
Gut Failure: A Review of the Pathophysiology and Therapeutic Potentials in the Gut–Heart Axis
 , ,
, ,
Abstract
:1. Introduction
2. Microbiota
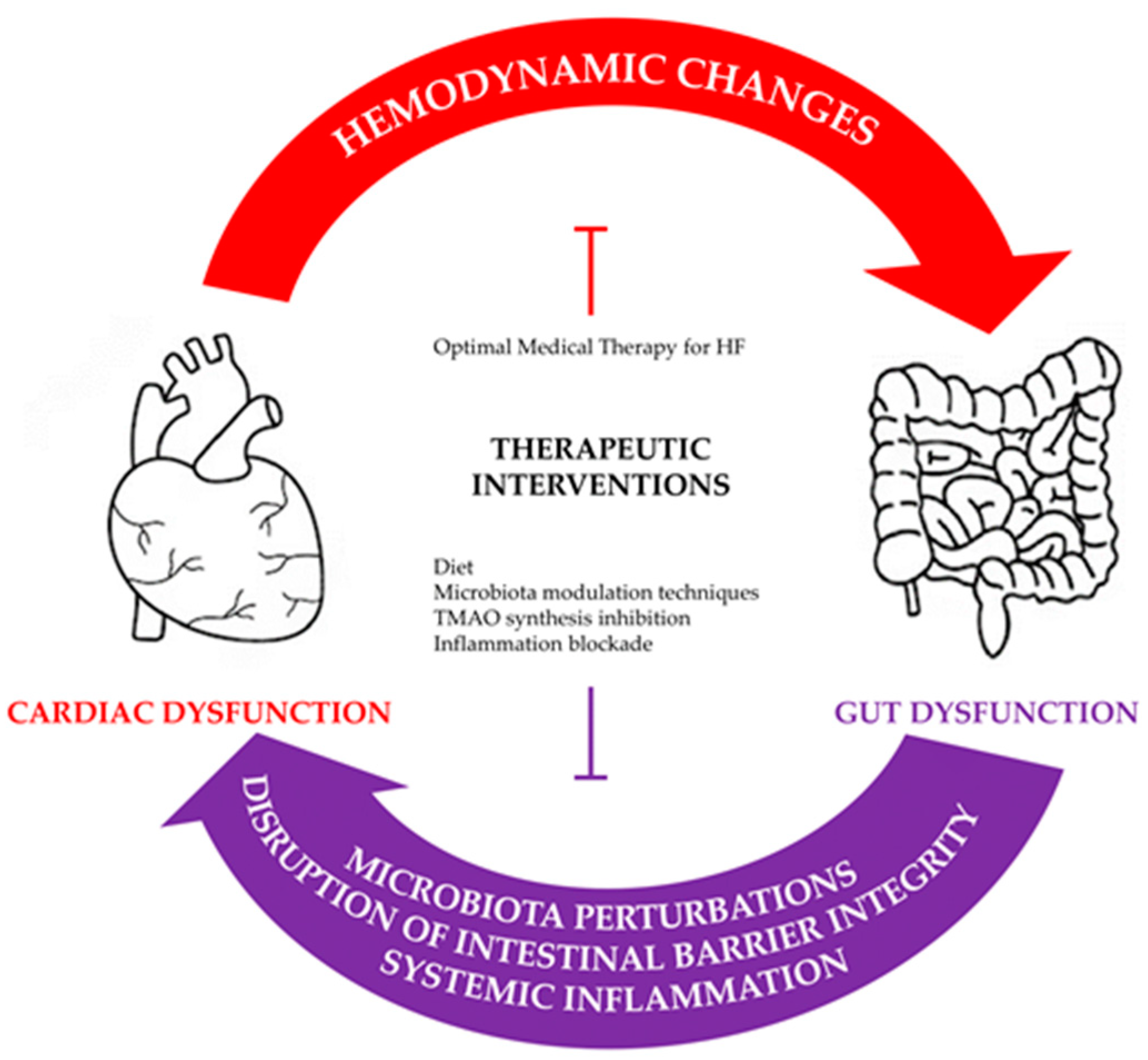
3. Pathophysiological Derangements in the Gut–Heart Axis
3.1. Intraluminal Derangements of the Microbiota Composition
3.1.1. Altered Microbiota Composition and SCFA Levels
3.1.2. TMAO
3.1.3. Amino Acids
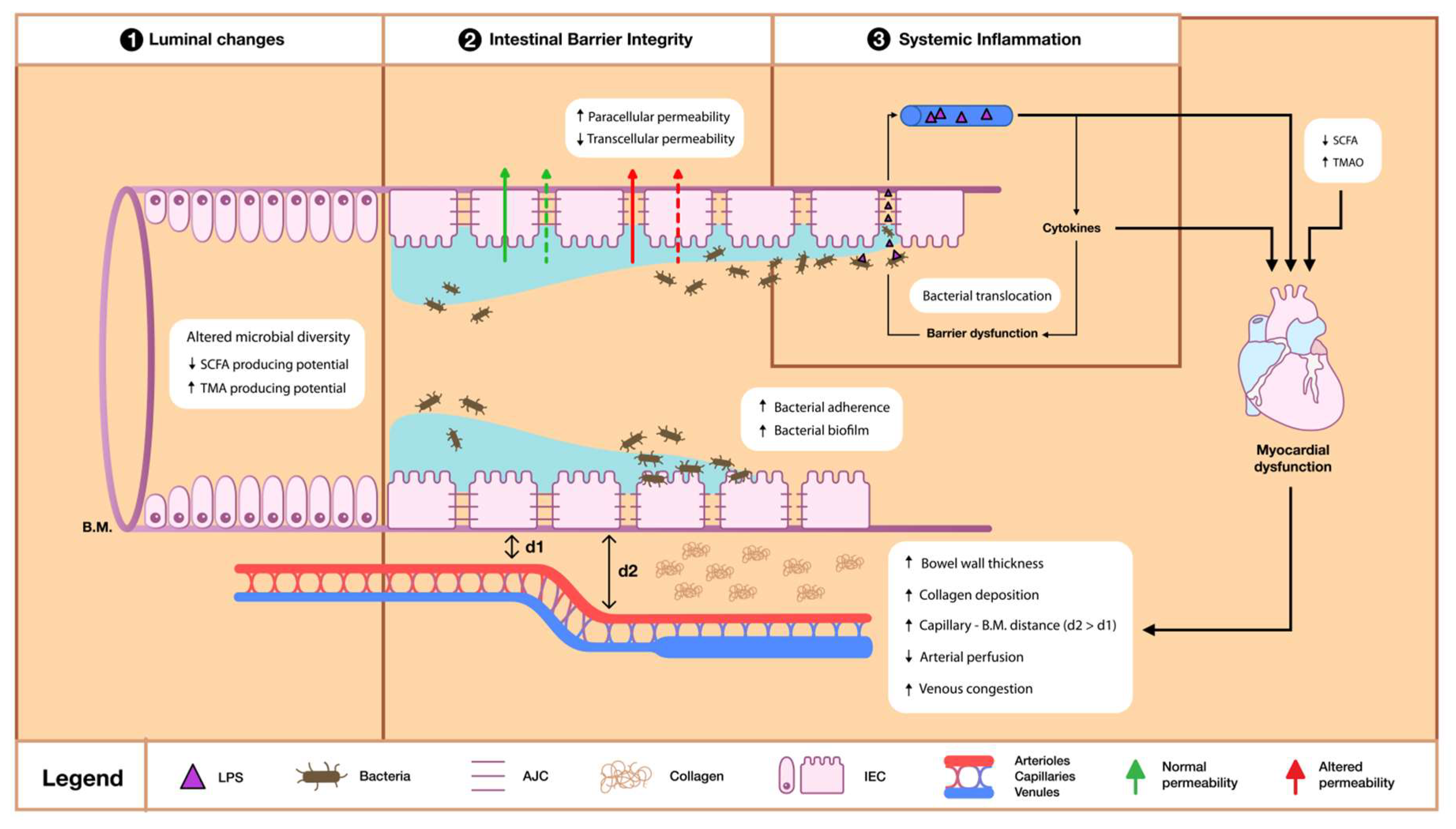
3.2. Intestinal Barrier Dysfunction
3.2.1. Bacterial Derangements
3.2.2. Permeability and Absorption Derangements
3.2.3. Trophic Changes
3.3. Systemic Inflammation
4. Therapeutic Potential
4.1. From the Standpoint of the Heart
4.2. From the Standpoint of the Gut
4.2.1. Diet
4.2.2. Lyases Inhibition
4.2.3. Microbiota Modification Techniques
4.2.4. Hepatic Flavin Inhibition
4.2.5. Inflammation Blockade
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, Regional, and National Incidence, Prevalence, and Years Lived with Disability for 354 Diseases and Injuries for 195 Countries and Territories, 1990-2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef] [Green Version]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; de Ferranti, S.; Després, J.-P.; Fullerton, H.J.; Howard, V.J.; et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics--2015 Update: A Report from the American Heart Association. Circulation 2015, 131, e29–e322. [Google Scholar] [CrossRef] [Green Version]
- Murad, K.; Goff, D.C.; Morgan, T.M.; Burke, G.L.; Bartz, T.M.; Kizer, J.R.; Chaudhry, S.I.; Gottdiener, J.S.; Kitzman, D.W. Burden of Comorbidities and Functional and Cognitive Impairments in Elderly Patients at the Initial Diagnosis of Heart Failure and Their Impact on Total Mortality. JACC Heart Fail. 2015, 3, 542–550. [Google Scholar] [CrossRef]
- Klabunde, R.E. Cardiovascular Physiology Concepts, 3rd ed.; Wolters Kluwer: Philadelphia, PA, USA, 2022. [Google Scholar]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Velavan, P.; Khan, N.K.; Goode, K.; Rigby, A.S.; Loh, P.H.; Komajda, M.; Follath, F.; Swedberg, K.; Madeira, H.; Cleland, J.G.F. Predictors of Short Term Mortality in Heart Failure-Insights from the Euro Heart Failure Survey. Int. J. Cardiol. 2010, 138, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kalman, J.; Mayer, L.; Fillit, H.M.; Packer, M. Elevated Circulating Levels of Tumor Necrosis Factor in Severe Chronic Heart Failure. N. Engl. J. Med. 1990, 323, 236–241. [Google Scholar] [CrossRef]
- Niebauer, J.; Volk, H.-D.; Kemp, M.; Dominguez, M.; Schumann, R.R.; Rauchhaus, M.; Poole-Wilson, P.A.; Coats, A.J.; Anker, S.D. Endotoxin and Immune Activation in Chronic Heart Failure: A Prospective Cohort Study. Lancet 1999, 353, 1838–1842. [Google Scholar] [CrossRef]
- Dominguez-Bello, M.G.; Costello, E.K.; Contreras, M.; Magris, M.; Hidalgo, G.; Fierer, N.; Knight, R. Delivery Mode Shapes the Acquisition and Structure of the Initial Microbiota across Multiple Body Habitats in Newborns. Proc. Natl. Acad. Sci. USA 2010, 107, 11971–11975. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.-M.; et al. Enterotypes of the Human Gut Microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The Role of the Gut Microbiota in Nutrition and Health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar] [CrossRef]
- Zhernakova, A.; Kurilshikov, A.; Bonder, M.J.; Tigchelaar, E.F.; Schirmer, M.; Vatanen, T.; Mujagic, Z.; Vila, A.V.; Falony, G.; Vieira-Silva, S.; et al. Population-Based Metagenomics Analysis Reveals Markers for Gut Microbiome Composition and Diversity. Science 2016, 352, 565–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Fan, Y.; Li, H.; Zhou, R.; Zhao, X.; Sun, Y.; Zhang, S. Impacts of Cigarette Smoking Status on Metabolomic and Gut Microbiota Profile in Male Patients With Coronary Artery Disease: A Multi-Omics Study. Front. Cardiovasc. Med. 2021, 8, 766739. [Google Scholar] [CrossRef] [PubMed]
- Maier, L.; Pruteanu, M.; Kuhn, M.; Zeller, G.; Telzerow, A.; Anderson, E.E.; Brochado, A.R.; Fernandez, K.C.; Dose, H.; Mori, H.; et al. Extensive Impact of Non-Antibiotic Drugs on Human Gut Bacteria. Nature 2018, 555, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Vital, M.; Howe, A.C.; Tiedje, J.M. Revealing the Bacterial Butyrate Synthesis Pathways by Analyzing (Meta)Genomic Data. mBio 2014, 5, e00889–e00914. [Google Scholar] [CrossRef] [Green Version]
- McNeil, N.I. The Contribution of the Large Intestine to Energy Supplies in Man. Am. J. Clin. Nutr. 1984, 39, 338–342. [Google Scholar] [CrossRef]
- Schönfeld, P.; Wojtczak, L. Short- and Medium-Chain Fatty Acids in Energy Metabolism: The Cellular Perspective. J. Lipid. Res. 2016, 57, 943–954. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, J.; Kasubuchi, M.; Nakajima, A.; Irie, J.; Itoh, H.; Kimura, I. The Role of Short-Chain Fatty Acid on Blood Pressure Regulation. Curr. Opin. Nephrol. Hypertens 2016, 25, 379–383. [Google Scholar] [CrossRef]
- Säemann, M.D.; Böhmig, G.A.; Österreicher, C.H.; Burtscher, H.; Parolini, O.; Diakos, C.; Stöckl, J.; Hörl, W.H.; Zlabinger, G.J. Anti-inflammatory Effects of Sodium Butyrate on Human Monocytes: Potent Inhibition of IL-12 and Up-regulation of IL-10 Production. FASEB J. 2000, 14, 2380–2382. [Google Scholar] [CrossRef]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly-Y, M.; Glickman, J.N.; Garrett, W.S. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites Produced by Commensal Bacteria Promote Peripheral Regulatory T-Cell Generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [Green Version]
