
Acute Haemostatic Depletion and Failure in Patients with Traumatic Brain Injury (TBI): Pathophysiological and Clinical Considerations

Abstract
:1. Introduction
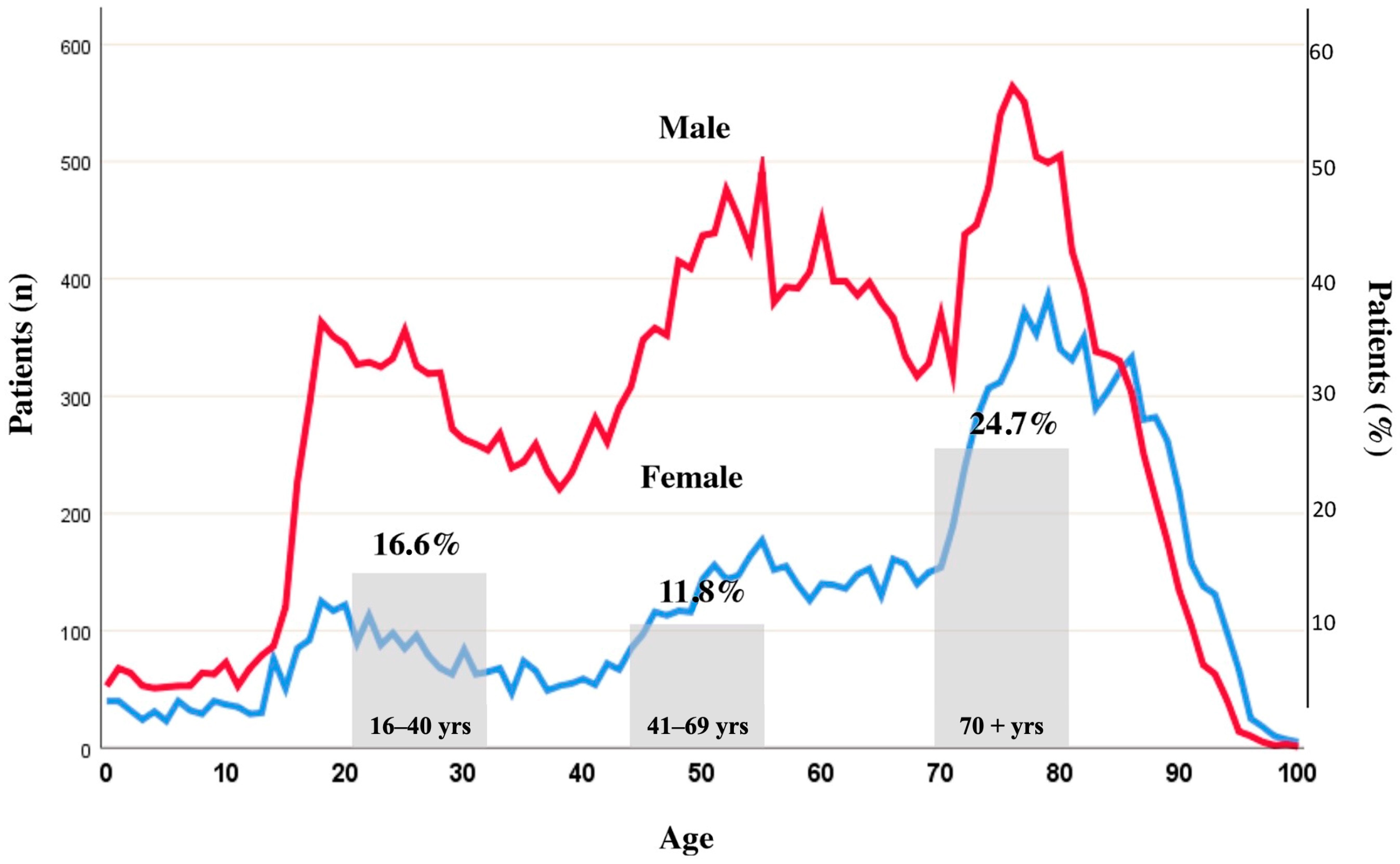
2. Frequency of Haemostatic Disorders after Isolated TBI upon Admission and the Role of Preinjury Anticoagulant and/or Antiplatelet Therapies
3. The Clinical Pattern of Coagulopathy after TBI
4. Coagulopathy as a Powerful Predictor for Prognosis
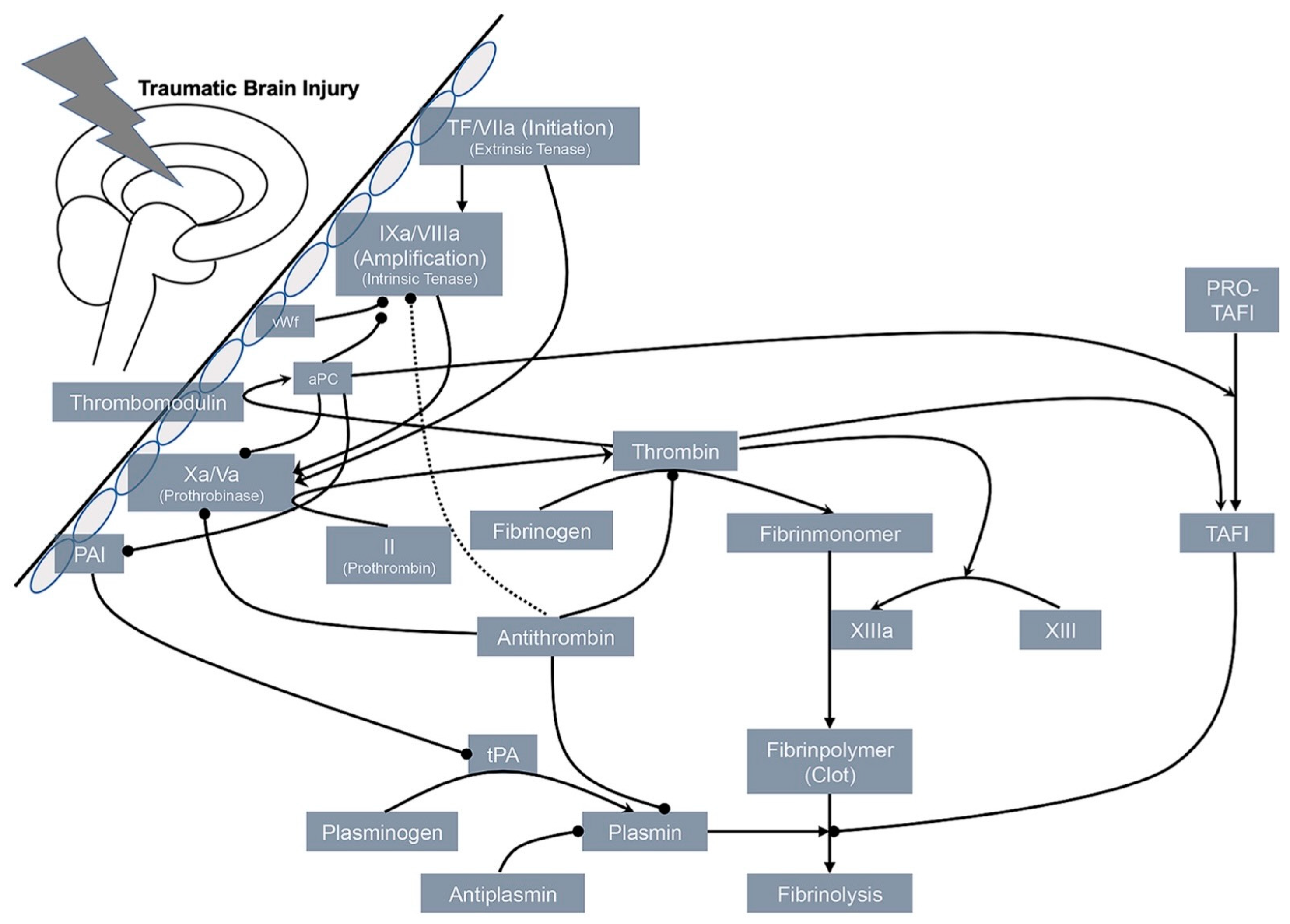
5. Potential Mechanisms of Hemorrhagic Progression and Coagulopathy after TBI
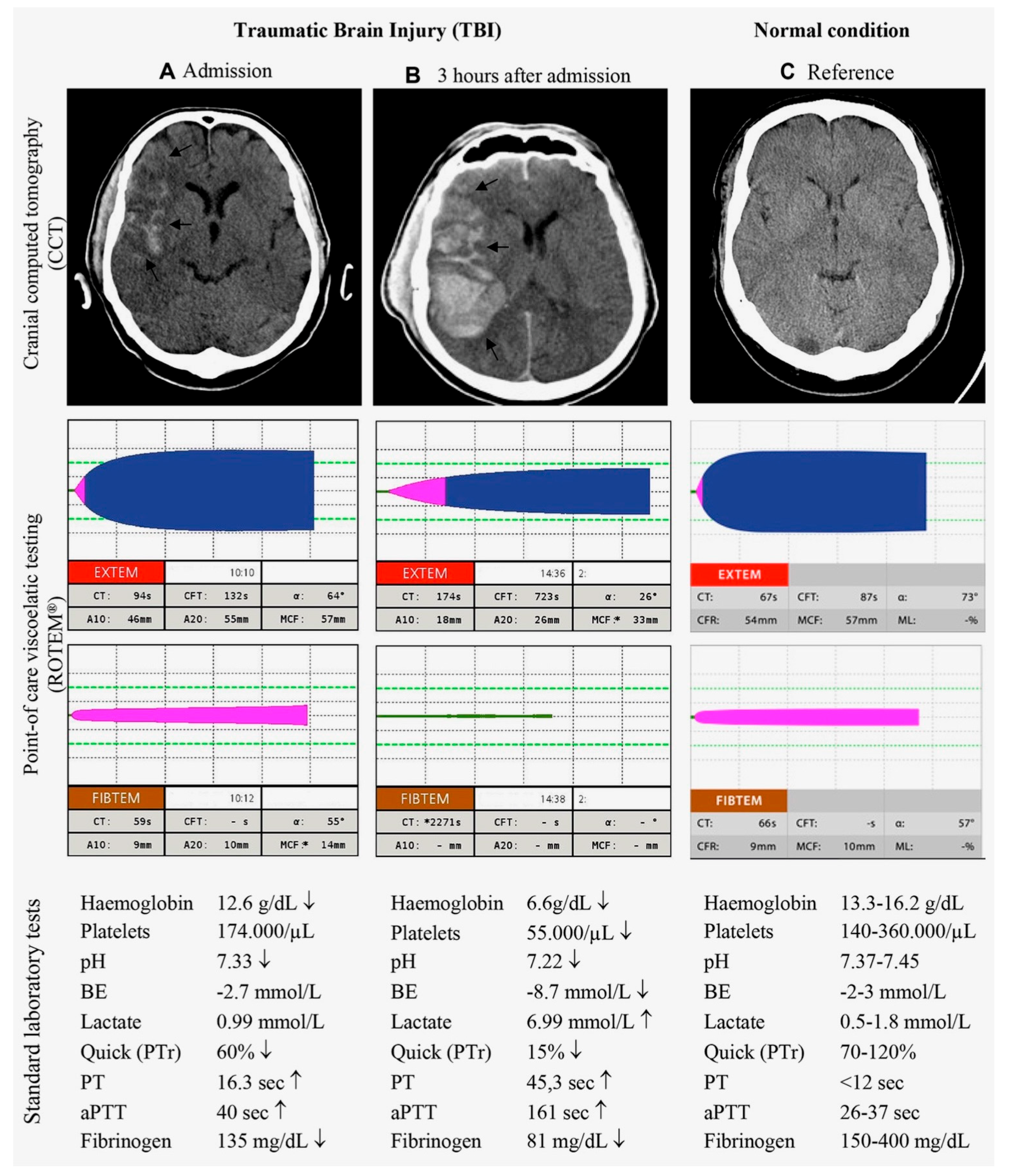
5.1. Hemorrhagic Progressions and New Lesions
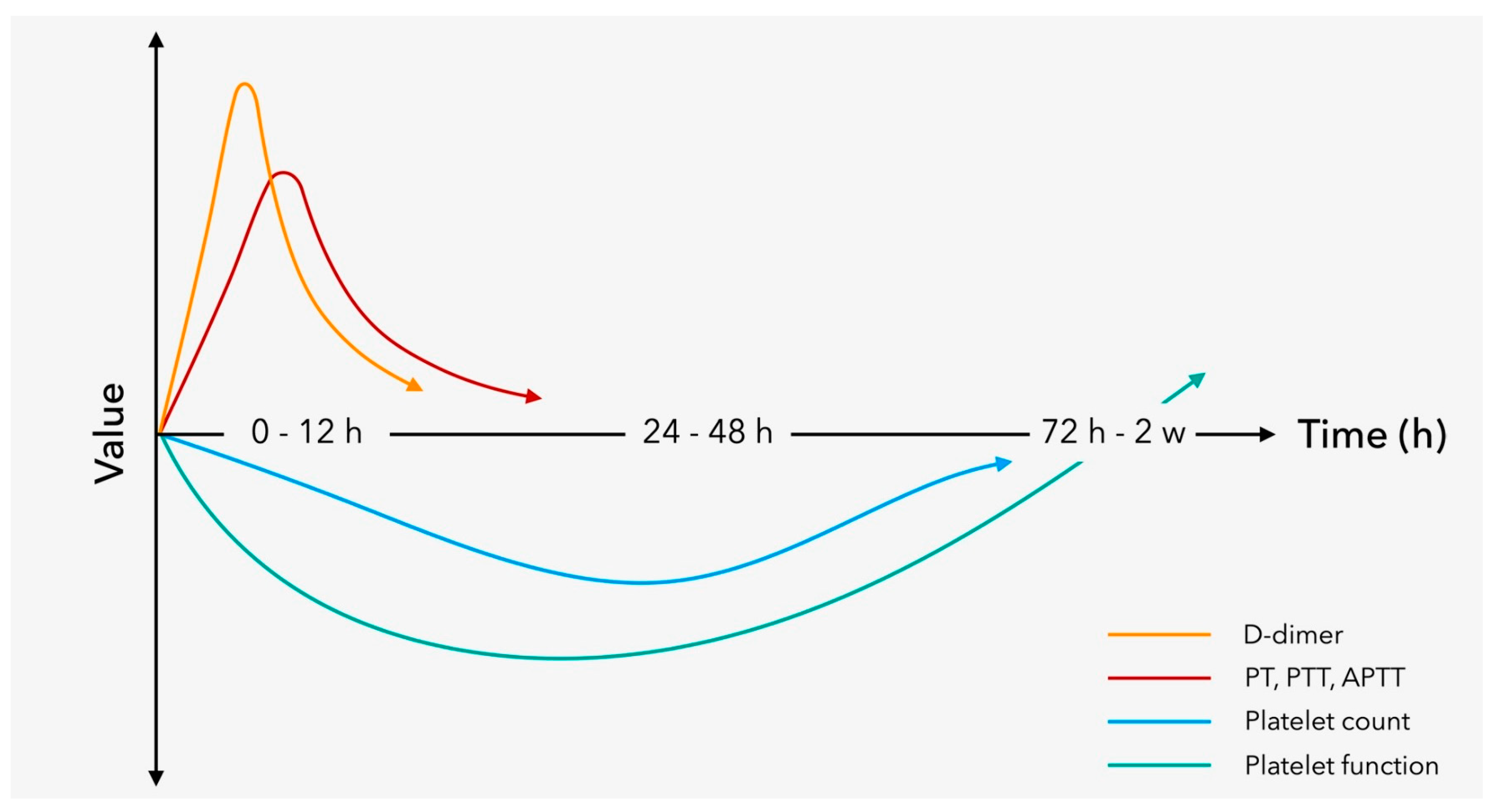
5.2. Coagulopathy after TBI
5.3. Traumatic Brain Injury and Shock
5.4. The Role of Platelets
5.5. Experimental Models
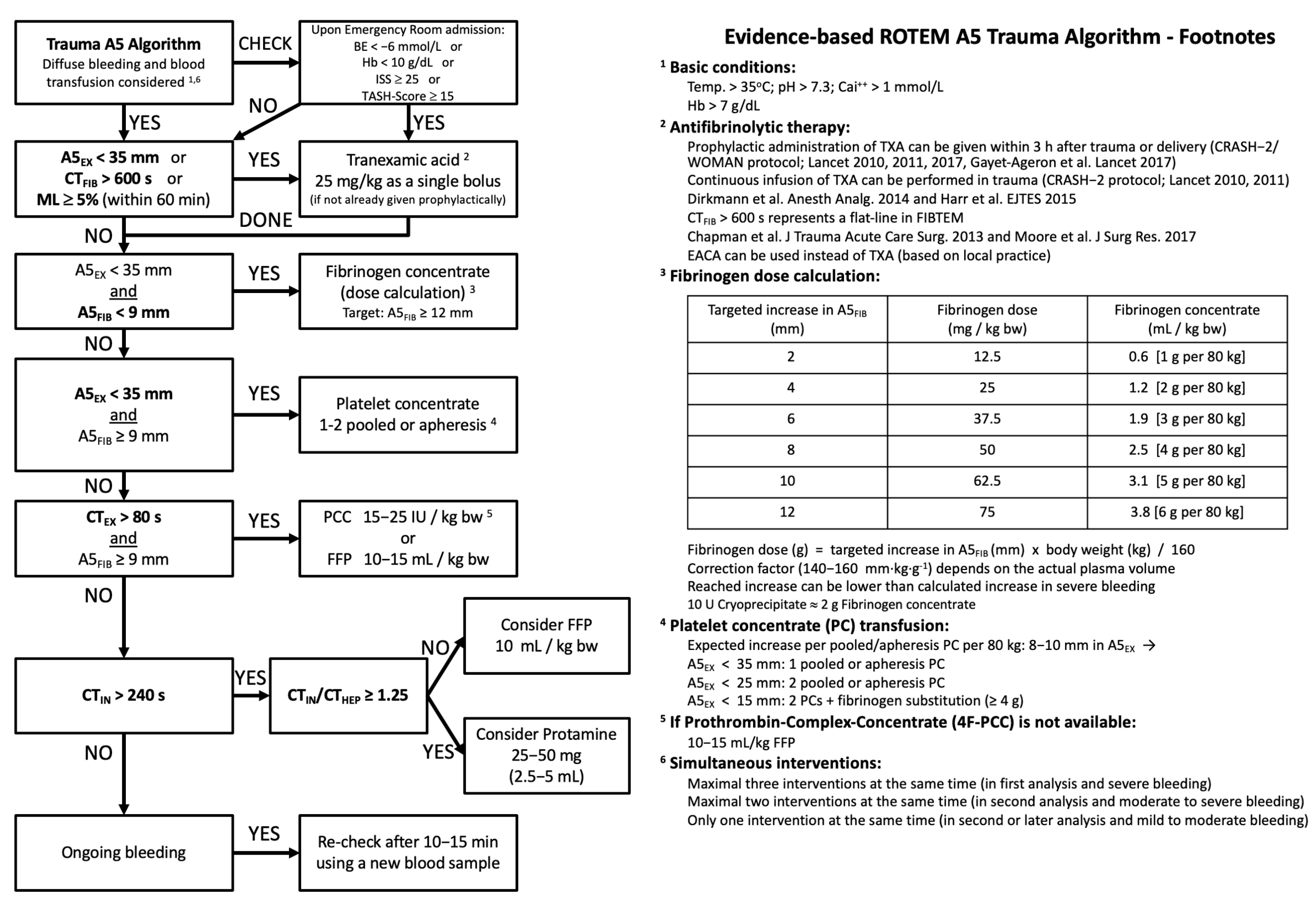
6. Novel Diagnostic Approaches to Coagulopathy
7. Treatment Approaches to Coagulopathy after TBI
7.1. Reversal of Iatrogenic Anticoagulation
7.2. Conventional Blood Products
7.3. Tranexamic Acid (TXA)
7.4. Goal-Directed Therapies
8. Limitations
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Büki, A.; Chesnut, R.M.; et al. Traumatic Brain Injury: Integrated Approaches to Improve Prevention, Clinical Care, and Research. Lancet Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the Global Incidence of Traumatic Brain Injury. J. Neurosurg. 2018, 130, 1080–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maegele, M.; Schöchl, H.; Menovsky, T.; Maréchal, H.; Marklund, N.; Buki, A.; Stanworth, S. Coagulopathy and Haemorrhagic Progression in Traumatic Brain Injury: Advances in Mechanisms, Diagnosis, and Management. Lancet Neurol. 2017, 16, 630–647. [Google Scholar] [CrossRef] [PubMed]
- Roozenbeek, B.; Maas, A.I.R.; Menon, D.K. Changing Patterns in the Epidemiology of Traumatic Brain Injury. Nat. Rev. Neurol. 2013, 9, 231–236. [Google Scholar] [CrossRef]
- Lok, J.; Leung, W.; Murphy, S.; Butler, W.; Noviski, N.; Lo, E.H. Intracranial Hemorrhage: Mechanisms of Secondary Brain Injury. Acta Neurochir. Suppl. 2011, 111, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Maegele, M.; Lefering, R.; Sakowitz, O.; Kopp, M.A.; Schwab, J.M.; Steudel, W.I.; Unterberg, A.; Hoffmann, R.; Uhl, E.; Marzi, I. The Incidence and Management of Moderate to Severe Head Injury. Dtsch. Arztebl. Int. 2019, 116, 167–173. [Google Scholar] [CrossRef]
- Maegele, M. Coagulopathy and Progression of Intracranial Hemorrhage in Traumatic Brain Injury. Neurosurgery 2021, 89, 954–966. [Google Scholar] [CrossRef]
- Böhm, J.K.; Güting, H.; Thorn, S.; Schäfer, N.; Rambach, V.; Schöchl, H.; Grottke, O.; Rossaint, R.; Stanworth, S.; Curry, N.; et al. Global Characterisation of Coagulopathy in Isolated Traumatic Brain Injury (ITBI): A CENTER-TBI Analysis. Neurocritical Care 2021, 35, 184–196. [Google Scholar] [CrossRef]
- Lustenberger, T.; Talving, P.; Kobayashi, L.; Inaba, K.; Lam, L.; Plurad, D.; Demetriades, D. Time Course of Coagulopathy in Isolated Severe Traumatic Brain Injury. Injury 2010, 41, 924–928. [Google Scholar] [CrossRef]
- Batchelor, J.S.; Grayson, A. A Meta-Analysis to Determine the Effect of Anticoagulation on Mortality in Patients with Blunt Head Trauma*. Br. J. Neurosurg. 2012, 26, 525–530. [Google Scholar] [CrossRef]
- Fabbri, A.; Servadei, F.; Marchesini, G.; Bronzoni, C.; Montesi, D.; Arietta, L. Antiplatelet Therapy and the Outcome of Subjects with Intracranial Injury: The Italian SIMEU Study. Crit. Care 2013, 17, R53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieu, F.; Güting, H.; Gravesteijn, B.; Monteiro, M.; Glocker, B.; Kornaropoulos, E.N.; Kamnistas, K.; Robertson, C.S.; Levin, H.; Whitehouse, D.P.; et al. Impact of Antithrombotic Agents on Radiological Lesion Progression in Acute Traumatic Brain Injury: A CENTER-TBI Propensity-Matched Cohort Analysis. J. Neurotrauma 2020, 37, 2069–2080. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, L.; Barmparas, G.; Bosarge, P.; Brown, C.V.; Bukur, M.; Carrick, M.M.; Catalano, R.D.; Holly-Nicolas, J.; Inaba, K.; Kaminski, S.; et al. Novel Oral Anticoagulants and Trauma: The Results of a Prospective American Association for the Surgery of Trauma Multi-Institutional Trial. J. Trauma Acute Care Surg. 2017, 82, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Zeeshan, M.; Jehan, F.; O’Keeffe, T.; Khan, M.; Zakaria, E.R.; Hamidi, M.; Gries, L.; Kulvatunyou, N.; Joseph, B. The Novel Oral Anticoagulants (NOACs) Have Worse Outcomes Compared with Warfarin in Patients with Intracranial Hemorrhage after TBI. J. Trauma Acute Care Surg. 2018, 85, 915–920. [Google Scholar] [CrossRef]
