
Lipoprotein (a), Inflammation, and Atherosclerosis
 ,
,  , , and
, , and
Abstract
:1. Introduction
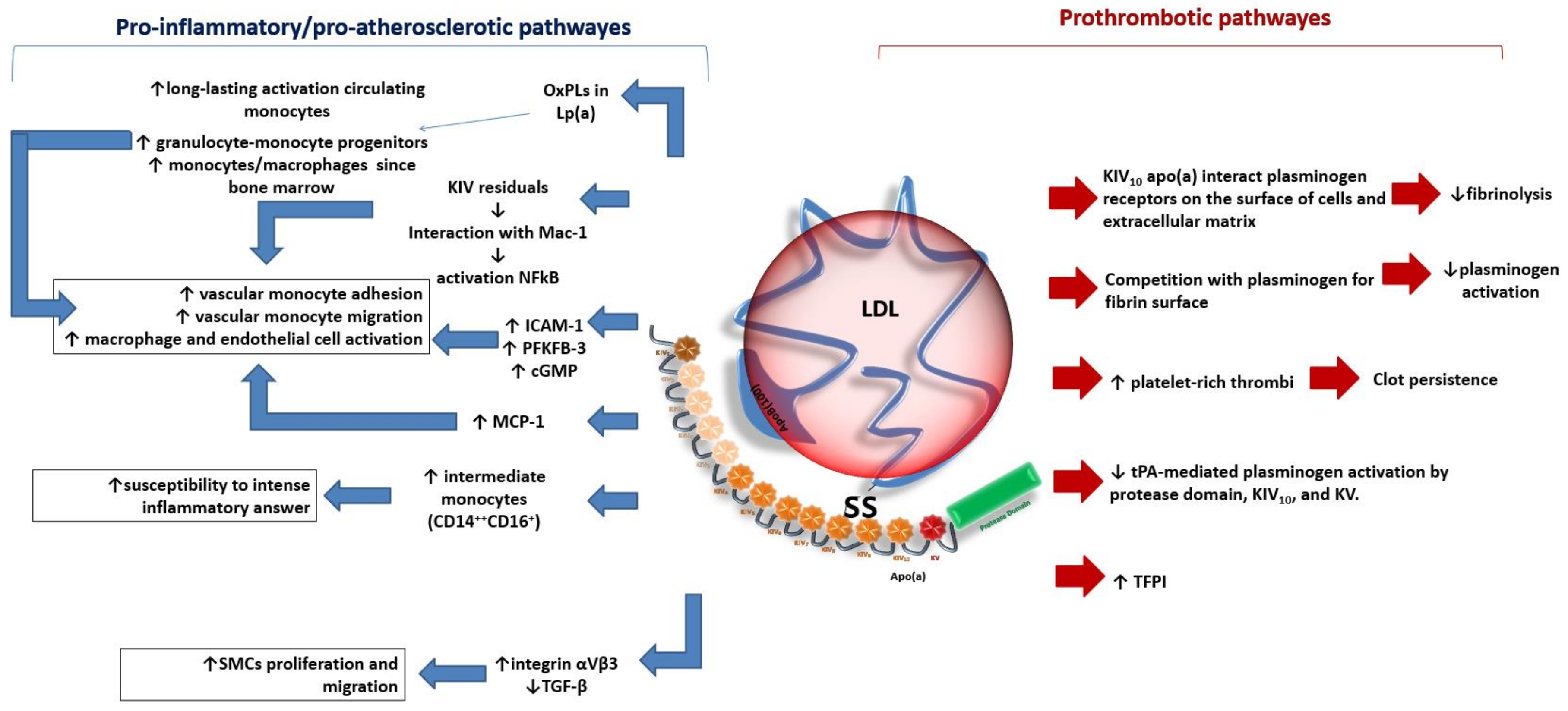
2. Lipoprotein (a) as an Atherogenic Factor
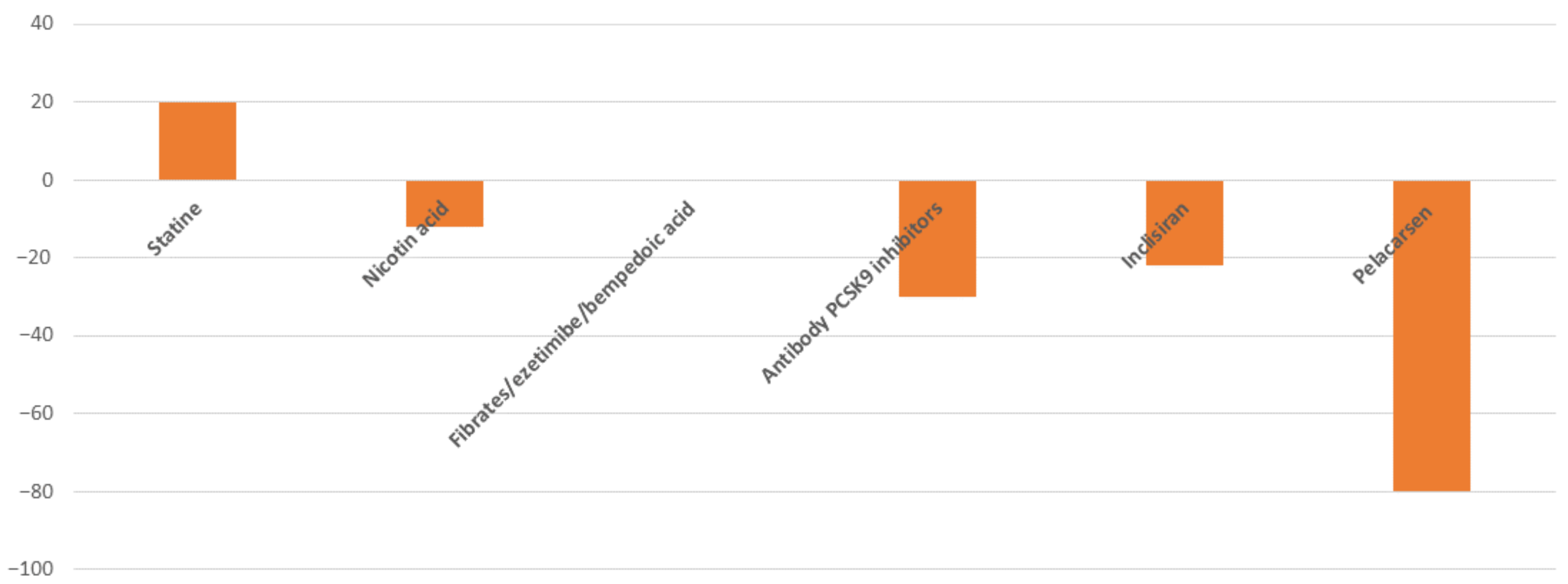
2.1. Clinical Evidence and Therapeutic Approach
2.2. How to Measure and Quantify the Risk Associated with High Lp(a) Levels
3. Inflammation and Atherosclerotic Cardiovascular Disease
3.1. Clinical Evidence and Therapeutic Approach
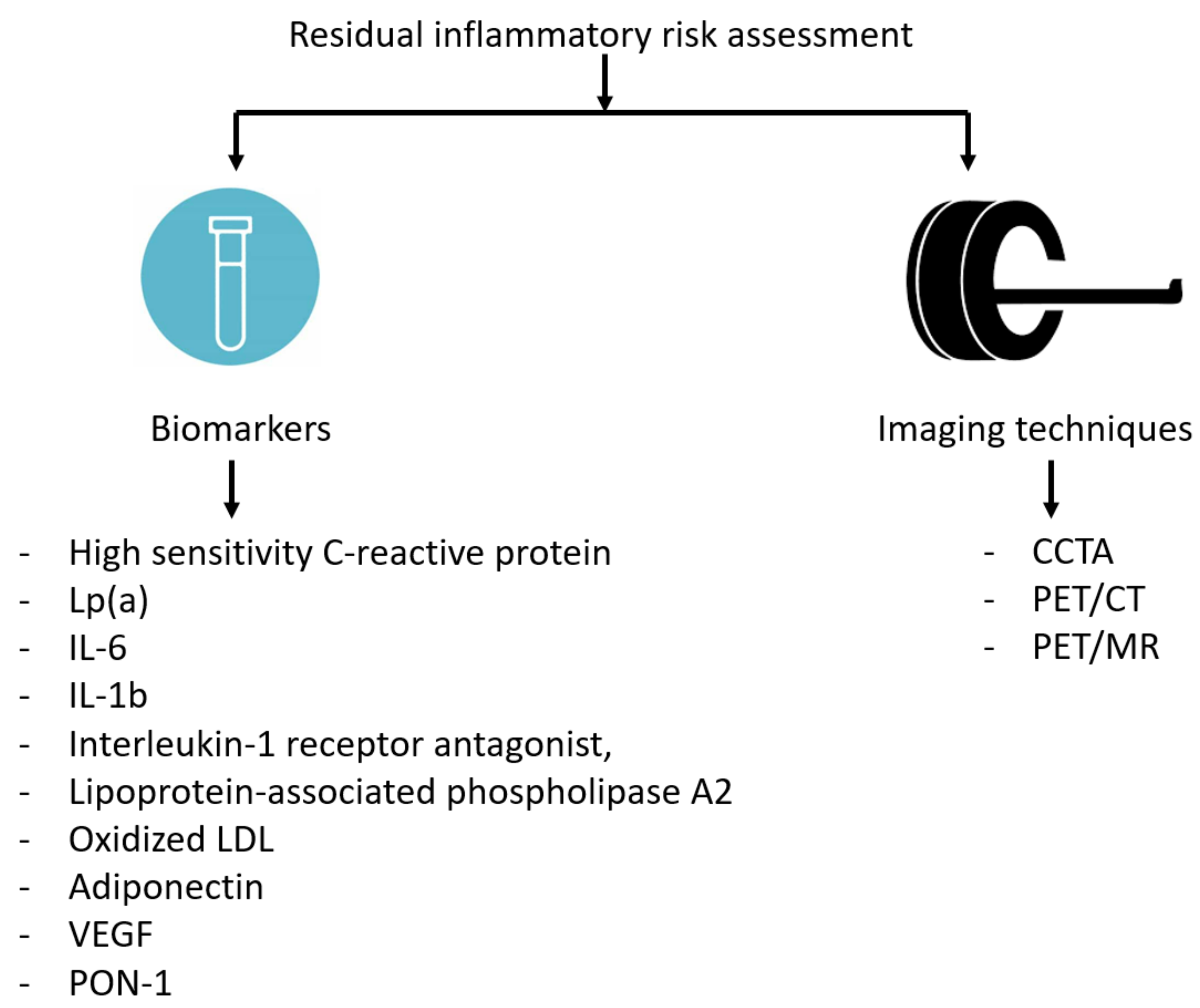
3.2. Circulating Biomarkers and Imaging to Assess Residual Inflammatory Risk
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Di Fusco, S.A.; Arca, M.; Scicchitano, P.; Alonzo, A.; Perone, F.; Gulizia, M.M.; Gabrielli, D.; Oliva, F.; Imperoli, G.; Colivicchi, F. Lipoprotein(a): A risk factor for atherosclerosis and an emerging therapeutic target. Heart 2022, 109, 18–25. [Google Scholar] [CrossRef]
- Schnitzler, J.G.; Hoogeveen, R.M.; Ali, L.; Prange, K.H.M.; Waissi, F.; van Weeghel, M.; Bachmann, J.C.; Versloot, M.; Borrelli, M.J.; Yeang, C.; et al. Atherogenic Lipoprotein(a) Increases Vascular Glycolysis, Thereby Facilitating Inflamma-tion and Leukocyte Extravasation. Circ. Res. 2020, 126, 1346–1359. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, S.N.; Orlova, V.V.; Al-Fakhri, N.; Ihanus, E.; Economopoulou, M.; Isermann, B.; Bdeir, K.; Nawroth, P.P.; Preissner, K.T.; Gahmberg, C.G.; et al. Lipoprotein(a) in atherosclerotic plaques recruits inflammatory cells through interaction with Mac-1 integrin. FASEB J. 2006, 20, 559–561. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, C.H.; Boffa, M.B.; Hancock, M.A.; Pickering, J.G.; Koschinsky, M.L. Stimulation of vascular smooth muscle cell proliferation and migration by apolipoprotein(a) is dependent on inhibition of transforming growth factor-beta activation and on the presence of kringle IV type 9. J. Biol. Chem. 2004, 279, 55187–55195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, S.; Green, M.A.; LoGrasso, P.V.; Boettcher, B.R.; Madison, E.L.; Curtiss, L.K.; Miles, L.A. Comparison of the effects of Apo(a) kringle IV-10 and plasminogen kringles on the inter-actions of lipoprotein(a) with regulatory molecules. Thromb. Haemost. 1999, 81, 428–435. [Google Scholar] [PubMed]