- Duncan, S.H.; Flint, H.J.; Stewart, C.S. Inhibitory Activity of Gut Bacteria against Escherichia Coli O157 Mediated by Dietary Plant Metabolites. FEMS Microbiol. Lett. 1998, 164, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Wang, Z.; Fan, Y.; Levison, B.; Hazen, J.E.; Donahue, L.M.; Wu, Y.; Hazen, S.L. Prognostic Value of Elevated Levels of Intestinal Microbe-Generated Metabolite Trimethylamine-N-Oxide in Patients With Heart Failure: Refining the Gut Hypothesis. J. Am. Coll. Cardiol. 2014, 64, 1908–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasini, E.; Aquilani, R.; Testa, C.; Baiardi, P.; Angioletti, S.; Boschi, F.; Verri, M.; Dioguardi, F. Pathogenic Gut Flora in Patients With Chronic Heart Failure. JACC Heart Fail. 2016, 4, 220–227. [Google Scholar] [CrossRef]
- Sun, W.; Du, D.; Fu, T.; Han, Y.; Li, P.; Ju, H. Alterations of the Gut Microbiota in Patients With Severe Chronic Heart Failure. Front. Microbiol. 2022, 12, 813289. [Google Scholar] [CrossRef]
- Kummen, M.; Mayerhofer, C.C.K.; Vestad, B.; Broch, K.; Awoyemi, A.; Storm-Larsen, C.; Ueland, T.; Yndestad, A.; Hov, J.R.; Trøseid, M. Gut Microbiota Signature in Heart Failure Defined From Profiling of 2 Independent Cohorts. J. Am. Coll. Cardiol. 2018, 71, 1184–1186. [Google Scholar] [CrossRef]
- Beale, A.L.; O’Donnell, J.A.; Nakai, M.E.; Nanayakkara, S.; Vizi, D.; Carter, K.; Dean, E.; Ribeiro, R.V.; Yiallourou, S.; Carrington, M.J.; et al. The Gut Microbiome of Heart Failure With Preserved Ejection Fraction. J. Am. Heart Assoc. 2021, 10, e020654. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Ye, L.; Li, J.; Jin, L.; Wang, W.; Li, S.; Bao, M.; Wu, S.; Li, L.; Geng, B.; et al. Metagenomic and Metabolomic Analyses Unveil Dysbiosis of Gut Microbiota in Chronic Heart Failure Patients. Sci. Rep. 2018, 8, 635. [Google Scholar] [CrossRef] [Green Version]
- Kamo, T.; Akazawa, H.; Suda, W.; Saga-Kamo, A.; Shimizu, Y.; Yagi, H.; Liu, Q.; Nomura, S.; Naito, A.T.; Takeda, N.; et al. Dysbiosis and Compositional Alterations with Aging in the Gut Microbiota of Patients with Heart Failure. PLoS ONE 2017, 12, e0174099. [Google Scholar] [CrossRef] [Green Version]
- Murashige, D.; Jang, C.; Neinast, M.; Edwards, J.J.; Cowan, A.; Hyman, M.C.; Rabinowitz, J.D.; Frankel, D.S.; Arany, Z. Comprehensive Quantification of Fuel Use by the Failing and Nonfailing Human Heart. Science 2020, 370, 364–368. [Google Scholar] [CrossRef]
- Carley, A.N.; Maurya, S.K.; Fasano, M.; Wang, Y.; Selzman, C.H.; Drakos, S.G.; Lewandowski, E.D. Short-Chain, Fatty Acids Outpace Ketone Oxidation in the Failing Heart. Circulation 2021, 143, 1797–1808. [Google Scholar] [CrossRef]
- Palm, C.L.; Nijholt, K.T.; Bakker, B.M.; Westenbrink, B.D. Short-Chain Fatty Acids in the Metabolism of Heart Failure—Rethinking the Fat Stigma. Front. Cardiovasc. Med. 2022, 9, 915102. [Google Scholar] [CrossRef] [PubMed]
- Streppel, M.T. Dietary Fiber and Blood Pressure: A Meta-Analysis of Randomized Placebo-Controlled Trials. Arch. Int. Med. 2005, 165, 150–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, E.S.; Viardot, A.; Psichas, A.; Morrison, D.J.; Murphy, K.G.; Zac-Varghese, S.E.K.; MacDougall, K.; Preston, T.; Tedford, C.; Finlayson, G.S.; et al. Effects of Targeted Delivery of Propionate to the Human Colon on Appetite Regulation, Body Weight Maintenance and Adiposity in Overweight Adults. Gut 2015, 64, 1744–1754. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Zhang, F.; Ding, X.; Wu, G.; Lam, Y.Y.; Wang, X.; Fu, H.; Xue, X.; Lu, C.; Ma, J.; et al. Gut Bacteria Selectively Promoted by Dietary Fibers Alleviate Type 2 Diabetes. Science 2018, 359, 1151–1156. [Google Scholar] [CrossRef] [Green Version]
- Salzano, A.; Cassambai, S.; Yazaki, Y.; Israr, M.Z.; Bernieh, D.; Wong, M.; Suzuki, T. The Gut Axis Involvement in Heart Failure. Heart Fail. Clin. 2020, 16, 23–31. [Google Scholar] [CrossRef]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Zhu, X.; Ran, L.; Lang, H.; Yi, L.; Mi, M. Trimethylamine-N-Oxide Induces Vascular Inflammation by Activating the NLRP3 Inflammasome Through the SIRT3-SOD2-mtROS Signaling Pathway. J. Am. Heart Assoc. 2017, 6, e006347. [Google Scholar] [CrossRef] [PubMed]
- Querio, G.; Antoniotti, S.; Geddo, F.; Levi, R.; Gallo, M.P. Trimethylamine N-Oxide (TMAO) Impairs Purinergic Induced Intracellular Calcium Increase and Nitric Oxide Release in Endothelial Cells. Int. J. Mol. Sci. 2022, 23, 3982. [Google Scholar] [CrossRef]
- Zullo, A.; Guida, R.; Sciarrillo, R.; Mancini, F.P. Redox Homeostasis in Cardiovascular Disease: The Role of Mitochondrial Sirtuins. Front. Endocrinol. (Lausanne) 2022, 13, 858330. [Google Scholar] [CrossRef]
- Wang, Z.; Bergeron, N.; Levison, B.S.; Li, X.S.; Chiu, S.; Jia, X.; Koeth, R.A.; Li, L.; Wu, Y.; Tang, W.H.W.; et al. Impact of Chronic Dietary Red Meat, White Meat, or Non-Meat Protein on Trimethylamine N-Oxide Metabolism and Renal Excretion in Healthy Men and Women. Eur. Heart J. 2019, 40, 583–594. [Google Scholar] [CrossRef]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal Microbiota Metabolism of L-Carnitine, a Nutrient in Red Meat, Promotes Atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trøseid, M.; Ueland, T.; Hov, J.R.; Svardal, A.; Gregersen, I.; Dahl, C.P.; Aakhus, S.; Gude, E.; Bjørndal, B.; Halvorsen, B.; et al. Microbiota-Dependent Metabolite Trimethylamine-N-Oxide Is Associated with Disease Severity and Survival of Patients with Chronic Heart Failure. J. Intern. Med. 2015, 277, 717–726. [Google Scholar] [CrossRef]
- Schuett, K.; Kleber, M.E.; Scharnagl, H.; Lorkowski, S.; März, W.; Niessner, A.; Marx, N.; Meinitzer, A. Trimethylamine-N-Oxide and Heart Failure With Reduced Versus Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2017, 70, 3202–3204. [Google Scholar] [CrossRef]