- Markou, M.; Pleger, B.; Grözinger, M.; Pintea, B.; Hamsen, U.; Könen, S.; Schildhauer, T.A.; Martínez, R.; Gousias, K. Intake of NOAC Is Associated with Hematoma Expansion of Intracerebral Hematomas after Traumatic Brain Injury. Eur. J. Trauma Emerg. Surg. 2021, 47, 565–571. [Google Scholar] [CrossRef]
- Harhangi, B.S.; Kompanje, E.J.O.; Leebeek, F.W.G.; Maas, A.I.R. Coagulation Disorders after Traumatic Brain Injury. Acta Neurochir. 2008, 150, 165–175. [Google Scholar] [CrossRef]
- Epstein, D.S.; Mitra, B.; O’Reilly, G.; Rosenfeld, J.V.; Cameron, P.A. Acute Traumatic Coagulopathy in the Setting of Isolated Traumatic Brain Injury: A Systematic Review and Meta-Analysis. Injury 2014, 45, 819–824. [Google Scholar] [CrossRef]
- Zehtabchi, S.; Soghoian, S.; Liu, Y.; Carmody, K.; Shah, L.; Whittaker, B.; Sinert, R. The Association of Coagulopathy and Traumatic Brain Injury in Patients with Isolated Head Injury. Resuscitation 2008, 76, 52–56. [Google Scholar] [CrossRef]
- Talving, P.; Benfield, R.; Hadjizacharia, P.; Inaba, K.; Chan, L.S.; Demetriades, D. Coagulopathy in Severe Traumatic Brain Injury: A Prospective Study. J. Trauma 2009, 66, 55–61. [Google Scholar] [CrossRef]
- Lustenberger, T.; Talving, P.; Kobayashi, L.; Barmparas, G.; Inaba, K.; Lam, L.; Branco, B.C.; Demetriades, D. Early Coagulopathy after Isolated Severe Traumatic Brain Injury: Relationship with Hypoperfusion Challenged. J. Trauma 2010, 69, 1410–1414. [Google Scholar] [CrossRef]
- Wafaisade, A.; Lefering, R.; Tjardes, T.; Wutzler, S.; Simanski, C.; Paffrath, T.; Fischer, P.; Bouillon, B.; Maegele, M. Acute Coagulopathy in Isolated Blunt Traumatic Brain Injury. Neurocritical Care 2010, 12, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, G.; Rangarajan, K.; Subramanian, A.; Agrawal, D.; Sharma, S.; Mukhopadhayay, A.K. Hypofibrinogenemia in Isolated Traumatic Brain Injury in Indian Patients. Neurol. India 2010, 58, 756–757. [Google Scholar] [CrossRef] [PubMed]
- Greuters, S.; van den Berg, A.; Franschman, G.; Viersen, V.A.; Beishuizen, A.; Peerdeman, S.M.; Boer, C.; The ALARM-BLEEDING Investigators. Acute and Delayed Mild Coagulopathy Are Related to Outcome in Patients with Isolated Traumatic Brain Injury. Crit. Care 2011, 15, R2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shehata, M.; Afify, M.; El-Shafie, M.; Khaled, M. Prevalence and Clinical Implications of Coagulopathy in Patients with Isolated Head Trauma. Med. J. Cairo Univ. 2011, 79, 131–137. [Google Scholar]
- Schöchl, H.; Solomon, C.; Traintinger, S.; Nienaber, U.; Tacacs-Tolnai, A.; Windhofer, C.; Bahrami, S.; Voelckel, W. Thromboelastometric (ROTEM) Findings in Patients Suffering from Isolated Severe Traumatic Brain Injury. J. Neurotrauma 2011, 28, 2033–2041. [Google Scholar] [CrossRef] [Green Version]
- Franschman, G.; Boer, C.; Andriessen, T.M.J.C.; van der Naalt, J.; Horn, J.; Haitsma, I.; Jacobs, B.; Vos, P.E. Multicenter Evaluation of the Course of Coagulopathy in Patients with Isolated Traumatic Brain Injury: Relation to CT Characteristics and Outcome. J. Neurotrauma 2012, 29, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Genét, G.F.; Johansson, P.I.; Meyer, M.A.S.; Sølbeck, S.; Sørensen, A.M.; Larsen, C.F.; Welling, K.L.; Windeløv, N.A.; Rasmussen, L.S.; Ostrowski, S.R. Trauma-Induced Coagulopathy: Standard Coagulation Tests, Biomarkers of Coagulopathy, and Endothelial Damage in Patients with Traumatic Brain Injury. J. Neurotrauma 2013, 30, 301–306. [Google Scholar] [CrossRef]
- Alexiou, G.A.; Lianos, G.; Fotakopoulos, G.; Michos, E.; Pachatouridis, D.; Voulgaris, S. Admission Glucose and Coagulopathy Occurrence in Patients with Traumatic Brain Injury. Brain Inj. 2014, 28, 438–441. [Google Scholar] [CrossRef]
- Joseph, B.; Aziz, H.; Zangbar, B.; Kulvatunyou, N.; Pandit, V.; O’Keeffe, T.; Tang, A.; Wynne, J.; Friese, R.S.; Rhee, P. Acquired Coagulopathy of Traumatic Brain Injury Defined by Routine Laboratory Tests: Which Laboratory Values Matter? J. Trauma Acute Care Surg. 2014, 76, 121–125. [Google Scholar] [CrossRef]
- de Oliveira Manoel, A.L.; Neto, A.C.; Veigas, P.V.; Rizoli, S. Traumatic Brain Injury Associated Coagulopathy. Neurocrit. Care 2015, 22, 34–44. [Google Scholar] [CrossRef]