- Hajjar, K.A.; Gavish, D.; Breslow, J.L.; Nachman, R.L. Lipoprotein(a) modulation of endothe-lial cell surface fibrinolysis and its potential role in atherosclerosis. Nature 1989, 339, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Caplice, N.M.; Panetta, C.; Peterson, T.E.; Kleppe, L.S.; Mueske, C.S.; Kostner, G.M.; Broze GJJr Simari, R.D. Lipoprotein (a) binds and inactivates tissue factor pathway inhibitor: A novel link between lipoproteins and thrombosis. Blood 2001, 98, 2980–2987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rikhi, R.; Hammoud, A.; Ashburn, N.; Snavely, A.C.; Michos, E.D.; Chevli, P.; Tsai, M.Y.; Herrington, D.; Shapiro, M.D. Relationship of low-density lipoprotein-cholesterol and lipoprotein(a) to cardiovascular risk: The Multi-Ethnic Study of Atherosclerosis (MESA). Atherosclerosis 2022, 363, 102–108. [Google Scholar] [CrossRef]
- Yao, Y.; Liu, J.; Wang, B.; Zhou, Z.; Lu, X.; Huang, Z.; Deng, J.; Yang, Y.; Tan, N.; Chen, S.; et al. Baseline Low-Density-Lipoprotein Cholesterol Modifies the Risk of All-Cause Death Associated with Elevated Lipoprotein(a) in Coronary Artery Disease Patients. Front. Cardiovasc. Med. 2022, 8, 817442. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, R.; Hoogeveen, R.M.; Langsted, A.; Stiekema, L.C.A.; Verweij, S.L.; Hovingh, G.K.; Wareham, N.J.; Khaw, K.T.; Boekholdt, S.M.; Nordestgaard, B.G.; et al. Cardiovascular disease risk associated with elevated lipoprotein(a) attenuates at low low-density lipoprotein cholesterol levels in a primary prevention setting. Eur. Heart J. 2018, 39, 2589–2596. [Google Scholar] [CrossRef] [Green Version]
- Afshar, M.; Pilote, L.; Dufresne, L.; Engert, J.C.; Thanassoulis, G. Lipoprotein(a) Interactions with Low-Density Lipoprotein Cholesterol and Other Cardiovascular Risk Factors in Premature Acute Coronary Syndrome (ACS). J. Am. Heart Assoc. 