- Suzuki, T.; Heaney, L.M.; Bhandari, S.S.; Jones, D.J.L.; Ng, L.L. Trimethylamine N -Oxide and Prognosis in Acute Heart Failure. Heart 2016, 102, 841–848. [Google Scholar] [CrossRef] [Green Version]
- Israr, M.Z.; Bernieh, D.; Salzano, A.; Cassambai, S.; Yazaki, Y.; Heaney, L.M.; Jones, D.J.L.; Ng, L.L.; Suzuki, T. Association of Gut-Related Metabolites with Outcome in Acute Heart Failure. Am. Heart J. 2021, 234, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Yazaki, Y.; Aizawa, K.; Israr, M.Z.; Negishi, K.; Salzano, A.; Saitoh, Y.; Kimura, N.; Kono, K.; Heaney, L.; Cassambai, S.; et al. Ethnic Differences in Association of Outcomes with Trimethylamine N-oxide in Acute Heart Failure Patients. ESC Heart Fail. 2020, 7, 2373–2378. [Google Scholar] [CrossRef] [PubMed]
- Melhem, N.J.; Taleb, S. Tryptophan: From Diet to Cardiovascular Diseases. Int. J. Mol. Sci. 2021, 22, 9904. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, R.; Forteza, M.J.; Ketelhuth, D.F.J. The Interplay Between Cytokines and the Kynurenine Pathway in Inflammation and Atherosclerosis. Cytokine 2019, 122, 154148. [Google Scholar] [CrossRef] [PubMed]
- Razquin, C.; Ruiz-Canela, M.; Toledo, E.; Hernández-Alonso, P.; Clish, C.B.; Guasch-Ferré, M.; Li, J.; Wittenbecher, C.; Dennis, C.; Alonso-Gómez, A.; et al. Metabolomics of the Tryptophan-Kynurenine Degradation Pathway and Risk of Atrial Fibrillation and Heart Failure: Potential Modification Effect of Mediterranean Diet. Am. J. Clin. Nutr. 2021, 114, 1646–1654. [Google Scholar] [CrossRef] [PubMed]
- Konishi, M.; Ebner, N.; Springer, J.; Schefold, J.C.; Doehner, W.; Dschietzig, T.B.; Anker, S.D.; von Haehling, S. Impact of Plasma Kynurenine Level on Functional Capacity and Outcome in Heart Failure—Results From Studies Investigating Co-morbidities Aggravating Heart Failure (SICA-HF). Circ. J. 2016, 81, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Lund, A.; Nordrehaug, J.E.; Slettom, G.; Solvang, S.H.; Pedersen, E.K.; Midttun, Ø.; Ulvik, A.; Ueland, P.M.; Nygård, O.; Giil, L.M. Plasma Kynurenines and Prognosis in Patients with Heart Failure. PLoS ONE 2020, 15, e0227365. [Google Scholar] [CrossRef]
- Imazu, M.; Takahama, H.; Shindo, K.; Hasegawa, T.; Kanzaki, H.; Anzai, T.; Asanuma, H.; Morita, T.; Asakura, M.; Kitakaze, M. A Pathophysiological Role of Plasma Indoxyl Sulfate in Patients with Heart Failure. Int. J. Gerontol. 2017, 11, 62–66. [Google Scholar] [CrossRef]
- Imazu, M.; Fukuda, H.; Kanzaki, H.; Amaki, M.; Hasegawa, T.; Takahama, H.; Hitsumoto, T.; Tsukamoto, O.; Morita, T.; Ito, S.; et al. Plasma Indoxyl Sulfate Levels Predict Cardiovascular Events in Patients with Mild Chronic Heart Failure. Sci. Rep. 2020, 10, 16528. [Google Scholar] [CrossRef]
- Shimazu, S.; Hirashiki, A.; Okumura, T.; Yamada, T.; Okamoto, R.; Shinoda, N.; Takeshita, K.; Kondo, T.; Niwa, T.; Murohara, T. Association Between Indoxyl Sulfate and Cardiac Dysfunction and Prognosis in Patients with Dilated Cardiomyopathy. Circ. J. 2013, 77, 390–396. [Google Scholar] [CrossRef] [Green Version]
- Vancamelbeke, M.; Vermeire, S. The Intestinal Barrier: A Fundamental Role in Health and Disease. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Wrzosek, L.; Miquel, S.; Noordine, M.L.; Bouet, S.; Chevalier-Curt, M.J.; Robert, V.; Philippe, C.; Bridonneau, C.; Cherbuy, C.; Robbe-Masselot, C.; et al. Bacteroides Thetaiotaomicron and Faecalibacterium Prausnitziiinfluence the Production of Mucus Glycans and the Development of Goblet Cells in the Colonic Epithelium of a Gnotobiotic Model Rodent. BMC Biol. 2013, 11, 61. [Google Scholar] [CrossRef] [Green Version]
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A.; et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 2016, 167, 1339–1353.e21. [Google Scholar] [CrossRef] [Green Version]
- Mollar, A.; Marrachelli, V.G.; Núñez, E.; Monleon, D.; Bodí, V.; Sanchis, J.; Navarro, D.; Núñez, J. Bacterial Metabolites Trimethylamine N-Oxide and Butyrate as Surrogates of Small Intestinal Bacterial Overgrowth in Patients with a Recent Decompensated Heart Failure. Sci. Rep. 2021, 11, 6110. [Google Scholar] [CrossRef]
- Song, Y.; Liu, Y.; Qi, B.; Cui, X.; Dong, X.; Wang, Y.; Han, X.; Li, F.; Shen, D.; Zhang, X.; et al. Association of Small Intestinal Bacterial Overgrowth With Heart Failure and Its Prediction for Short-Term Outcomes. J. Am. Heart Assoc. 2021, 10, e015292. [Google Scholar] [CrossRef]
- Sandek, A.; Bauditz, J.; Swidsinski, A.; Buhner, S.; Weber-Eibel, J.; von Haehling, S.; Schroedl, W.; Karhausen, T.; Doehner, W.; Rauchhaus, M.; et al. Altered Intestinal Function in Patients With Chronic Heart Failure. J. Am. Coll Cardiol. 2007, 50, 1561–1569. [Google Scholar] [CrossRef] [Green Version]
- Kaye, D.M.; Shihata, W.A.; Jama, H.A.; Tsyganov, K.; Ziemann, M.; Kiriazis, H.; Horlock, D.; Vijay, A.; Giam, B.; Vinh, A.; et al. Deficiency of Prebiotic Fiber and Insufficient Signaling Through Gut Metabolite-Sensing Receptors Leads to Cardiovascular Disease. Circulation 2020, 141, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Odenwald, M.A.; Turner, J.R. The Intestinal Epithelial Barrier: A Therapeutic Target? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.-J.; Hylemon, P.B. Bile Salt Biotransformations by Human Intestinal Bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenman, L.K. High-Fat-Induced Intestinal Permeability Dysfunction Associated with Altered Fecal Bile Acids. World J. Gastroenterol. 2012, 18, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Mayerhofer, C.C.K.; Ueland, T.; Broch, K.; Vincent, R.P.; Cross, G.F.; Dahl, C.P.; Aukrust, P.; Gullestad, L.; Hov, J.R.; Trøseid, M. Increased Secondary/Primary Bile Acid Ratio in Chronic Heart Failure. J. Card. Fail. 2017, 23, 666–671. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.C. Splanchnic and Overall Cardiovascular Hemodynamics during Eating and Digestion. Fed. Proc. 1983, 42, 1658–1661. [Google Scholar]