- Dekker, S.E.; Duvekot, A.; de Vries, H.M.; Geeraedts, L.M.G.; Peerdeman, S.M.; de Waard, M.C.; Boer, C.; Schober, P. Relationship between Tissue Perfusion and Coagulopathy in Traumatic Brain Injury. J. Surg. Res. 2016, 205, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Yu, J.; Wu, X.; Sun, Y.R.; Li, Z.Q.; Du, Z.Y.; Wu, X.H.; Hu, J. Prognostic Value of Coagulation Tests for In-Hospital Mortality in Patients with Traumatic Brain Injury. Scand. J. Trauma Resusc. Emerg. Med. 2018, 26, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, V.; Arulselvi, S.; Agrawal, D.; Pati, H.P.; Pandey, R.M. Early Posttraumatic Changes in Coagulation and Fibrinolysis Systems in Isolated Severe Traumatic Brain Injury Patients and Its Influence on Immediate Outcome. Hematol. Oncol. Stem Cell Ther. 2019, 12, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Lin, A.; Hilliard, C.; Fu, R.; Lennox, T.; Barbosa, R.; Schreiber, M.; Rowell, S. The Utility of Thromboelastography for Predicting The Risk of Progression of Intracranial Hemorrhage in Traumatic Brain Injury Patients. Neurosurgery 2017, 64, 182–187. [Google Scholar] [CrossRef] [PubMed]
- van Gent, J.A.N.; van Essen, T.A.; Bos, M.H.A.; Cannegieter, S.C.; van Dijck, J.T.J.M.; Peul, W.C. Coagulopathy after Hemorrhagic Traumatic Brain Injury, an Observational Study of the Incidence and Prognosis. Acta Neurochir. 2020, 162, 329–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homnick, A.; Sifri, Z.; Yonclas, P.; Mohr, A.; Livingston, D. The Temporal Course of Intracranial Haemorrhage Progression: How Long Is Observation Necessary? Injury 2012, 43, 2122–2125. [Google Scholar] [CrossRef]
- Fletcher-Sandersjöö, A.; Thelin, E.P.; Maegele, M.; Svensson, M.; Bellander, B.-M. Time Course of Hemostatic Disruptions After Traumatic Brain Injury: A Systematic Review of the Literature. Neurocrit. Care 2021, 34, 635–656. [Google Scholar] [CrossRef]
- Folkerson, L.E.; Sloan, D.; Cotton, B.A.; Holcomb, J.B.; Tomasek, J.S.; Wade, C.E. Predicting Progressive Hemorrhagic Injury from Isolated Traumatic Brain Injury and Coagulation. Surgery 2015, 158, 655–661. [Google Scholar] [CrossRef]
- Yuan, Q.; Sun, Y.R.; Wu, X.; Yu, J.; Li, Z.Q.; Du, Z.Y.; Wu, X.H.; Zhou, L.F.; Hu, J. Coagulopathy in Traumatic Brain Injury and Its Correlation with Progressive Hemorrhagic Injury: A Systematic Review and Meta-Analysis. J. Neurotrauma 2016, 33, 1279–1291. [Google Scholar] [CrossRef]
- Mayer, S.A.; Lignelli, A.; Fink, M.E.; Kessler, D.B.; Thomas, C.E.; Swarup, R.; van Heertum, R.L. Perilesional Blood Flow and Edema Formation in Acute Intracerebral Hemorrhage: A SPECT Study. Stroke 1998, 29, 1791–1798. [Google Scholar] [CrossRef] [Green Version]
- Fisher, C.M. Pathological Observations in Hypertensive Cerebral Hemorrhage. J. Neuropathol. Exp. Neurol. 1971, 30, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Fujii, Y.; Tanaka, R.; Takeuchi, S.; Koike, T.; Minakawa, T.; Sasaki, O. Hematoma Enlargement in Spontaneous Intracerebral Hemorrhage. J. Neurosurg. 1994, 80, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Takasugi, S.; Ueda, S.; Matsumoto, K. Chronological Changes in Spontaneous Intracerebral Hematoma--an Experimental and Clinical Study. Stroke 1985, 16, 651–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim-Hing, K.; Rincon, F. Secondary Hematoma Expansion and Perihemorrhagic Edema after Intracerebral Hemorrhage: From Bench Work to Practical Aspects. Front. Neurol. 2017, 8, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurland, D.; Hong, C.; Aarabi, B.; Gerzanich, V.; Simard, J.M. Hemorrhagic Progression of a Contusion after Traumatic Brain Injury: A Review. J. Neurotrauma 2012, 29, 19–31. [Google Scholar] [CrossRef]
- Simard, J.M.; Kilbourne, M.; Tsymbalyuk, O.; Tosun, C.; Caridi, J.; Ivanova, S.; Keledjian, K.; Bochicchio, G.; Gerzanich, V. Key Role of Sulfonylurea Receptor 1 in Progressive Secondary Hemorrhage after Brain Contusion. J. Neurotrauma 2009, 26, 2257–2267. [Google Scholar] [CrossRef]
- Stein, S.C.; Smith, D.H. Coagulopathy in Traumatic Brain Injury. Neurocrit. Care 2004, 1, 479–488. [Google Scholar] [CrossRef]
- Wada, T.; Gando, S.; Maekaw, K.; Katabami, K.; Sageshima, H.; Hayakawa, M.; Sawamura, A. Disseminated Intravascular Coagulation with Increased Fibrinolysis during the Early Phase of Isolated Traumatic Brain Injury. Crit. Care 2017, 21, 219. [Google Scholar] [CrossRef] [Green Version]
- Keimowitz, R.M.; Annis, B.L. Disseminated Intravascular Coagulation Associated with Massive Brain Injury. J. Neurosurg. 1973, 39, 178–180. [Google Scholar] [CrossRef]
