
Vinorelbine and Intermittent Cyclophosphamide Sensitize an Aggressive Myc-Driven B-Cell Lymphoma to Anti-PD-1 by an Immunological Memory Effective against Tumor Re-Challenge
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Cell Line Infection
2.3. In Vivo Experiments
2.4. In Vivo Therapy
2.5. Flow Cytometry
2.6. T Cell Receptor Clonality
2.7. Statistical Analysis
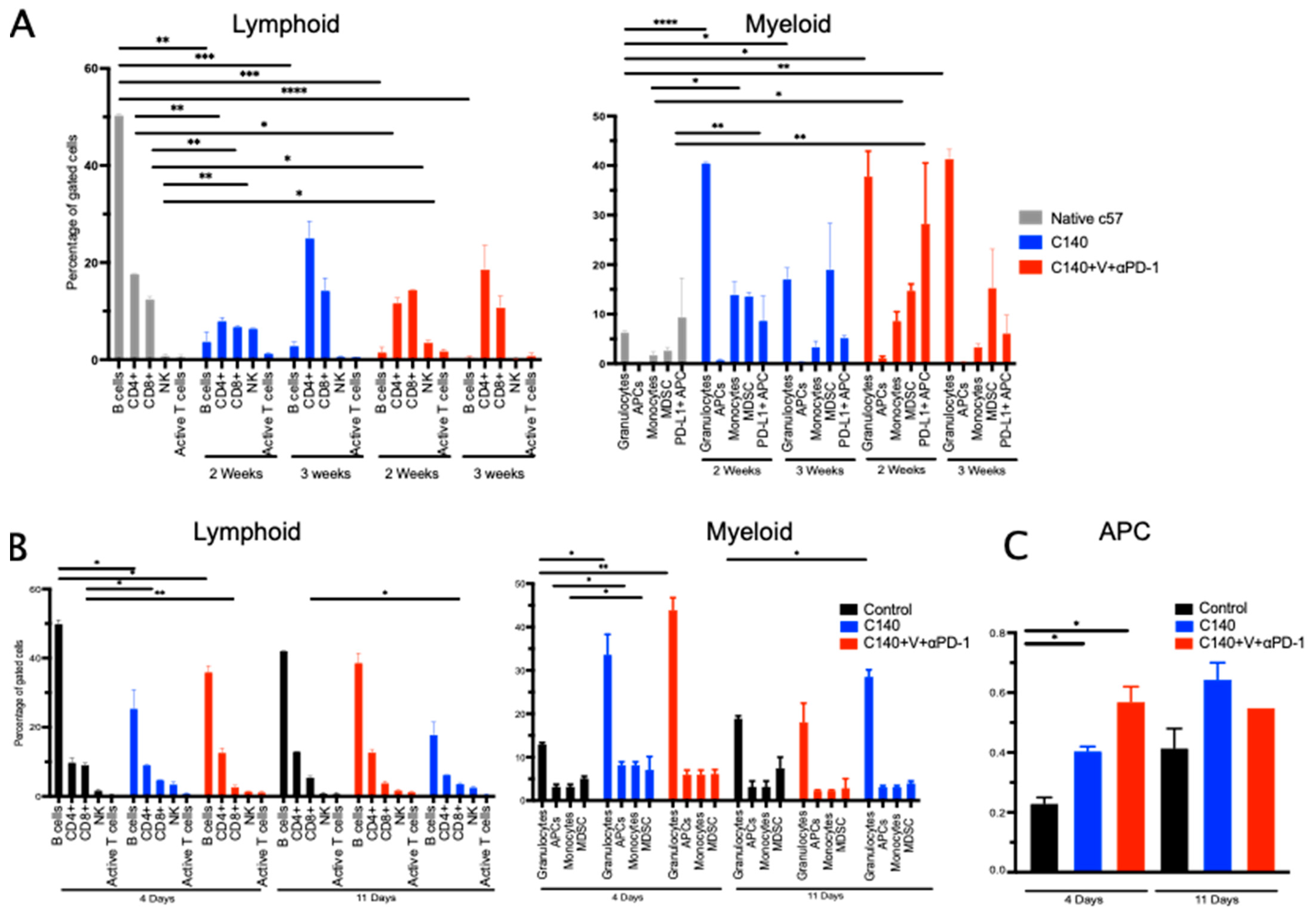
3. Results
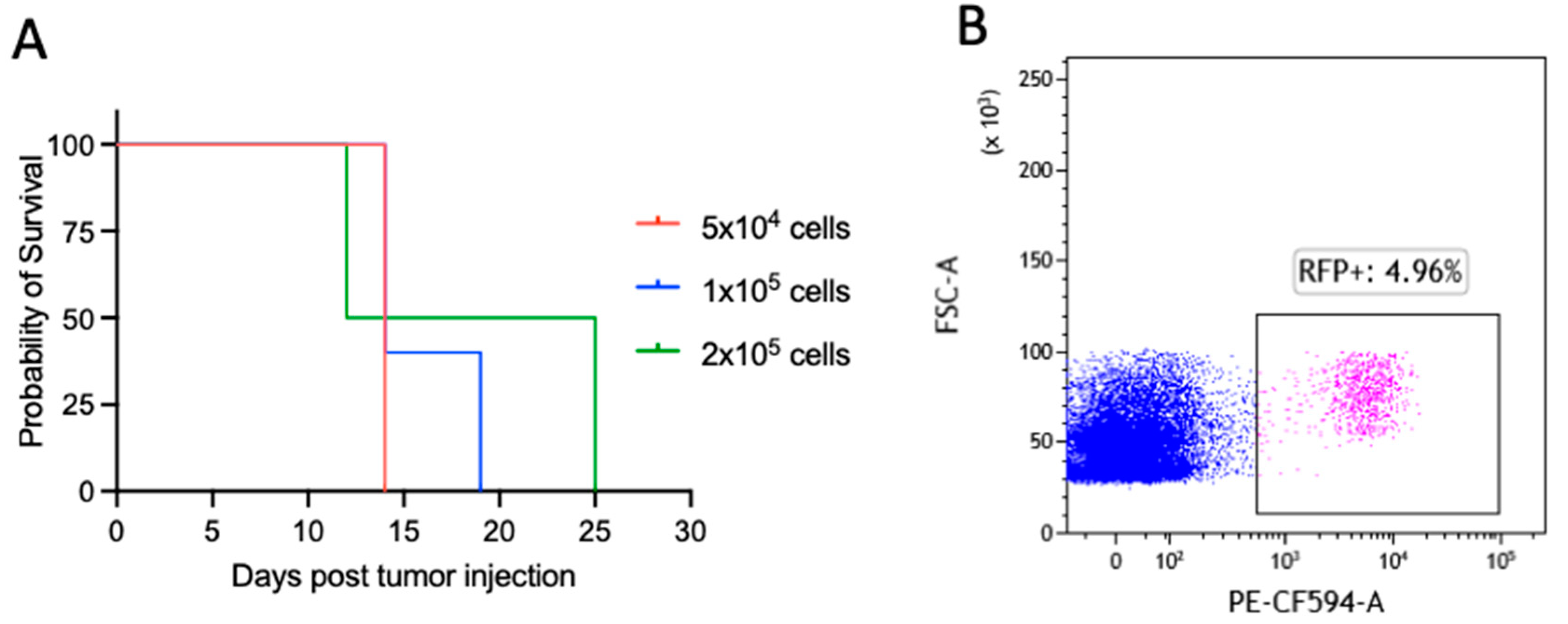
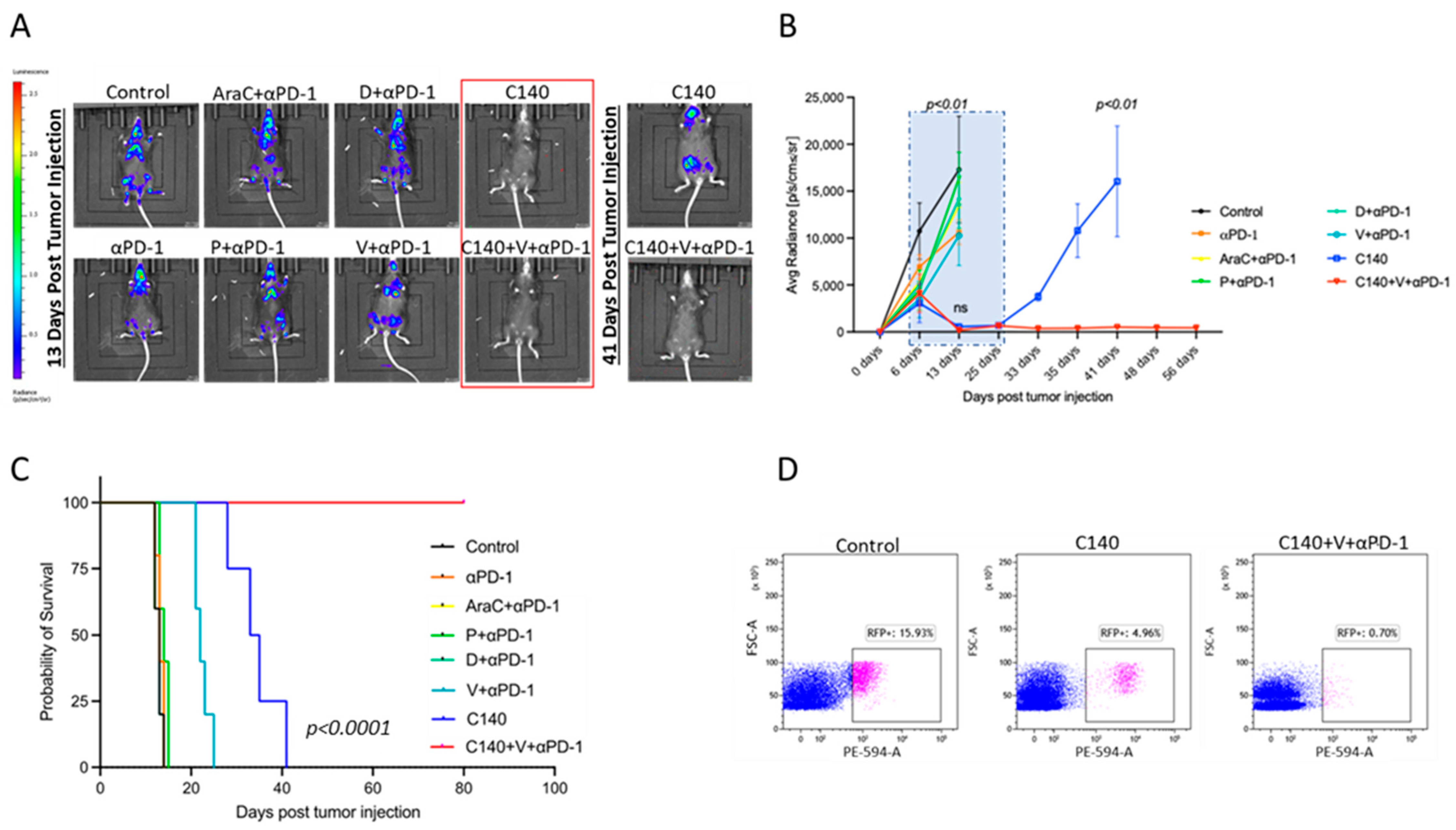
3.1. TT Sensitizes an Aggressive Myc-Driven B-Cell Lymphoma to Anti-PD-1 and Prevents Lymphoma Growth In Vivo
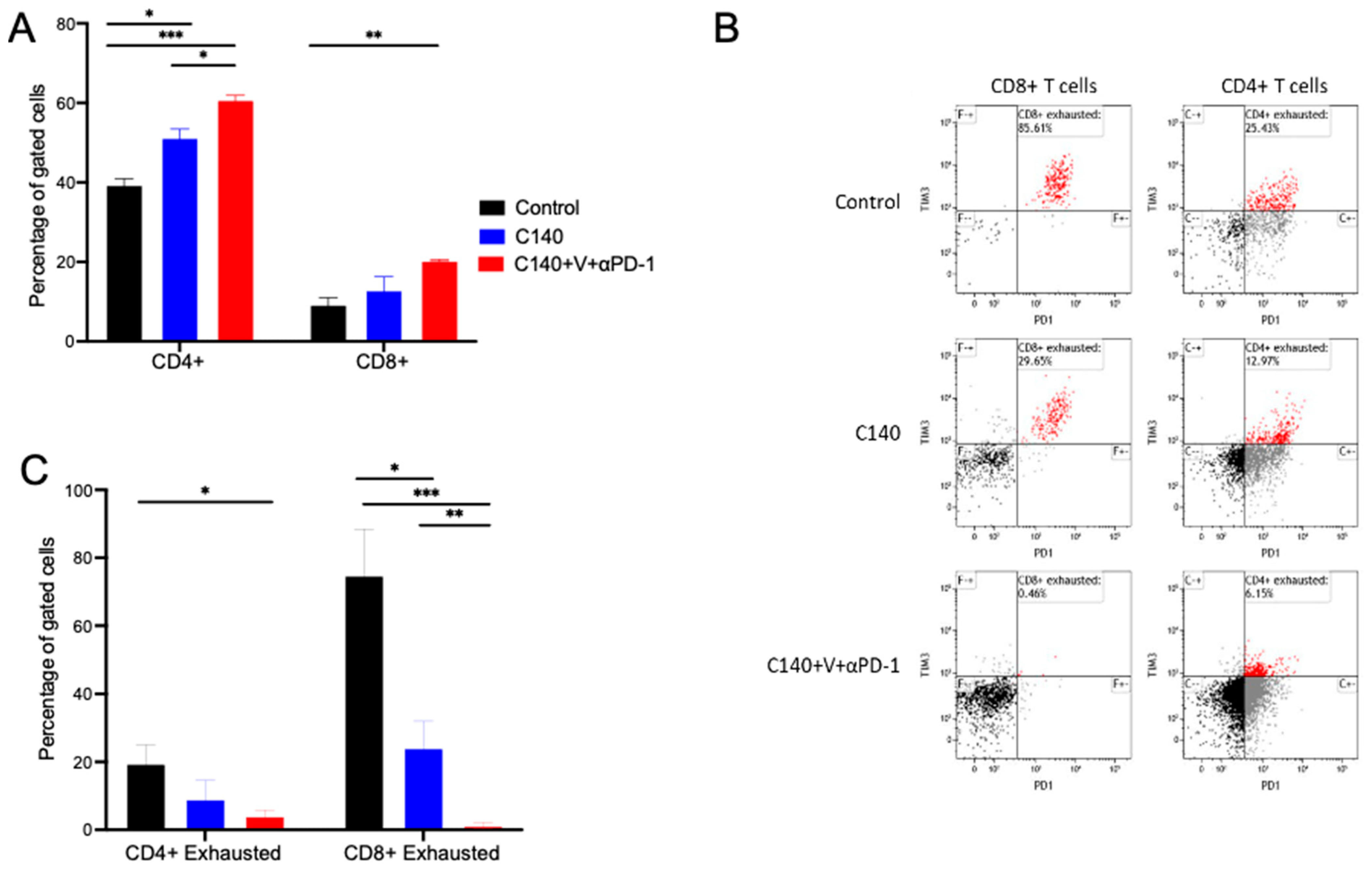
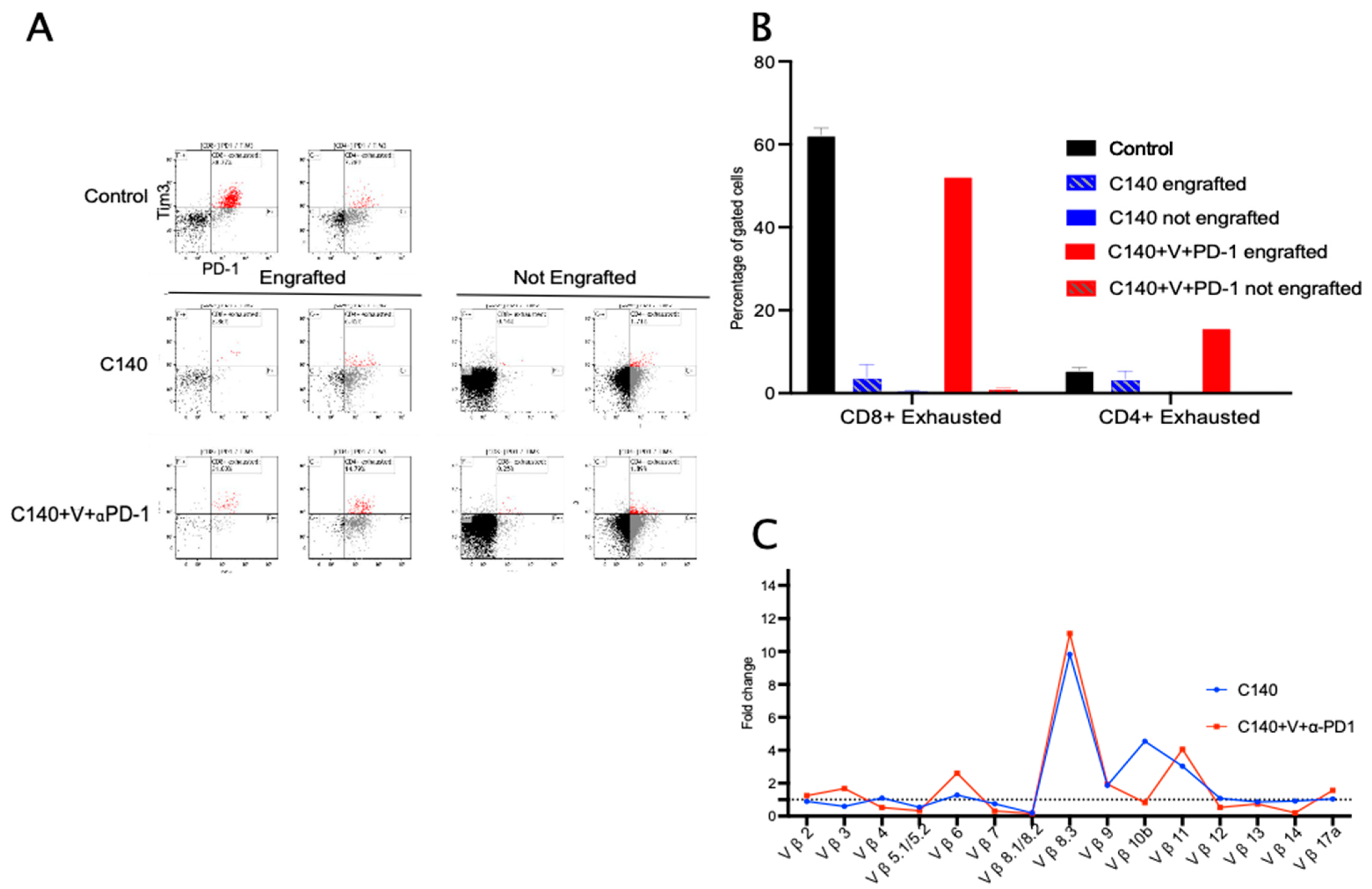
3.2. TT Reshapes the Intratumoral Immune Cell Landscape and Targets Terminally Exhausted T Cells
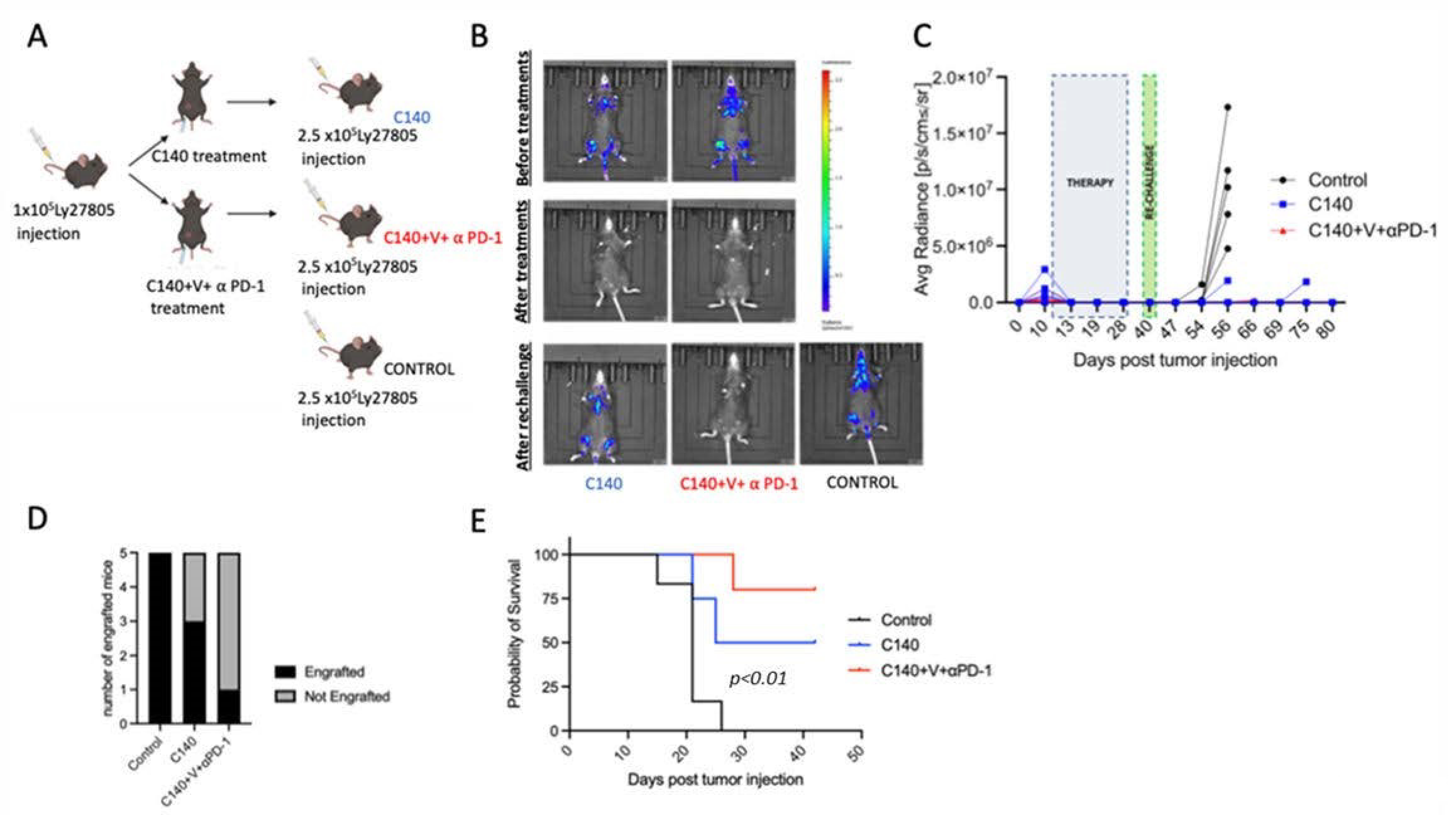
3.3. TT Orchestrates the Activation of T Cell Antitumor Memory in Mice
3.4. The Immune Cell Landscape in the Peripheral Blood Is Altered after Lymphoma Re-Challenge