- Takala, J. Determinants of Splanchnic Blood Flow. Br. J. Anaesth 1996, 77, 50–58. [Google Scholar] [CrossRef]
- Krack, A.; Richartz, B.M.; Gastmann, A.; Greim, K.; Lotze, U.; Anker, S.D.; Figulla, H.R. Studies on Intragastric PCO 2 at Rest and during Exercise as a Marker of Intestinal Perfusion in Patients with Chronic Heart Failure. Eur. J. Heart Fail. 2004, 6, 403–407. [Google Scholar] [CrossRef]
- Parks, D.A.; Jacobson, E.D. Physiology of the Splanchnic Circulation. Arch Intern Med. 1985, 145, 1278–1281. [Google Scholar] [CrossRef]
- Sandek, A.; Swidsinski, A.; Schroedl, W.; Watson, A.; Valentova, M.; Herrmann, R.; Scherbakov, N.; Cramer, L.; Rauchhaus, M.; Grosse-Herrenthey, A.; et al. Intestinal Blood Flow in Patients With Chronic Heart Failure. J. Am. Coll Cardiol. 2014, 64, 1092–1102. [Google Scholar] [CrossRef] [Green Version]
- Arutyunov, G.P.; Kostyukevich, O.I.; Serov, R.A.; Rylova, N.V.; Bylova, N.A. Collagen Accumulation and Dysfunctional Mucosal Barrier of the Small Intestine in Patients with Chronic Heart Failure. Int. J. Cardiol. 2008, 125, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Yndestad, A.; Kristian Damås, J.; Øie, E.; Ueland, T.; Gullestad, L.; Aukrust, P. Systemic Inflammation in Heart Failure—The Whys and Wherefores. Heart Fail. Rev. 2006, 11, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Park, B.S.; Lee, J.O. Recognition of Lipopolysaccharide Pattern by TLR4 Complexes. Exp. Mol. Med. 2013, 45, e66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.B.; Mark, M.R.; Gray, A.; Huang, A.; Xie, M.H.; Zhang, M.; Goddard, A.; Wood, W.I.; Gurney, A.L.; Godowski, P.J. Toll-like Receptor-2 Mediates Lipopolysaccharide-Induced Cellular Signalling. Nature 1998, 395, 284–288. [Google Scholar] [CrossRef]
- Charalambous, B.M.; Stephens, R.C.M.; Feavers, I.M.; Montgomery, H.E. Role of Bacterial Endotoxin in Chronic Heart Failure: The Gut of the Matter. Shock 2007, 28, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Cicco, N.A.; Lindemann, A.; Content, J.; Vandenbussche, P.; Lübbert, M.; Gauss, J.; Mertelsmann, R.; Herrmann, F. Inducible Production of Interleukin-6 by Human Polymorphonuclear Neutrophils: Role of Granulocyte-Macrophage Colony-Stimulating Factor and Tumor Necrosis Factor-Alpha. Blood 1990, 75, 2049–2052. [Google Scholar] [CrossRef] [Green Version]
- Bailly, S.; Ferrua, B.; Fay, M.; Gougerot-Pocidalo, M.A. Differential Regulation of IL 6, IL 1 A, IL 1β and TNFα Production in LPS-Stimulated Human Monocytes: Role of Cyclic AMP. Cytokine 1990, 2, 205–210. [Google Scholar] [CrossRef]
- van Deventer, S.J.; Büller, H.R.; ten Cate, J.W.; Aarden, L.A.; Hack, C.E.; Sturk, A. Experimental Endotoxemia in Humans: Analysis of Cytokine Release and Coagulation, Fibrinolytic, and Complement Pathways. Blood 1990, 76, 2520–2526. [Google Scholar] [CrossRef] [Green Version]
- Oral, H.; Kapadia, S.; Nakano, M.; Torre-Amione, G.; Lee, J.; Lee-Jackson, D.; Young, J.B.; Mann, D.L. Tumor Necrosis Factor-Alpha and the Failing Human Heart. Clin. Cardiol. 1995, 18 (Suppl. S4), IV20–IV27. [Google Scholar] [CrossRef]
- Ferrari, R. The Role of TNF in Cardiovascular Disease. Pharmacol. Res. 1999, 40, 97–105. [Google Scholar] [CrossRef]
- Torre-Amione, G.; Kapadia, S.; Benedict, C.; Oral, H.; Young, J.B.; Mann, D.L. Proinflammatory Cytokine Levels in Patients with Depressed Left Ventricular Ejection Fraction: A Report from the Studies of Left Ventricular Dysfunction (SOLVD). J. Am. Coll Cardiol. 1996, 27, 1201–1206. [Google Scholar] [CrossRef] [Green Version]
- Anker, S.D.; Sharma, R. The Syndrome of Cardiac Cachexia. Int. J. Cardiol. 2002, 85, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Frantz, S.; Kobzik, L.; Kim, Y.-D.; Fukazawa, R.; Medzhitov, R.; Lee, R.T.; Kelly, R.A. Toll4 (TLR4) Expression in Cardiac Myocytes in Normal and Failing Myocardium. J. Clin. Investig. 1999, 104, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Kälsch, T.; Elmas, E.; Nguyen, X.D.; Suvajac, N.; Klüter, H.; Borggrefe, M.; Dempfle, C.-E. Endotoxin-Induced Effects on Platelets and Monocytes in an in Vivo Model of Inflammation. Basic Res. Cardiol. 2007, 102, 460–466. [Google Scholar] [CrossRef]
- Bierhaus, A.; Chen, J.; Liliensiek, B.; Nawroth, P.P. LPS and Cytokine-Activated Endothelium. Semin. Thromb. Hemost. 2000, 26, 571–588. [Google Scholar] [CrossRef] [PubMed]
- Suffredini, A.F.; Fromm, R.E.; Parker, M.M.; Brenner, M.; Kovacs, J.A.; Wesley, R.A.; Parrillo, J.E. The Cardiovascular Response of Normal Humans to the Administration of Endotoxin. N. Engl. J. Med. 1989, 321, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Yücel, G.; Zhao, Z.; El-Battrawy, I.; Lan, H.; Lang, S.; Li, X.; Buljubasic, F.; Zimmermann, W.-H.; Cyganek, L.; Utikal, J.; et al. Lipopolysaccharides Induced Inflammatory Responses and Electrophysiological Dysfunctions in Human-Induced Pluripotent Stem Cell Derived Cardiomyocytes. Sci. Rep. 2017, 7, 2935. [Google Scholar] [CrossRef] [Green Version]
- Sattler, K.; El-Battrawy, I.; Cyganek, L.; Lang, S.; Lan, H.; Li, X.; Zhao, Z.; Utikal, J.; Wieland, T.; Borggrefe, M.; et al. TRPV1 Activation and Internalization Is Part of the LPS-Induced Inflammation in Human IPSC-Derived Cardiomyocytes. Sci. Rep. 2021, 11, 14689. [Google Scholar] [CrossRef]