- Herbert, J.P.; Guillotte, A.R.; Hammer, R.D.; Scott Litofsky, N. Coagulopathy in the Setting of Mild Traumatic Brain Injury: Truths and Consequences. Brain Sci. 2017, 7, 92. [Google Scholar] [CrossRef]
- Nakae, R.; Murai, Y.; Morita, A.; Yokobori, S. Coagulopathy and Traumatic Brain Injury: Overview of New Diagnostic and Therapeutic Strategies. Neurol. Med. Chirurgica. 2022, 62, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Morel, N.; Morel, O.; Petit, L.; Hugel, B.; Cochard, J.F.; Freyssinet, J.M.; Sztark, F.; Dabadie, P. Generation of Procoagulant Microparticles in Cerebrospinal Fluid and Peripheral Blood after Traumatic Brain Injury. J. Trauma 2008, 64, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Kai, Y.; Maeda, Y.; Sasaki, T.; Kanaide, H.; Hirano, K. Basic and Translational Research on Proteinase-Activated Receptors: The Role of Thrombin Receptor in Cerebral Vasospasm in Subarachnoid Hemorrhage. J. Pharmacol. Sci. 2008, 108, 426–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhang, F.; Dong, J.F. Coagulopathy Induced by Traumatic Brain Injury: Systemic Manifestation of a Localized Injury. Blood 2018, 131, 2001–2006. [Google Scholar] [CrossRef] [Green Version]
- Hijazi, N.; Fanne, R.A.; Abramovitch, R.; Yarovoi, S.; Higazi, M.; Abdeen, S.; Basheer, M.; Maraga, E.; Cines, D.B.; Higazi, A.A.R. Endogenous Plasminogen Activators Mediate Progressive Intracerebral Hemorrhage after Traumatic Brain Injury in Mice. Blood 2015, 125, 2558–2567. [Google Scholar] [CrossRef] [Green Version]
- Foley, J.H.; Conway, E.M. Cross Talk Pathways Between Coagulation and Inflammation. Circ. Res. 2016, 118, 1392–1408. [Google Scholar] [CrossRef]
- Nekludov, M.; Bellander, B.M.; Blombäck, M.; Wallen, H.N. Platelet Dysfunction in Patients with Severe Traumatic Brain Injury. J. Neurotrauma 2007, 24, 1699–1706. [Google Scholar] [CrossRef]
- Wohlauer, M.V.; Moore, E.E.; Thomas, S.; Sauaia, A.; Evans, E.; Harr, J.; Silliman, C.C.; Ploplis, V.; Castellino, F.J.; Walsh, M. Early Platelet Dysfunction: An Unrecognized Role in the Acute Coagulopathy of Trauma. J. Am. Coll. Surg. 2012, 214, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Estrada-Rojo, F.; Martínez-Tapia, R.J.; Estrada-Bernal, F.; Martínez-Vargas, M.; Perez-Arredondo, A.; Flores-Avalos, L.; Navarro, L. Models Used in the Study of Traumatic Brain Injury. Rev. Neurosci. 2018, 29, 139–149. [Google Scholar] [CrossRef]
- Alder, J.; Fujioka, W.; Lifshitz, J.; Crockett, D.P.; Thakker-Varia, S. Lateral Fluid Percussion: Model of Traumatic Brain Injury in Mice. J. Vis. Exp. 2011, e3063. [Google Scholar] [CrossRef] [Green Version]
- Acosta, S.A.; Tajiri, N.; Shinozuka, K.; Ishikawa, H.; Grimmig, B.; Diamond, D.; Sanberg, P.R.; Bickford, P.C.; Kaneko, Y.; Borlongan, C.V. Long-Term Upregulation of Inflammation and Suppression of Cell Proliferation in the Brain of Adult Rats Exposed to Traumatic Brain Injury Using the Controlled Cortical Impact Model. PLoS ONE 2013, 8, e53376. [Google Scholar] [CrossRef]
- Flierl, M.A.; Stahel, P.F.; Beauchamp, K.M.; Morgan, S.J.; Smith, W.R.; Shohami, E. Mouse Closed Head Injury Model Induced by a Weight-Drop Device. Nat. Protoc. 2009, 4, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Maqsood, M.I.; Matin, M.M.; Bahrami, A.R.; Ghasroldasht, M.M. Immortality of Cell Lines: Challenges and Advantages of Establishment. Cell Biol. Int. 2013, 37, 1038–1045. [Google Scholar] [CrossRef] [PubMed]
- Morrison, B.; Cater, H.L.; Benham, C.D.; Sundstrom, L.E. An in Vitro Model of Traumatic Brain Injury Utilising Two-Dimensional Stretch of Organotypic Hippocampal Slice Cultures. J. Neurosci. Methods 2006, 150, 192–201. [Google Scholar] [CrossRef]
- Daviaud, N.; Garbayo, E.; Schiller, P.C.; Perez-Pinzon, M.; Montero-Menei, C.N. Organotypic Cultures as Tools for Optimizing Central Nervous System Cell Therapies. Exp. Neurol. 2013, 248, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Gratz, J.; Güting, H.; Thorn, S.; Brazinova, A.; Görlinger, K.; Schäfer, N.; Schöchl, H.; Stanworth, S.; Maegele, M. Protocolised Thromboelastometric-guided Haemostatic Management in Patients with Traumatic Brain Injury: A Pilot Study. Anaesthesia 2019, 74, 883–890. [Google Scholar] [CrossRef]
- Bradbury, J.L.; Thomas, S.G.; Sorg, N.R.; Mjaess, N.; Berquist, M.R.; Brenner, T.J.; Langford, J.H.; Marsee, M.K.; Moody, A.N.; Bunch, C.M.; et al. Viscoelastic Testing and Coagulopathy of Traumatic Brain Injury. J. Clin. Med. 2021, 10, 5039. [Google Scholar] [CrossRef]