3.5. TT Decreases Terminally Exhausted T Cells upon Tumor Re-Challenge and Induces Specific T-Cell Clonal Selection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef] [PubMed]
- Beaver, J.A.; Pazdur, R. The wild west of checkpoint inhibitor development. N. Engl. J. Med. 2022, 386, 1297–1301. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, F.; Sukhatme, V.P.; Bouche, G. Drug repurposing in oncology—Patient and health systems opportunities. Nat. Rev. Clin. Oncol. 2015, 12, 732–742. [Google Scholar] [CrossRef]
- Falvo, P.; Orecchioni, S.; Hillje, R.; Raveane, A.; Mancuso, P.; Camisaschi, C.; Luzi, L.; Pelicci, P.; Bertolini, F. Cyclophosphamide and vinorelbine activate stem-like CD8+ T cells and improve anti-PD-1 efficacy in triple-negative breast cancer. Cancer Res. 2021, 81, 685–697. [Google Scholar] [CrossRef]
- Falvo, P.; Orecchioni, S.; Raveane, A.; Mitola, G.; Bertolini, F. A “two-hit”(chemo) therapy to improve checkpoint inhibition in cancer. Oncoscience 2021, 8, 55. [Google Scholar] [CrossRef]
- Carpen, L.; Falvo, P.; Orecchioni, S.; Mitola, G.; Hillje, R.; Mazzara, S.; Mancuso, P.; Pileri, S.; Raveane, A.; Bertolini, F. A single-cell transcriptomic landscape of innate and adaptive intratumoral immunity in triple negative breast cancer during chemo-and immunotherapies. Cell Death Discov. 2022, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.; Gritti, I.; Nicoli, P.; Giorgio, M.; Doni, M.; Conti, A.; Bianchi, V.; Casoli, L.; Sabò, A.; Mironov, A. The mitochondrial translation machinery as a therapeutic target in Myc-driven lymphomas. Oncotarget 2016, 7, 72415. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Waxman, D.J. Metronomic cyclophosphamide eradicates large implanted GL261 gliomas by activating antitumor Cd8+ T-cell responses and immune memory. Oncoimmunology 2015, 4, e1005521. [Google Scholar] [CrossRef] [Green Version]