- Hietbrink, F.; Besselink, M.G.H.; Renooij, W.; de Smet, M.B.M.; Draisma, A.; van der Hoeven, H.; Pickkers, P. Systemic Inflammation Increases Intestinal Permeability During Experimental Human Endotoxemia. Shock 2009, 32, 374–378. [Google Scholar] [CrossRef]
- O’Dwyer, S.T. A Single Dose of Endotoxin Increases Intestinal Permeability in Healthy Humans. Arch. Surg. 1988, 123, 1459. [Google Scholar] [CrossRef]
- Anker, S.D.; Egerer, K.R.; Volk, H.D.; Kox, W.J.; Poole-Wilson, P.A.; Coats, A.J.S. Elevated Soluble CD14 Receptors and Altered Cytokines in Chronic Heart Failure. Am. J. Cardiol. 1997, 79, 1426–1430. [Google Scholar] [CrossRef]
- Genth-Zotz, S.; von Haehling, S.; Bolger, A.P.; Kalra, P.R.; Wensel, R.; Coats, A.J.S.; Anker, S.D. Pathophysiologic Quantities of Endotoxin-Induced Tumor Necrosis Factor-Alpha Release in Whole Blood from Patients with Chronic Heart Failure. Am. J. Cardiol. 2002, 90, 1226–1230. [Google Scholar] [CrossRef]
- Peschel, T.; Schönauer, M.; Thiele, H.; Anker, S.; Schuler, G.; Niebauer, J. Invasive Assessment of Bacterial Endotoxin and Inflammatory Cytokines in Patients with Acute Heart Failure. Eur. J. Heart Fail. 2003, 5, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Vonhof, S.; Brost, B.; Stille-Siegener, M.; Grumbach, I.M.; Kreuzer, H.; Figulla, H.R. Monocyte Activation in Congestive Heart Failure Due to Coronary Artery Disease and Idiopathic Dilated Cardiomyopathy. Int. J. Cardiol. 1998, 63, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Krüger, S.; Kunz, D.; Graf, J.; Stickel, T.; Merx, M.W.; Hanrath, P.; Janssens, U. Endotoxin Sensitivity and Immune Competence in Chronic Heart Failure. Clin. Chim. Acta 2004, 343, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, H.; Kuroiwa-Matsumoto, M.; Takeshita, A. Cytokine Generation Capacities of Monocytes Are Reduced in Patients with Severe Heart Failure. Am. Heart J. 1998, 136, 991–1002. [Google Scholar] [CrossRef]
- Sharma, R.; Bolger, A.P.; Rauchhaus, M.; von Haehling, S.; Doehner, W.; Adcock, I.M.; Barnes, P.J.; Poole-Wilson, P.A.; Volk, H.-D.; Coats, A.J.S.; et al. Cellular Endotoxin Desensitization in Patients with Severe Chronic Heart Failure. Eur. J. Heart Fail. 2005, 7, 865–868. [Google Scholar] [CrossRef] [Green Version]
- Velavan, P.; Huan Loh, P.; Clark, A.; Cleland, J.G.F. The Cholesterol. Paradox in Heart Failure. Congest. Heart Fail. 2007, 13, 336–341. [Google Scholar] [CrossRef]
- Rauchhaus, M.; Coats, A.J.; Anker, S.D. The Endotoxin-Lipoprotein Hypothesis. Lancet 2000, 356, 930–933. [Google Scholar] [CrossRef]
- Sandek, A.; Utchill, S.; Rauchhaus, M. The Endotoxin-Lipoprotein Hypothesis—An Update. Arch. Med. Sci. 2007, 3, S81–S90. [Google Scholar]
- Yin, L.; Laevsky, G.; Giardina, C. Butyrate Suppression of Colonocyte NF-ΚB Activation and Cellular Proteasome Activity. J. Biol. Chem. 2001, 276, 44641–44646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Yu, T.; Huang, X.; Bilotta, A.J.; Xu, L.; Lu, Y.; Sun, J.; Pan, F.; Zhou, J.; Zhang, W.; et al. Intestinal Microbiota-Derived Short-Chain Fatty Acids Regulation of Immune Cell IL-22 Production and Gut Immunity. Nat. Commun. 2020, 11, 4457. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Elson, C.O. Adaptive Immune Education by Gut Microbiota Antigens. Immunology 2018, 154, 28–37. [Google Scholar] [CrossRef] [Green Version]
- Carrillo-Salinas, F.J.; Ngwenyama, N.; Anastasiou, M.; Kaur, K.; Alcaide, P. Heart Inflammation: Immune Cell Roles and Roads to the Heart. Am. J. Pathol. 2019, 189, 1482–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Prabhu, S.D.; Bansal, S.S. CD4+ T-Lymphocytes Exhibit Biphasic Kinetics Post-Myocardial Infarction. Front. Cardiovasc. Med. 2022, 9, 992653. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, T.; Soejima, H.; Irie, A.; Sugamura, K.; Oe, Y.; Tanaka, T.; Nagayoshi, Y.; Kaikita, K.; Sugiyama, S.; Yoshimura, M.; et al. Relation Between CD4+ T-Cell Activation and Severity of Chronic Heart Failure Secondary to Ischemic or Idiopathic Dilated Cardiomyopathy. Am. J. Cardiol. 2007, 100, 483–488. [Google Scholar] [CrossRef]
- Carrillo-Salinas, F.J.; Anastasiou, M.; Ngwenyama, N.; Kaur, K.; Tai, A.; Smolgovsky, S.A.; Jetton, D.; Aronovitz, M.; Alcaide, P. Gut Dysbiosis Induced by Cardiac Pressure Overload Enhances Adverse Cardiac Remodeling in a T Cell-Dependent Manner. Gut Microbes 2020, 12, 1–20. [Google Scholar] [CrossRef]
- Rungoe, C.; Basit, S.; Ranthe, M.F.; Wohlfahrt, J.; Langholz, E.; Jess, T. Risk of Ischaemic Heart Disease in Patients with Inflammatory Bowel Disease: A Nationwide Danish Cohort Study. Gut 2013, 62, 689–694. [Google Scholar] [CrossRef]
- Kristensen, S.L.; Ahlehoff, O.; Lindhardsen, J.; Erichsen, R.; Lamberts, M.; Khalid, U.; Nielsen, O.H.; Torp-Pedersen, C.; Gislason, G.H.; Hansen, P.R. Inflammatory Bowel Disease Is Associated With an Increased Risk of Hospitalization for Heart Failure: A Danish Nationwide Cohort Study. Circ. Heart Fail. 2014, 7, 717–722. [Google Scholar] [CrossRef] [Green Version]
- Prasada, S.; Rivera, A.; Nishtala, A.; Pawlowski, A.E.; Sinha, A.; Bundy, J.D.; Chadha, S.A.; Ahmad, F.S.; Khan, S.S.; Achenbach, C.; et al. Differential Associations of Chronic Inflammatory Diseases With Incident Heart Failure. JACC Heart Fail. 2020, 8, 489–498. [Google Scholar] [CrossRef]
- Polyzogopoulou, E.; Boultadakis, A.; Ikonomidis, I.; Parissis, J. It’s Not All about Echocardiography. Open the Lung Window for the Cardiac Emergencies. Medicina (Kaunas) 2021, 57, 69. [Google Scholar] [CrossRef] [PubMed]