- Cannon, J.W.; Dias, J.D.; Kumar, M.A.; Walsh, M.; Thomas, S.G.; Cotton, B.A.; Schuster, J.M.; Evans, S.L.; Schreiber, M.A.; Adam, E.H.; et al. Use of Thromboelastography in the Evaluation and Management of Patients with Traumatic Brain Injury: A Systematic Review and Meta-Analysis. Crit. Care Explor. 2021, 3, e0526. [Google Scholar] [CrossRef]
- Oberladstätter, D.; Voelckel, W.; Schlimp, C.; Zipperle, J.; Ziegler, B.; Grottke, O.; Schöchl, H. A Prospective Observational Study of the Rapid Detection of Clinically-Relevant Plasma Direct Oral Anticoagulant Levels Following Acute Traumatic Injury. Anaesthesia 2021, 76, 373–380. [Google Scholar] [CrossRef]
- Davis, P.K.; Musunuru, H.; Walsh, M.; Cassady, R.; Yount, R.; Losiniecki, A.; Moore, E.E.; Wohlauer, M.V.; Howard, J.; Ploplis, V.A.; et al. Platelet Dysfunction Is an Early Marker for Traumatic Brain Injury-Induced Coagulopathy. Neurocrit. Care 2013, 18, 201–208. [Google Scholar] [CrossRef]
- Epstein, D.S.; Mitra, B.; Cameron, P.A.; Fitzgerald, M.; Rosenfeld, J.V. Normalization of Coagulopathy Is Associated with Improved Outcome after Isolated Traumatic Brain Injury. J. Clin. Neurosci. 2016, 29, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Spahn, D.R.; Bouillon, B.; Cerny, V.; Duranteau, J.; Filipescu, D.; Hunt, B.J.; Komadina, R.; Maegele, M.; Nardi, G.; Riddez, L.; et al. The European Guideline on Management of Major Bleeding and Coagulopathy Following Trauma: Fifth Edition. Crit. Care 2019, 23, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frontera, J.A.; Lewin, J.J.; Rabinstein, A.A.; Aisiku, I.P.; Alexandrov, A.W.; Cook, A.M.; del Zoppo, G.J.; Kumar, M.A.; Peerschke, E.I.B.; Stiefel, M.F.; et al. Guideline for Reversal of Antithrombotics in Intracranial Hemorrhage: A Statement for Healthcare Professionals from the Neurocritical Care Society and Society of Critical Care Medicine. Neurocritical Care 2016, 24, 6–46. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.R.; Cornutt, D. Reversal Agents for Direct Oral Anticoagulants: Considerations for Hospital Physicians and Intensivists. Hosp. Pract. 2019, 47, 113–122. [Google Scholar] [CrossRef]
- Anglin, C.O.; Spence, J.S.; Warner, M.A.; Paliotta, C.; Harper, C.; Moore, C.; Sarode, R.; Madden, C.; Diaz-Arrastia, R. Effects of Platelet and Plasma Transfusion on Outcome in Traumatic Brain Injury Patients with Moderate Bleeding Diatheses. J. Neurosurg. 2013, 118, 676–686. [Google Scholar] [CrossRef] [Green Version]
- Baharoglu, M.I.; Cordonnier, C.; Salman, R.A.-S.; de Gans, K.; Koopman, M.M.; Brand, A.; Majoie, C.B.; Beenen, L.F.; Marquering, H.A.; Vermeulen, M.; et al. Platelet Transfusion versus Standard Care after Acute Stroke Due to Spontaneous Cerebral Haemorrhage Associated with Antiplatelet Therapy (PATCH): A Randomised, Open-Label, Phase 3 Trial. Lancet 2016, 387, 2605–2613. [Google Scholar] [CrossRef]
- Thorn, S.; Güting, H.; Mathes, T.; Schäfer, N.; Maegele, M. The Effect of Platelet Transfusion in Patients with Traumatic Brain Injury and Concomitant Antiplatelet Use: A Systematic Review and Meta-Analysis. Transfusion 2019, 59, 3536–3544. [Google Scholar] [CrossRef] [Green Version]
- Lelubre, C.; Bouzat, P.; Crippa, I.A.; Taccone, F.S. Anemia Management after Acute Brain Injury. Crit. Care 2016, 20, 152. [Google Scholar] [CrossRef] [Green Version]
- Robertson, C.S.; Hannay, H.J.; Yamal, J.M.; Gopinath, S.; Goodman, J.C.; Tilley, B.C.; Baldwin, A.; Lara, L.R.; Saucedo-Crespo, H.; Ahmed, O.; et al. Effect of Erythropoietin and Transfusion Threshold on Neurological Recovery after Traumatic Brain Injury: A Randomized Clinical Trial. JAMA 2014, 312, 36–47. [Google Scholar] [CrossRef]
- Vedantam, A.; Yamal, J.M.; Rubin, M.L.; Robertson, C.S.; Gopinath, S.P. Progressive Hemorrhagic Injury after Severe Traumatic Brain Injury: Effect of Hemoglobin Transfusion Thresholds. J. Neurosurg. 2016, 125, 1229–1234. [Google Scholar] [CrossRef] [Green Version]
- Sillesen, M.; Bambakidis, T.; Dekker, S.E.; Li, Y.; Alam, H.B. Fresh Frozen Plasma Modulates Brain Gene Expression in a Swine Model of Traumatic Brain Injury and Shock: A Network Analysis. J. Am. Coll. Surg. 2017, 224, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Nikolian, V.C.; Dekker, S.E.; Bambakidis, T.; Higgins, G.A.; Dennahy, I.S.; Georgoff, P.E.; Williams, A.M.; Andjelkovic, A.V.; Alam, H.B. Improvement of Blood-Brain Barrier Integrity in Traumatic Brain Injury and Hemorrhagic Shock Following Treatment with Valproic Acid and Fresh Frozen Plasma. Crit. Care Med. 2018, 46, e59–e66. [Google Scholar] [CrossRef] [PubMed]