2016, 5, e003012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, S.Y.; Liu, J.; Jiang, H.X.; Hu, B.L.; Zhou, Y.; Olkkonen, V.M. Association between baseline lipoprotein (a) levels and restenosis after coronary stenting: Meta-analysis of 9 cohort studies. Atherosclerosis 2013, 227, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Obisesan, O.H.; Kou, M.; Wang, F.M.; Boakye, E.; Honda, Y.; Uddin, S.M.I.; Dzaye, O.; Osei, A.D.; Orimoloye, O.A.; Howard-Claudio, C.M.; et al. Lipoprotein(a) and Subclinical Vascular and Valvular Calcification on Cardiac Computed Tomography: The Atherosclerosis Risk in Communities Study. J. Am. Heart Assoc. 2022, 11, e024870. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zha, L.; Chen, J.; Du, D.; Liu, D.; Zhong, M.; Shang, R.; Sun, D.; Sun, C.; Jin, E. The relationship between lipoprotein(a) and risk of cardiovascular disease: A Mendelian randomization analysis. Eur. J. Med. Res. 2022, 27, 211. [Google Scholar] [CrossRef] [PubMed]
- Colivicchi, F.; Di Fusco, S.A.; Arca, M.; Leggio, M.; Caldarola, P.; Murrone, A.; Valente, S.; Urbinati, S.; Roncon, L.; Amodeo, V.; et al. Non-high-density lipoprotein cholesterol versus low-density lipoprotein cholesterol in clinical practice: ANMCO position paper. J. Cardiovasc. Med. 2021, 22, 609–617. [Google Scholar] [CrossRef]
- De Boer, L.M.; Oorthuys, A.O.J.; Wiegman, A.; Langendam, M.W.; Kroon, J.; Spijker, R.; Zwinderman, A.H.; Hutten, B.A. Statin therapy and lipoprotein(a) levels: A systematic review and meta-analysis. Eur. J. Prev. Cardiol. 2022, 29, 779–792. [Google Scholar] [CrossRef]
- Tsimikas, S.; Plsm, G.; Nora, C.; Yeang, C.; Jl, W. Statin therapy increases lipoprotein(a) levels. Eur. Heart J. 2020, 41, 2275–2284. [Google Scholar] [CrossRef]
- Pirillo, A.; Catapano, A.L. Statins increase Lp(a) plasma level: Is this clinically relevant? Eur. Heart J. 2020, 41, 2285–2287. [Google Scholar] [CrossRef]
- Wang, X.; Luo, S.; Gan, X.; He, C.; Huang, R. Safety and efficacy of ETC-1002 in hypercholesterolaemic patients: A meta-analysis of randomised controlled trials. Kardiol. Pol. 2019, 77, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Sahebkar, A.; Simental-Mendía, L.E.; Pirro, M.; Banach, M.; Watts, G.F.; Sirtori, C.; Al-Rasadi, K.; Atkin, S.L. Impact of ezetimibe on plasma lipoprotein(a) concentrations as monotherapy or in combination with statins: A systematic review and meta-analysis of randomized controlled trials. Sci. Rep. 2018, 8, 17887. [Google Scholar] [CrossRef] [Green Version]