- Orecchioni, S.; Talarico, G.; Labanca, V.; Calleri, A.; Mancuso, P.; Bertolini, F. Vinorelbine, cyclophosphamide and 5-FU effects on the circulating and intratumoural landscape of immune cells improve anti-PD-L1 efficacy in preclinical models of breast cancer and lymphoma. Br. J. Cancer 2018, 118, 1329–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfirschke, C.; Engblom, C.; Rickelt, S.; Cortez-Retamozo, V.; Garris, C.; Pucci, F.; Yamazaki, T.; Poirier-Colame, V.; Newton, A.; Redouane, Y. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity 2016, 44, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Budak-Alpdogan, T.; Alpdogan, O.; Banerjee, D.; Wang, E.; Moore, M.A.; Bertino, J.R. Methotrexate and cytarabine inhibit progression of human lymphoma in NOD/SCID mice carrying a mutant dihydrofolate reductase and cytidine deaminase fusion gene. Mol. Ther. 2004, 10, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Visan, I. TCF-1+ progenitors. Nat. Immunol. 2022, 23, 988. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fang, J.; Wen, S.; Li, Q.; Wang, J.; Yang, L.; Dai, W.; Lu, H.; Guo, J.; Shan, Z. A comprehensive profile of TCF1+ progenitor and TCF1− terminally exhausted PD-1+ CD8+ T cells in head and neck squamous cell carcinoma: Implications for prognosis and immunotherapy. Int. J. Oral Sci. 2022, 14, 8. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Matsushima, H.; Mizumoto, N.; Takashima, A. Classification of chemotherapeutic agents based on their differential in vitro effects on dendritic cells. Cancer Res. 2009, 69, 6978–6986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zsiros, E.; Lynam, S.; Attwood, K.M.; Wang, C.; Chilakapati, S.; Gomez, E.C.; Liu, S.; Akers, S.; Lele, S.; Frederick, P.J. Efficacy and safety of pembrolizumab in combination with bevacizumab and oral metronomic cyclophosphamide in the treatment of recurrent ovarian cancer: A phase 2 nonrandomized clinical trial. JAMA Oncol. 2021, 7, 78–85. [Google Scholar] [CrossRef]