- Dekker, S.E.; Nikolian, V.C.; Sillesen, M.; Bambakidis, T.; Schober, P.; Alam, H.B. Different Resuscitation Strategies and Novel Pharmacologic Treatment with Valproic Acid in Traumatic Brain Injury. J. Neurosci. Res. 2018, 96, 711–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, R.; Folkerson, L.E.; Sloan, D.; Tomasek, J.S.; Kitagawa, R.S.; Choi, H.A.; Wade, C.E.; Holcomb, J.B. Early Plasma Transfusion Is Associated with Improved Survival after Isolated Traumatic Brain Injury in Patients with Multifocal Intracranial Hemorrhage. Surgery 2017, 161, 538–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruen, D.S.; Guyette, F.X.; Brown, J.B.; Okonkwo, D.O.; Puccio, A.M.; Campwala, I.K.; Tessmer, M.T.; Daley, B.J.; Miller, R.S.; Harbrecht, B.G.; et al. Association of Prehospital Plasma with Survival in Patients with Traumatic Brain Injury: A Secondary Analysis of the PAMPer Cluster Randomized Clinical Trial. JAMA Netw. Open 2020, 3, e2016869. [Google Scholar] [CrossRef] [PubMed]
- Etemadrezaie, H.; Baharvahdat, H.; Shariati, Z.; Lari, S.M.; Shakeri, M.T.; Ganjeifar, B. The Effect of Fresh Frozen Plasma in Severe Closed Head Injury. Clin. Neurol. Neurosurg. 2007, 109, 166–171. [Google Scholar] [CrossRef]
- Roberts, I.; Shakur-Still, H.; Aeron-Thomas, A.; Belli, A.; Brenner, A.; Chaudary, M.A.; Chaudhri, R.; Jamaluddin, S.F.B.; Frimley, L.; Javaid, K.; et al. Effects of Tranexamic Acid on Death, Disability, Vascular Occlusive Events and Other Morbidities in Patients with Acute Traumatic Brain Injury (CRASH-3): A Randomised, Placebo-Controlled Trial. Lancet 2019, 394, 1713–1723. [Google Scholar] [CrossRef] [Green Version]
- Perel, P.; Al-Shahi Salman, R.; Kawahara, T.; Morris, Z.; Prieto-Merino, D.; Roberts, I.; Sandercock, P.; Shakur, H.; Wardlaw, J. CRASH-2 (Clinical Randomisation of an Antifibrinolytic in Significant Haemorrhage) Intracranial Bleeding Study: The Effect of Tranexamic Acid in Traumatic Brain Injury--A Nested Randomised, Placebo-Controlled Trial. Health Technol. Assess. 2012, 16, 1–54. [Google Scholar] [CrossRef] [Green Version]
- Rowell, S.E.; Meier, E.N.; McKnight, B.; Kannas, D.; May, S.; Sheehan, K.; Bulger, E.M.; Idris, A.H.; Christenson, J.; Morrison, L.J.; et al. Effect of Out-of-Hospital Tranexamic Acid vs Placebo on 6-Month Functional Neurologic Outcomes in Patients With Moderate or Severe Traumatic Brain Injury. JAMA 2020, 324, 961–974. [Google Scholar] [CrossRef]
- Bossers, S.M.; Loer, S.A.; Bloemers, F.W.; den Hartog, D.; van Lieshout, E.M.M.; Hoogerwerf, N.; van der Naalt, J.; Absalom, A.R.; Peerdeman, S.M.; Schwarte, L.A.; et al. Association Between Prehospital Tranexamic Acid Administration and Outcomes of Severe Traumatic Brain Injury. JAMA Neurol. 2021, 78, 328–345. [Google Scholar] [CrossRef]
- Inaba, K.; Rizoli, S.; Veigas, P.V.; Callum, J.; Davenport, R.; Hess, J.; Maegele, M. 2014 Consensus Conference on Viscoelastic Test-Based Transfusion Guidelines for Early Trauma Resuscitation: Report of the Panel. J. Trauma Acute Care Surg. 2015, 78, 1220–1229. [Google Scholar] [CrossRef] [Green Version]
- Kozek-Langenecker, S.A.; Ahmed, A.B.; Afshari, A.; Albaladejo, P.; Aldecoa, C.; Barauskas, G.; de Robertis, E.; Faraoni, D.; Filipescu, D.C.; Fries, D.; et al. Management of Severe Perioperative Bleeding: Guidelines from the European Society of Anaesthesiology: First Update 2016. Eur. J. Anaesthesiol. 2017, 34, 332–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baksaas-Aasen, K.; Gall, L.S.; Stensballe, J.; Juffermans, N.P.; Curry, N.; Maegele, M.; Brooks, A.; Rourke, C.; Gillespie, S.; Murphy, J.; et al. Viscoelastic Haemostatic Assay Augmented Protocols for Major Trauma Haemorrhage (ITACTIC): A Randomized, Controlled Trial. Intensive Care Med. 2021, 47, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.; Moore, E.E.; Moore, H.B.; Chapman, M.P.; Chin, T.L.; Ghasabyan, A.; Wohlauer, M.V.; Barnett, C.C.; Bensard, D.D.; Biffl, W.L.; et al. Goal-Directed Hemostatic Resuscitation of Trauma-Induced Coagulopathy: A Pragmatic Randomized Clinical Trial Comparing a Viscoelastic Assay to Conventional Coagulation Assays. Ann. Surg. 2016, 263, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Rimaitis, M.; Bilskienė, D.; Tamošuitis, T.; Vilcinis, R.; Rimaitis, K.; Macas, A. Implementation of Thromboelastometry for Coagulation Management in Isolated Traumatic Brain Injury Patients Undergoing Craniotomy. Med. Sci. Monit. 2020, 26, e922879. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | No. of Patients | Definition of TBI | Definition of Coagulopathy | Prevalence of Coagulopathy (%) | Mortality with Coagulopathy (%) |