- Sahebkar, A.; Reiner, Ž.; Simental-Mendía, L.E.; Ferretti, G.; Cicero, A.F. Effect of extended-release niacin on plasma lipoprotein(a) levels: A systematic review and meta-analysis of randomized placebo-controlled trials. Metabolism 2016, 65, 1664–1678. [Google Scholar] [CrossRef] [PubMed]
- Farmakis, I.; Doundoulakis, I.; Pagiantza, A.; Zafeiropoulos, S.; Antza, C.; Karvounis, H.; Giannakoulas, G. Lipoprotein(a) Reduction with Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitors: A Systematic Review and Meta-analysis. J. Cardiovasc. Pharmacol. 2021, 77, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K.; Balog, C.; Swerdlow, D.I.; Scrimgeour, A.C.; Rambaran, C.; Wilson, R.J.; Boyce, M.; Ray, K.K.; Cho, L.; et al. Single Ascending Dose Study of a Short Interfering RNA Targeting Lipoprotein(a) Production in Individuals with Elevated Plasma Lipoprotein(a) Levels. JAMA 2022, 327, 1679–1687. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.L.; Rosenson, R.S.; Gencer, B.; López, J.A.G.; Lepor, N.E.; Baum, S.J.; Stout, E.; Gaudet, D.; Knusel, B.; Kuder, J.F.; et al. Small Interfering RNA to Reduce Lipopro-tein(a) in Cardiovascular Disease. N. Engl. J. Med. 2022, 387, 1855–1864. [Google Scholar] [CrossRef]
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.C.; Baum, S.J.; Steinhagen-Thiessen, E.; Shapiro, M.D.; Stroes, E.S.; Moriarty, P.M.; Nordestgaard, B.G.; et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Stiekema, L.C.A.; Prange, K.H.M.; Hoogeveen, R.M.; Verweij, S.L.; Kroon, J.; Schnitzler, J.G.; Dzobo, K.E.; Cupido, A.J.; Tsimikas, S.; Stroes, E.S.G.; et al. Potent lipoprotein(a) lowering following apolipoprotein(a) antisense treatment reduces the pro-inflammatory activation of circulating monocytes in patients with elevated lipo-protein(a). Eur. Heart J. 2020, 41, 2262–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roeseler, E.; Julius, U.; Heigl, F.; Spitthoever, R.; Heutling, D.; Breitenberger, P.; Leebmann, J.; Lehmacher, W.; Kamstrup, P.R.; Nordestgaard, B.G.; et al. Lipoprotein Apheresis for Lipoprotein(a)-Associated Cardiovascular Disease: Prospective 5 Years of Follow-Up and Apolipoprotein(a) Characterization. Arter. Thromb. Vasc. Biol. 2016, 36, 2019–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, S.; Ference, B.A.; Staley, J.R.; Freitag, D.F.; Mason, A.M.; Nielsen, S.F.; Willeit, P.; Young, R.; Surendran, P.; Karthikeyan, S.; et al. Association of LPA Variants with Risk of Coronary Disease and the Implications for Lipoprotein(a)-Lowering Therapies: A Mendelian Randomization Analysis. JAMA Cardiol. 