- Tilsed, C.M.; Principe, N.; Kidman, J.; Chin, W.L.; Morales, M.L.O.; Zemek, R.M.; Chee, J.; Islam, R.; Fear, V.S.; Forbes, C. CD4+ T cells drive an inflammatory, TNF-α/IFN-rich tumor microenvironment responsive to chemotherapy. Cell Rep. 2022, 41, 111874. [Google Scholar] [CrossRef]
- Røssevold, A.H.; Andresen, N.K.; Bjerre, C.A.; Gilje, B.; Jakobsen, E.H.; Raj, S.X.; Falk, R.S.; Russnes, H.G.; Jahr, T.; Mathiesen, R.R.; et al. Atezolizumab plus anthracycline-based chemotherapy in metastatic triple-negative breast cancer: The randomized, double-blind phase 2b ALICE trial. Nat. Med. 2022, 28, 2573–2583. [Google Scholar] [CrossRef]
- Davoodi-Moghaddam, Z.; Jafari-Raddani, F.; Noori, M.; Bashash, D. A systematic review and meta-analysis of immune checkpoint therapy in relapsed or refractory non-Hodgkin lymphoma; a friend or foe? Transl. Oncol. 2023, 30, 101636. [Google Scholar] [CrossRef]
- Bocci, G.; Pelliccia, S.; Orlandi, P.; Caridi, M.; Banchi, M.; Musuraca, G.; Di Napoli, A.; Bianchi, M.P.; Patti, C.; Anticoli-Borza, P.; et al. Remarkable Remission Rate and Long-Term Efficacy of Upfront Metronomic Chemotherapy in Elderly and Frail Patients, with Diffuse Large B-Cell Lymphoma. J. Clin. Med. 2022, 11, 7162. [Google Scholar] [CrossRef]
- Ye, X.; Wang, L.; Nie, M.; Wang, Y.; Dong, S.; Ren, W.; Li, G.; Li, Z.M.; Wu, K.; Pan-Hammarström, Q. A single-cell atlas of diffuse large B cell lymphoma. Cell Rep. 2022, 39, 110713. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.; Mo, H.; Hu, X.; Gao, R.; Zhao, Y.; Liu, B.; Niu, L.; Sun, X.; Yu, X.; et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell 2021, 39, 1578–1593. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orecchioni, S.; Falvo, P.; Talarico, G.; Mitola, G.; Bravetti, G.; Mancuso, P.; Nicoli, P.; Bertolini, F. Vinorelbine and Intermittent Cyclophosphamide Sensitize an Aggressive Myc-Driven B-Cell Lymphoma to Anti-PD-1 by an Immunological Memory Effective against Tumor Re-Challenge. J. Clin. Med. 2023, 12, 2535. https://doi.org/10.3390/jcm12072535
Orecchioni S, Falvo P, Talarico G, Mitola G, Bravetti G, Mancuso P, Nicoli P, Bertolini F. Vinorelbine and Intermittent Cyclophosphamide Sensitize an Aggressive Myc-Driven B-Cell Lymphoma to Anti-PD-1 by an Immunological Memory Effective against Tumor Re-Challenge. Journal of Clinical Medicine. 2023; 12(7):2535. https://doi.org/10.3390/jcm12072535
Chicago/Turabian StyleOrecchioni, Stefania, Paolo Falvo, Giovanna Talarico, Giulia Mitola, Giulia Bravetti, Patrizia Mancuso, Paola Nicoli, and Francesco Bertolini. 2023. "Vinorelbine and Intermittent Cyclophosphamide Sensitize an Aggressive Myc-Driven B-Cell Lymphoma to Anti-PD-1 by an Immunological Memory Effective against Tumor Re-Challenge" Journal of Clinical Medicine 12, no. 7: 2535. https://doi.org/10.3390/jcm12072535