|---|---|---|---|---|---|
| Harhangi [16] * | 5357 | Heterogeneous | Heterogeneous | 32.7 (10–97.5) | 51 (25–93) |
| Epstein [17] ** | 7037 | Heterogeneous | Heterogeneous | 35.2 (7–86.1) | 17–86 |
| Zehtabchi [18] | 224 | AIShead > 2 and/or any intracranial hematoma on CT | INR > 1.3 or PTT > 34 s | 17 (8–30) | - |
| Talving [19] | 387 | AIShead ≥ 3 and extracranial AIS < 3 | Platelets < 100,000 mm3 or INR > 1.1 or aPTT > 36 s | 34 | 34.7 |
| Lustenberger [9] | 278 | AIShead ≥ 3 and extracranial AIS < 3 | Platelets < 100,000 mm3 and/or INR > 1.4 and/oraPTT > 36 s | 45.7 | 40.9 |
| Lustenberger [20] | 132 | AIShead ≥ 3 and extracranial AIS < 3 | Platelets < 100,000 mm3 or INR > 1.2 or aPTT > 36 s | 36.4 | 32.5 |
| Wafaisade [21] | 3114 | AIShead ≥ 3 and extracranial AIS < 3 | Quick (PTR) < 70% and/or platelets < 100,000/mL | 22.7 | 50.4 |
| Chhabra [22] | 100 | GCS < 13 | Fibrinogen < 200 mg/dL | 7 | - |
| Greuters [23] | 107 | Brain tissue injury on CT and extracranial AIS < 3 | aPTT > 40 s and/or INR > 1.2 and/or platelets < 120 × 109/L | 24 (54 #) | 41 |
| Shehata [24] | 101 | iTBI on admission brain CT | INR ≥ 1.2, PT > 13 s, d-dimer positive, platelets < 100 × 103/CC | 63 | 36 |
| Schöchl [25] | 88 | AIShead ≥ 3 and extracranial AIS < 3 | Quick (PTR) < 70% and/or aPTT > 35 s and/or fibrinogen < 150 mg/Dl and/or platelets < 100 × 109/L | 15.8 | 50 |
| Franschman [26] | 226 | iTBI on CT and extracranial AIS <3 | aPTT > 40 s and/or PT > 1.2 and/or platelets < 120 × 109/L | 25 (44 #) | 33 |
| Genet [27] | 23 | AIShead ≥ 3 and extracranial AIS < 3 | aPTT > 35 sand/orINR > 1.2 | 13 | 22 |
| Alexiou [28] | 149 | iTBI with exclusion of multisystem trauma | aPTT > 40 s and/or INR > 1.2 and/or platelets < 120 × 109/L | 14.8 (22.8 #) | - |
| Joseph [29] | 591 | AIShead ≥ 3 and extracranial AIS < 3 | INR ≥ 1.5 and/or PTT ≥ 35 s and/or platelets ≤ 100 × 103/mL | 13.3 | 23 |
| Epstein [17] | 1718 | AIShead ≥ 3 and extracranial AIS < 3 | INR ≥ 1.3 | 7.7 | 45.1 |
| De Oliveira Manoel [30] | 48 | AIShead ≥ 3 and extracranial AIS < 3 | INR ≥ 1.5 and/or aPTT ≥ 60 s and/or platelets < 100 × 103/mm3 § | 12.5 | 66 |
| Dekker [31] | 52 | AIShead ≥ 3 | INR ≥ 1.2 and/or aPTT ≥ 40 s and/or platelets < 120 × 109/L | 42 | 45.5 |
| Yuan [32] | 2319 | Intracranial injury on CT and extracranial AIS < 3 | INR > 1.25 and/or PT > 14 s and/or aPTT > 36 s and/or platelets < 100 × 109/L | 18.6 | 17.6 |
| Albert [33] | 561 | iTBI on admission brain CT | INR ≥ 1.27 and/or PT ≥ 16.7 s and/or aPTT > 28.8 s | 41.6% | 61.1% |
| Böhm [8] | 598 | iTBI on CT and no extracranial injuries | INR > 1.2 and/or aPTT > 35 s and/or fibrinogen < 150 mg/dL and/or platelets < 100 × 103/nL | 19.6 | - |
| Antithrombotic | Strong Recommendation with Moderate-to-High Quality Evidence | Conditional Recommendation with Low-to-Moderate Quality Evidence |
|---|---|---|
| Vitamin K antagonists (VKAs) | Vitamin K for INR reversal in VKA-associated ICH as soon as possible or with other reversal agents 3- and 4-factor PCC iv be preferred to FFP in VKA-associated ICH and INR ≥ 1.4; dosing on weight, INR and type of PCC. Monitoring via repeated INR after PCC administration (15–60 min) and every 6–8 h for 24–48 h; subsequent treatment according to follow-up INR as repeated dosing may increase thrombotic and DIC risk (Good practice statement) Treatment with vitamin K and FFP is recommended over no treatment! | If INR ≥ 1.4 vitamin K 10 mg iv with subsequent treatment according to follow-up INR; if repeated INR ≥ 1.4 within 24–48 h redosing with vitamin K 10 mg iv (Good practice statement) or if treated with PCC and repeated INR ≥ 1.4 within first 24–48 h further correction with FFP Fresh frozen plasma/FFP (10–15 mL/kg iv) along with one dose of vitamin K 10 mg iv if PCC is not available/contraindicated |
| Direct factor Xa inhibitors | Andexanet alfa (400–800 mg as an initial bolus followed by 4–8 mg/min over 120 min (480–960 mg); approved for adults treated with direct factor Xa inhibitors apixaban and rivaroxaban if rapid reversal is indicated due to life-threatening or uncontrolled bleeding! | 4-factor PCC or activated PCC (4-factor PCC 50 U/kg iv or aPCC (FEIBA) 50 U/kg iv) if ICH occurred within 3–5 terminal half-lives of drug exposure or in the context of liver failure Activated charcoal (50 g) within 2 h of drug ingestion to intubated ICH patients with enteral access and/or low risk of aspiration |