2018, 3, 619–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen, C.M.; Kamstrup, P.R.; Langsted, A.; Varbo, A.; Nordestgaard, B.G. Lipoprotein(a)-Lowering by 50 mg/dL (105 nmol/L) May Be Needed to Reduce Cardiovascular Disease 20% in Secondary Prevention: A Population-Based Study. Arter. Thromb. Vasc. Biol. 2020, 40, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G.; Ference, B.A.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.; Lambert, G.; Mach, F.; et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: A European Atherosclerosis Society consensus statement. Eur. Heart J. 2022, 43, 3925–3946. [Google Scholar] [CrossRef]
- Boerwinkle, E.; Leffert, C.C.; Lin, J.; Lackner, C.; Chiesa, G.; Hobbs, H.H. Apolipoprotein(a) gene accounts for greater than 90% of the variation in plasma lipoprotein(a) concentrations. J. Clin. Investig. 1992, 90, 52–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcovina, S.M.; Albers, J.J. Lipoprotein (a) measurements for clinical application. J. Lipid Res. 2016, 57, 526–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcovina, S.M.; Clouet-Foraison, N.; Koschinsky, M.L.; Lowenthal, M.S.; Orquillas, A.; Boffa, M.B.; Hoofnagle, A.N.; Vaisar, T. Development of an LC-MS/MS Proposed Candidate Reference Method for the Standardization of Analytical Methods to Measure Lipoprotein(a). Clin. Chem. 2021, 67, 490–499. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef]
- Corrado, E.; Rizzo, M.; Tantillo, R.; Muratori, I.; Bonura, F.; Vitale, G.; Novo, S. Markers of inflammation and infection influence the outcome of patients with baseline asymptomatic carotid lesions: A 5-year follow-up study. Stroke 2006, 37, 482–486. [Google Scholar] [CrossRef] [Green Version]
- Alfaddagh, A.; Martin, S.S.; Leucker, T.M.; Michos, E.D.; Blaha, M.J.; Lowenstein, C.J.; Jones, S.R.; Toth, P.P. Inflammation and cardiovascular disease: From mechanisms to therapeutics. Am. J. Prev. Cardiol. 2020, 4, 100130. [Google Scholar] [CrossRef]
