; Piva, E.; Tolomeo, A.M.; Ferro, D.; Grapputo, A.; Santovito, G.; Irato, P. Molecular Evolution of Metallothioneins of Antarctic Fish: A Physiological Adaptation to Peculiar Seawater Chemical Characteristics. J. Mar. Sci. Eng. 2022, 10, 1592. https://doi.org/10.3390/jmse10111592
Bakiu R, Boldrin F, Pacchini S, Schumann S, Piva E, Tolomeo AM, Ferro D, Grapputo A, Santovito G, Irato P. Molecular Evolution of Metallothioneins of Antarctic Fish: A Physiological Adaptation to Peculiar Seawater Chemical Characteristics. Journal of Marine Science and Engineering. 2022; 10(11):1592. https://doi.org/10.3390/jmse10111592
Chicago/Turabian StyleBakiu, Rigers, Francesco Boldrin, Sara Pacchini, Sophia Schumann, Elisabetta Piva, Anna Maria Tolomeo, Diana Ferro, Alessandro Grapputo, Gianfranco Santovito, and Paola Irato. 2022. "Molecular Evolution of Metallothioneins of Antarctic Fish: A Physiological Adaptation to Peculiar Seawater Chemical Characteristics" Journal of Marine Science and Engineering 10, no. 11: 1592. https://doi.org/10.3390/jmse10111592