| Direct thrombin inhibitors (DTIs) | Idarucizumab (2 × 2.5 g/50 mL) to ICH associated with dabigatran when administered within 3–5 half-lives and no renal failure and in renal failure with continued drug exposure beyond normal 3–5 half-lives | If idarucizumab is not available or in case of overdose, consider hemodialysis; consider redosing idarucizumab and repeated hemodialysis in ongoing bleeding 4-factor PCC or activated PCC (4-factor PCC 50 U/kg iv or aPCC (FEIBA) 50 U/kg iv) if idarucizumab is not available or if ICH with DTIs other than dabigatran and if DTI was administered within 3–5 half-lives and absence of renal failure or in renal failure with continued drug exposure beyond normal 3–5 half-lives Activated charcoal (50 g) within 2 h of drug ingestion to intubated ICH patients with enteral access and/or low risk of aspiration |
| Unfractionated heparin | Protamine sulfate iv for heparin reversal with dosing according to heparin dose over the preceding 2–3 h; protamine sulfate 1 mg for every 100 U heparin given over the preceding 2–3 h with a maximum single dose of 50 mg | If aPTT remains elevated, repeated protamine sulfate at 0.5 mg per 100 U of heparin |
| Low Molecular Weight Heparin (LMWHs) | Protamine sulfate slowly IV over 10 min in the following dosing, if (a.) enoxaparin was given within 8 h 1 mg protamine per 1 mg of enoxaparin administered (maximum single dose 50 mg), if (b.) enoxaparin was given within 8–12 h 0.5 mg protamine per 1 mg enoxaparin (maximum single dose 50 mg). The following dosing applies for dalteparin, nadroparin and tinzaparin: Protamine sulfate 1 mg per 100 anti Xa U of LMWH administered during the past 3–5 half-lives with maximum single dose 50 mg. Only minimal effect on reversal > 12 h from dosing! | Redosing protamine sulfate (0.5 mg per 100 anti-Xa U of LMWH or per 1 mg of enoxaparin) if life-threatening bleeding continous or in renal insufficiency Recombinant factor VIIa (rFVIIa 90 μg/kg iv) if protamine is contraindicated Reversal of danaparoid with rFVIIa in the context of ICH |
| Pentasaccharides | No recommendation | Activated PCC (aPCC (FEIBA) 20 U/kg iv) for pentasaccharide reversal Recombinant factor VIIa (rFVIIa 90 μg/kg iv) if aPCC is contraindicated/not available |
| Thrombolytic agents (Plasminogen activators) | No recommendation | Cryoprecipitate (initial dose 10 U iv) in thrombolytic agent-associated symptomatic ICH if administered within the previous 24 h. If contraindicated/not available antifibrinolytic agent (tranexamic acid 10–15 mg/kg iv over 20 min or ε-aminocaproic acid 4–5 g iv). If fibrinogen levels < 150 mg/dL, administration of additional cryoprecipitate. Although substitution with fibrinogen concentrate, if available, may be reasonable, there is no recommendation at this time! |
| Antiplatelet agents | Platelet function testing prior to platelet transfusion is recommended; if laboratory-documented platelet function is within normal ranges or documented platelet resistance. Recommendation against platelet transfusion! | Desmopressin (0.4 μg/kg iv × 1) in ICH with aspirin/cyclooxygenase-1 or adenosine diphosphate (ADP) inhibitors Platelet concentrates in aspirin- or ADP inhibitor-associated ICH in case of neurosurgical intervention. An initial dose of one single donor apheresis unit of platelets. Platelet testing is suggested prior to repeated platelet transfusion and repeated dosing only if persisting abnormalities! |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kockelmann, F.; Maegele, M. Acute Haemostatic Depletion and Failure in Patients with Traumatic Brain Injury (TBI): Pathophysiological and Clinical Considerations. J. Clin. Med. 2023, 12, 2809. https://doi.org/10.3390/jcm12082809
Kockelmann F, Maegele M. Acute Haemostatic Depletion and Failure in Patients with Traumatic Brain Injury (TBI): Pathophysiological and Clinical Considerations. Journal of Clinical Medicine. 2023; 12(8):2809. https://doi.org/10.3390/jcm12082809
Chicago/Turabian StyleKockelmann, Fabian, and Marc Maegele. 2023. "Acute Haemostatic Depletion and Failure in Patients with Traumatic Brain Injury (TBI): Pathophysiological and Clinical Considerations" Journal of Clinical Medicine 12, no. 8: 2809. https://doi.org/10.3390/jcm12082809