- Ridker, P.M.; Rifai, N.; Rose, L.; Buring, J.E.; Cook, N.R. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N. Engl. J. Med. 2002, 347, 1557–1565. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Zhou, S.; Dreyer, R.P.; Spatz, E.S.; Geda, M.; Lorenze, N.P.; D’Onofrio, G.; Lichtman, J.H.; Spertus, J.A.; Ridker, P.M.; et al. Sex Differences in Inflammatory Markers and Health Status Among Young Adults With Acute Myocardial Infarction: Results From the VIRGO (Variation in Recovery: Role of Gender on Outcomes of Young Acute Myocardial Infarction Patients) Study. Circ. Cardiovasc. Qual. Outcomes 2017, 10, e003470. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Cannon, C.P.; Morrow, D.; Rifai, N.; Rose, L.M.; McCabe, C.H.; Pfeffer, M.A.; Braunwald, E.; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 (PROVE IT-TIMI 22) Investigators. C-reactive protein levels and outcomes after statin therapy. N. Engl. J. Med. 2005, 352, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Bohula, E.A.; Giugliano, R.P.; Cannon, C.P.; Zhou, J.; Murphy, S.A.; White, J.A.; Tershakovec, A.M.; Blazing, M.A.; Braunwald, E. Achievement of dual low-density lipoprotein cholesterol and high-sensitivity C-reactive protein targets more frequent with the addition of ezetimibe to simvastatin and associated with better outcomes in IMPROVE-IT. Circulation 2015, 132, 1224–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto AMJr Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; Macfadyen, J.G.; Nordestgaard, B.G.; et al. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: A prospective study of the JUPITER trial. Lancet 2009, 373, 1175–1182. [Google Scholar] [CrossRef]
- Bohula, E.A.; Giugliano, R.P.; Leiter, L.A.; Verma, S.; Park, J.G.; Sever, P.S.; Lira Pineda, A.; Honarpour, N.; Wang, H.; Murphy, S.A.; et al. Inflammatory and Cholesterol Risk in the FOURIER Trial. Circulation 2018, 138, 131–140. [Google Scholar] [CrossRef]