- Volpe, M.; Carnovali, M.; Mastromarino, V. The Natriuretic Peptides System in the Pathophysiology of Heart Failure: From Molecular Basis to Treatment. Clin. Sci. (Lond) 2015, 130, 57–77. [Google Scholar] [CrossRef] [Green Version]
- Boorsma, E.M.; ter Maaten, J.M.; Damman, K.; Dinh, W.; Gustafsson, F.; Goldsmith, S.; Burkhoff, D.; Zannad, F.; Udelson, J.E.; Voors, A.A. Congestion in Heart Failure: A Contemporary Look at Physiology, Diagnosis and Treatment. Nat. Rev. Cardiol. 2020, 17, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yazaki, Y.; Voors, A.A.; Jones, D.J.L.; Chan, D.C.S.; Anker, S.D.; Cleland, J.G.; Dickstein, K.; Filippatos, G.; Hillege, H.L.; et al. Association with Outcomes and Response to Treatment of Trimethylamine N-oxide in Heart Failure: Results from BIOSTAT-CHF. Eur. J. Heart Fail. 2019, 21, 877–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerley, C.P. Dietary Patterns and Components to Prevent and Treat Heart Failure: A Comprehensive Review of Human Studies. Nutr. Res. Rev. 2019, 32, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Marques, F.Z.; Nelson, E.; Chu, P.-Y.; Horlock, D.; Fiedler, A.; Ziemann, M.; Tan, J.K.; Kuruppu, S.; Rajapakse, N.W.; El-Osta, A.; et al. High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation 2017, 135, 964–977. [Google Scholar] [CrossRef]
- Mayerhofer, C.C.K.; Kummen, M.; Holm, K.; Broch, K.; Awoyemi, A.; Vestad, B.; Storm-Larsen, C.; Seljeflot, I.; Ueland, T.; Bohov, P.; et al. Low Fibre Intake Is Associated with Gut Microbiota Alterations in Chronic Heart Failure. ESC Heart Fail. 2020, 7, 456–466. [Google Scholar] [CrossRef] [Green Version]
- Davis, C.; Bryan, J.; Hodgson, J.; Murphy, K. Definition of the Mediterranean Diet; A Literature Review. Nutrients 2015, 7, 9139–9153. [Google Scholar] [CrossRef] [Green Version]
- De Filippis, F.; Pellegrini, N.; Vannini, L.; Jeffery, I.B.; La Storia, A.; Laghi, L.; Serrazanetti, D.I.; Di Cagno, R.; Ferrocino, I.; Lazzi, C.; et al. High-Level Adherence to a Mediterranean Diet Beneficially Impacts the Gut Microbiota and Associated Metabolome. Gut 2016, 65, 1812–1821. [Google Scholar] [CrossRef]
- Ghosh, T.S.; Rampelli, S.; Jeffery, I.B.; Santoro, A.; Neto, M.; Capri, M.; Giampieri, E.; Jennings, A.; Candela, M.; Turroni, S.; et al. Mediterranean Diet Intervention Alters the Gut Microbiome in Older People Reducing Frailty and Improving Health Status: The NU-AGE 1-Year Dietary Intervention across Five European Countries. Gut 2020, 69, 1218–1228. [Google Scholar] [CrossRef] [Green Version]
- Estruch, R.; Ros, E.; Salas-Salvadó, J.; Covas, M.-I.; Corella, D.; Arós, F.; Gómez-Gracia, E.; Ruiz-Gutiérrez, V.; Fiol, M.; Lapetra, J.; et al. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet Supplemented with Extra-Virgin Olive Oil or Nuts. N. Engl. J. Med. 2018, 378, e34. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Tektonidis, T.G.; Gigante, B.; Åkesson, A.; Wolk, A. Healthy Lifestyle and Risk of Heart Failure: Results From 2 Prospective Cohort Studies. Circ. Heart Fail. 2016, 9, e002855. [Google Scholar] [CrossRef] [Green Version]
- Tektonidis, T.G.; Åkesson, A.; Gigante, B.; Wolk, A.; Larsson, S.C. Adherence to a Mediterranean Diet Is Associated with Reduced Risk of Heart Failure in Men: Mediterranean Diet and HF Risk. Eur. J. Heart Fail. 2016, 18, 253–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrysohoou, C.; Metallinos, G.; Aggelopoulos, P.; Kastorini, C.; Athanasopoulou, S.; Pitsavos, C.; Panagiotakos, D.B.; Stefanadis, C. Abstract 810: Long-Term Adherence to the Traditional Mediterranean Diet Is Associated With Improved Biventricular Systolic Function, in Chronic Heart Failure Patients. Circulation 2009, 120 (Suppl. S18), S395. [Google Scholar] [CrossRef]
- Levitan, E.B.; Wolk, A.; Mittleman, M.A. Consistency With the DASH Diet and Incidence of Heart Failure. Arch. Intern. Med. 2009, 169, 851. [Google Scholar] [CrossRef]
- Rifai, L.; Pisano, C.; Hayden, J.; Sulo, S.; Silver, M.A. Impact of the Dash Diet on Endothelial Function, Exercise Capacity, and Quality of Life in Patients with Heart Failure. Proc. (Bayl. Univ. Med. Cent.) 2015, 28, 151–156. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, J.; Skye, S.M.; Graham, B.; Zabell, A.; Li, X.S.; Li, L.; Shelkay, S.; Fu, X.; Neale, S.; O’Laughlin, C.; et al. Dietary Choline Supplements, but Not Eggs, Raise Fasting TMAO Levels in Participants with Normal Renal Function: A Randomized Clinical Trial. Am. J. Med. 2021, 134, 1160–1169.e3. [Google Scholar] [CrossRef]
- Rath, S.; Heidrich, B.; Pieper, D.H.; Vital, M. Uncovering the Trimethylamine-Producing Bacteria of the Human Gut Microbiota. Microbiome 2017, 5, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabr, M.; Świderek, K. Discovery of a Histidine-Based Scaffold as an Inhibitor of Gut Microbial Choline Trimethylamine-Lyase. ChemMedChem 2020, 15, 2273–2279. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; Gu, X.; Buffa, J.A.; Hurd, A.G.; Wang, Z.; Zhu, W.; Gupta, N.; Skye, S.M.; Cody, D.B.; Levison, B.S.; et al. Development of a Gut Microbe–Targeted Nonlethal Therapeutic to Inhibit Thrombosis Potential. Nat. Med. 2018, 24, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- Organ, C.L.; Li, Z.; Sharp, T.E.; Polhemus, D.J.; Gupta, N.; Goodchild, T.T.; Tang, W.H.W.; Hazen, S.L.; Lefer, D.J. Nonlethal Inhibition of Gut Microbial Trimethylamine N-oxide Production Improves Cardiac Function and Remodeling in a Murine Model of Heart Failure. J. Am. Heart Assoc. 2020, 9, e016223. [Google Scholar] [CrossRef]