- Van’t Klooster, C.C.; van der Graaf, Y.; Ridker, P.M.; Westerink, J.; Hjortnaes, J.; Sluijs, I.; Asselbergs, F.W.; Bots, M.L.; Kappelle, L.J.; Visseren, F.L.J.; et al. The relation between healthy lifestyle changes and decrease in systemic inflammation in patients with stable cardiovascular disease. Atherosclerosis 2020, 301, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Rifai, N.; Clearfield, M.; Downs, J.R.; Weis, S.E.; Miles, J.S.; Gotto, A.M., Jr.; Air Force/Texas Coronary Atherosclerosis Prevention Study Investigators. Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N. Engl. J. Med. 2001, 344, 1959–1965. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Eikelboom, J.W.; Budgeon, C.A.; Thompson, P.L. Low-dose colchicine for secondary prevention of cardiovascular disease. J. Am. Coll Cardiol. 2013, 61, 404–410. [Google Scholar] [CrossRef] [Green Version]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Kontos, M.C.; Abouzaki, N.A.; Melchior, R.D.; Thomas, C.; Van Tassell, B.W.; Oddi, C.; Carbone, S.; Trankle, C.R.; Roberts, C.S.; et al. Comparative safety of interleukin-1 blockade with anakinra in patients with ST-segment elevation acute myocardial in-farction (from the VCU-ART and VCU-ART2 pilot studies). Am. J. Cardiol. 2015, 115, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Trankle, C.R.; Buckley, L.F.; Lipinski, M.J.; Appleton, D.; Kadariya, D.; Canada, J.M.; Carbone, S.; Roberts, C.S.; Abouzaki, N.; et al. Interleukin-1 Blockade Inhibits the Acute Inflammatory Response in Patients With ST-Segment-Elevation Myocardial Infarction. J. Am. Heart Assoc. 2020, 9, e014941. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Devalaraja, M.; Baeres, F.M.M.; Engelmann, M.D.M.; Hovingh, G.K.; Ivkovic, M.; Lo, L.; Kling, D.; Pergola, P.; Raj, D.; et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 2060–2069. [Google Scholar] [CrossRef]
- Kleveland, O.; Kunszt, G.; Bratlie, M.; Ueland, T.; Broch, K.; Holte, E.; Michelsen, A.E.; Bendz, B.; Amundsen, B.H.; Espevik, T.; et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: A double-blind, randomized, placebo-controlled phase 2 trial. Eur. Heart J. 2016, 37, 2406–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broch, K.; Anstensrud, A.K.; Woxholt, S.; Sharma, K.; Tøllefsen, I.M.; Bendz, B.; Aakhus, S.; Ueland, T.; Amundsen, B.H.; Damås, J.K.; et al. Randomized Trial of Interleukin-6 Receptor Inhibition in Patients with Acute ST-Segment Elevation Myocardial Infarction. J. Am. Coll Cardiol. 2021, 77, 1845–1855. [Google Scholar] [CrossRef]