- Probiotics in Food: Health and Nutritional Properties and Guidelines for Evaluation; Food and Agriculture Organization of the United Nations, World Health Organization (Ed.) Food and Agriculture Organization of the United Nations: Rome, Italy; World Health Organization: Rome, Italy, 2006. [Google Scholar]
- Bindels, L.B.; Delzenne, N.M.; Cani, P.D.; Walter, J. Towards a More Comprehensive Concept for Prebiotics. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 303–310. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, M.; Liu, Y.; Xu, M.; Shi, L.; Li, X.; Zhao, J.; Zhang, H.; Wang, G.; Chen, W. Bifidobacterium Breve and Bifidobacterium Longum Attenuate Choline-Induced Plasma Trimethylamine N-Oxide Production by Modulating Gut Microbiota in Mice. Nutrients 2022, 14, 1222. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Tao, X.; Xiong, H.; Yu, J.; Wei, H. Lactobacillus plantarum ZDY04 Exhibits a Strain-Specific Property of Lowering TMAO via the Modulation of Gut Microbiota in Mice. Food Funct. 2018, 9, 4299–4309. [Google Scholar] [CrossRef]
- Gan, X.T.; Ettinger, G.; Huang, C.X.; Burton, J.P.; Haist, J.V.; Rajapurohitam, V.; Sidaway, J.E.; Martin, G.; Gloor, G.B.; Swann, J.R.; et al. Probiotic Administration Attenuates Myocardial Hypertrophy and Heart Failure After Myocardial Infarction in the Rat. Circ. Heart Fail. 2014, 7, 491–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costanza, A.C.; Moscavitch, S.D.; Faria Neto, H.C.C.; Mesquita, E.T. Probiotic Therapy with Saccharomyces Boulardii for Heart Failure Patients: A Randomized, Double-Blind, Placebo-Controlled Pilot Trial. Int. J. Cardiol. 2015, 179, 348–350. [Google Scholar] [CrossRef] [Green Version]
- Awoyemi, A.; Mayerhofer, C.; Felix, A.S.; Hov, J.R.; Moscavitch, S.D.; Lappegård, K.T.; Hovland, A.; Halvorsen, S.; Halvorsen, B.; Gregersen, I.; et al. Rifaximin or Saccharomyces Boulardii in Heart Failure with Reduced Ejection Fraction: Results from the Randomized GutHeart Trial. EBioMedicine 2021, 70, 103511. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.P.; Martin, J.C.; Duncan, S.H.; Flint, H.J. Prebiotic Stimulation of Human Colonic Butyrate-Producing Bacteria and Bifidobacteria, in Vitro. FEMS Microbiol. Ecol. 2014, 87, 30–40. [Google Scholar] [CrossRef] [Green Version]
- Neyrinck, A.M.; Possemiers, S.; Druart, C.; Van de Wiele, T.; De Backer, F.; Cani, P.D.; Larondelle, Y.; Delzenne, N.M. Prebiotic Effects of Wheat Arabinoxylan Related to the Increase in Bifidobacteria, Roseburia and Bacteroides/Prevotella in Diet-Induced Obese Mice. PLoS ONE 2011, 6, e20944. [Google Scholar] [CrossRef] [Green Version]
- De Vuyst, L.; Leroy, F. Cross-Feeding between Bifidobacteria and Butyrate-Producing Colon Bacteria Explains Bifdobacterial Competitiveness, Butyrate Production, and Gas Production. Int. J. Food Microbiol. 2011, 149, 73–80. [Google Scholar] [CrossRef]
- Chen, M.; Yi, L.; Zhang, Y.; Zhou, X.; Ran, L.; Yang, J.; Zhu, J.; Zhang, Q.; Mi, M. Resveratrol. Attenuates Trimethylamine- N -Oxide (TMAO)-Induced Atherosclerosis by Regulating TMAO Synthesis and Bile Acid Metabolism via Remodeling of the Gut Microbiota. mBio 2016, 7, e02210–e02215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; DuGar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.-M.; et al. Gut Flora Metabolism of Phosphatidylcholine Promotes Cardiovascular Disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conraads, V.M.; Jorens, P.G.; De Clerck, L.S.; Van Saene, H.K.; Ieven, M.M.; Bosmans, J.M.; Schuerwegh, A.; Bridts, C.H.; Wuyts, F.; Stevens, W.J.; et al. Selective Intestinal Decontamination in Advanced Chronic Heart Failure: A Pilot Trial. Eur. J. Heart Fail. 2004, 6, 483–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, V.; Su, J.; Koprowski, S.; Hsu, A.; Tweddell, J.S.; Rafiee, P.; Gross, G.J.; Salzman, N.H.; Baker, J.E. Intestinal Microbiota Determine Severity of Myocardial Infarction in Rats. FASEB J. 2012, 26, 1727–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nace, C. Effect of Methimazole, an FMO Substrate and Competitive Inhibitor, on the Neurotoxicity of 3,3′-Iminodipropionitrile in Male Rats. Fundam. Appl. Toxicol. 1997, 37, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Cashman, J.R.; Xiong, Y.; Lin, J.; Verhagen, H.; van Poppel, G.; van Bladeren, P.J.; Larsen-Su, S.; Williams, D.E. In Vitro and in Vivo Inhibition of Human Flavin-Containing Monooxygenase Form 3 (FMO3) in the Presence of Dietary Indoles. Biochem. Pharmacol. 1999, 58, 1047–1055. [Google Scholar] [CrossRef]
- Zhu, W.; Buffa, J.A.; Wang, Z.; Warrier, M.; Schugar, R.; Shih, D.M.; Gupta, N.; Gregory, J.C.; Org, E.; Fu, X.; et al. Flavin Monooxygenase 3, the Host Hepatic Enzyme in the Metaorganismal Trimethylamine N-Oxide-Generating Pathway, Modulates Platelet Responsiveness and Thrombosis Risk. J. Thromb. Haemost. 2018, 16, 1857–1872. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, Y.; Shimizu, I.; Shimada, A.; Nakahara, K.; Yanagisawa, S.; Kubo, M.; Fukuda, S.; Ishii, C.; Yamamoto, H.; Ishikawa, T.; et al. Brown Adipose Tissue Dysfunction Promotes Heart Failure via a Trimethylamine N-Oxide-Dependent Mechanism. Sci. Rep. 2022, 12, 14883. [Google Scholar] [CrossRef]
- Shi, C.; Pei, M.; Wang, Y.; Chen, Q.; Cao, P.; Zhang, L.; Guo, J.; Deng, W.; Wang, L.; Li, X.; et al. Changes of Flavin-Containing Monooxygenases and Trimethylamine-N-Oxide May Be Involved in the Promotion of Non-Alcoholic Fatty Liver Disease by Intestinal Microbiota Metabolite Trimethylamine. Biochem. Biophy. Res. Commun. 2022, 594, 1–7. [Google Scholar] [CrossRef]
- Cashman, J.; Camp, K.; Fakharzadeh, S.; Fennessey, P.; Hines, R.; Mamer, O.; Mitchell, S.; Preti, G.; Schlenk, D.; Smith, R. Biochemical and Clinical Aspects of the Human Flavin-Containing Monooxygenase Form 3 (FMO3) Related to Trimethylaminuria. Curr. Drug Metabol. 2003, 4, 151–170. [Google Scholar] [CrossRef]
- Deswal, A.; Bozkurt, B.; Seta, Y.; Parilti-Eiswirth, S.; Hayes, F.A.; Blosch, C.; Mann, D.L. Safety and Efficacy of a Soluble P75 Tumor Necrosis Factor Receptor (Enbrel, Etanercept) in Patients With Advanced Heart Failure. Circulation 1999, 99, 3224–3226. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Torre-Amione, G.; Warren, M.S.; Whitmore, J.; Soran, O.Z.; Feldman, A.M.; Mann, D.L. Results of Targeted Anti–Tumor Necrosis Factor Therapy With Etanercept (ENBREL) in Patients With Advanced Heart Failure. Circulation 2001, 103, 1044–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, D.L.; McMurray, J.J.V.; Packer, M.; Swedberg, K.; Borer, J.S.; Colucci, W.S.; Djian, J.; Drexler, H.; Feldman, A.; Kober, L.; et al. Targeted Anticytokine Therapy in Patients With Chronic Heart Failure: Results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation 2004, 109, 1594–1602. [Google Scholar] [CrossRef] [Green Version]
- Chung, E.S.; Packer, M.; Lo, K.H.; Fasanmade, A.A.; Willerson, J.T. Randomized, Double-Blind, Placebo-Controlled, Pilot Trial of Infliximab, a Chimeric Monoclonal Antibody to Tumor Necrosis Factor-α, in Patients With Moderate-to-Severe Heart Failure: Results of the Anti-TNF Therapy Against Congestive Heart Failure (ATTACH) Trial. Circulation 2003, 107, 3133–3140. [Google Scholar] [CrossRef] [Green Version]
- Van Tassell, B.W.; Trankle, C.R.; Canada, J.M.; Carbone, S.; Buckley, L.; Kadariya, D.; Del Buono, M.G.; Billingsley, H.; Wohlford, G.; Viscusi, M.; et al. IL-1 Blockade in Patients With Heart Failure With Preserved Ejection Fraction: Results From DHART2. Circ. Heart Fail. 2018, 11, e005036. [Google Scholar] [CrossRef]
- Van Tassell, B.W.; Canada, J.; Carbone, S.; Trankle, C.; Buckley, L.; Oddi Erdle, C.; Abouzaki, N.A.; Dixon, D.; Kadariya, D.; Christopher, S.; et al. Interleukin-1 Blockade in Recently Decompensated Systolic Heart Failure: Results From REDHART (Recently Decompensated Heart Failure Anakinra Response Trial). Circ. Heart Fail. 2017, 10, e004373. [Google Scholar] [CrossRef] [PubMed]
- Van Tassell, B.; Mihalick, V.; Thomas, G.; Marawan, A.; Talasaz, A.H.; Lu, J.; Kang, L.; Ladd, A.; Damonte, J.I.; Dixon, D.L.; et al. Rationale and Design of Interleukin-1 Blockade in Recently Decompensated Heart Failure (REDHART2): A Randomized, Double Blind, Placebo Controlled, Single Center, Phase 2 Study. J. Transl. Med. 2022, 20, 270. [Google Scholar] [CrossRef]
- von Haehling, S.; Schefold, J.C.; Jankowska, E.A.; Springer, J.; Vazir, A.; Kalra, P.R.; Sandek, A.; Fauler, G.; Stojakovic, T.; Trauner, M.; et al. Ursodeoxycholic Acid in Patients With Chronic Heart Failure: A Double-Blind, Randomized, Placebo-Controlled, Crossover Trial. J. Am. Coll Cardiol. 2012, 59, 585–592. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Kumar, V.; Gupta, S.; Bermeo-Blanco, O.; Stratton, M.S.; Gumina, R.J.; Bansal, S.S. Estrogen Receptor-β Agonists Modulate T-Lymphocyte Activation and Ameliorate Left Ventricular Remodeling During Chronic Heart Failure. Circ. Heart Fail. 2022, 15, e008997. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Target | Intervention | Effect | Study Type |
|---|---|---|---|
| Diet | MD | Reduced the incidence of HF | Human interventional [120,121] |
| Improved LV diastolic filling in CHF | Human interventional [122] | ||
| DASH | Reduced the incidence of HF Reduced mortality in female patients with HF Better quality of life | Human interventional [123] | |
| Microbiota modulation | Probiotics | Attenuated HF development | Animal interventional [133] |
| Conflicting results reported regarding LVEF in patients with HF (either improved LVEF or had no effect on LVEF) | Human interventional [134,135] | ||
| Prebiotics | Stimulated SCFA-producing bacteria | Animal interventional [137] | |
| Decreased TMAO levels | Animal interventional [139] | ||
| Antibiotics | Decreased intestinal concentrations of LPS and cytokines | Human interventional [141] | |
| No effect on LVEF | Human interventional [135] | ||
| TMAO synthesis inhibition | Lyases inhibition | Decreased TMAO levels | Animal interventional [127] |
| Improved cardiac function in murine HF models | Animal interventional [128] | ||
| FMO-3 inhibition | Decreased TMAO levels | Animal interventional [145] | |
| Ameliorated cardiac fibrosis in induced HF models | Animal interventional [146] | ||
| Inflammation blockade | Anti-IL-1 | Improved exercise capacity in AHF and CHF | Human interventional [153,154] |
| ER-β agonists | Ameliorate LV remodelling in HF-induced mouse model | Animal interventional [161] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsiras, D.; Bezati, S.; Ventoulis, I.; Verras, C.; Parissis, J.; Polyzogopoulou, E. Gut Failure: A Review of the Pathophysiology and Therapeutic Potentials in the Gut–Heart Axis. J. Clin. Med. 2023, 12, 2567. https://doi.org/10.3390/jcm12072567
Matsiras D, Bezati S, Ventoulis I, Verras C, Parissis J, Polyzogopoulou E. Gut Failure: A Review of the Pathophysiology and Therapeutic Potentials in the Gut–Heart Axis. Journal of Clinical Medicine. 2023; 12(7):2567. https://doi.org/10.3390/jcm12072567
Chicago/Turabian StyleMatsiras, Dionysis, Sofia Bezati, Ioannis Ventoulis, Christos Verras, John Parissis, and Effie Polyzogopoulou. 2023. "Gut Failure: A Review of the Pathophysiology and Therapeutic Potentials in the Gut–Heart Axis" Journal of Clinical Medicine 12, no. 7: 2567. https://doi.org/10.3390/jcm12072567