- Di Fusco, S.A.; Di Michele, S.; Colivicchi, F. Prevenzione dell’aterosclerosi: Nuove strategie terapeutiche all’orizzonte. G. Ital. Cardiol. 2021, 22, 922–931. (In Italian) [Google Scholar] [CrossRef]
- Ridker, P.M. From C-Reactive Protein to Interleukin-6 to Interleukin-1: Moving Upstream to Identify Novel Targets for Atheroprotection. Circ. Res. 2016, 118, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Antoniades, C.; Antonopoulos, A.S.; Deanfield, J. Imaging residual inflammatory cardiovascular risk. Eur. Heart J. 2020, 41, 748–758. [Google Scholar] [CrossRef] [Green Version]
- Maurovich-Horvat, P.; Ferencik, M.; Voros, S.; Merkely, B.; Hoffmann, U. Comprehensive plaque assessment by coronary CT angiography. Nat. Rev. Cardiol. 2014, 11, 390–402. [Google Scholar] [CrossRef]
- Conte, E.; Andreini, D.; Magnoni, M.; Masson, S.; Mushtaq, S.; Berti, S.; Canestrari, M.; Casolo, G.; Gabrielli, D.; Latini, R.; et al. Association of high-risk coronary atherosclerosis at CCTA with clinical and circulating biomarkers: Insight from CAPIRE study. J. Cardiovasc. Comput. Tomogr. 2021, 15, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulos, A.S.; Sanna, F.; Sabharwal, N.; Thomas, S.; Oikonomou, E.K.; Herdman, L.; Margaritis, M.; Shirodaria, C.; Kampoli, A.M.; Akoumianakis, I.; et al. Detecting human coronary inflammation by imaging perivascular fat. Sci. Transl. Med. 2017, 9, eaal2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, N.V.; Vesey, A.T.; Williams, M.C.; Shah, A.S.; Calvert, P.A.; Craighead, F.H.; Yeoh, S.E.; Wallace, W.; Salter, D.; Fletcher, A.M.; et al. 18F-fluoride positron emission tomography for identification of ruptured and high-risk coronary atherosclerotic plaques: A prospective clinical trial. Lancet 2014, 383, 705–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarkin, J.M.; Joshi, F.R.; Evans, N.R.; Chowdhury, M.M.; Figg, N.L.; Shah, A.V.; Starks, L.T.; Martin-Garrido, A.; Manavaki, R.; Yu, E.; et al. Detection of Atherosclerotic Inflammation by 68Ga-DOTATATE PET Compared to [18F]FDG PET Imaging. J. Am. Coll Cardiol. 2017, 69, 1774–1791. [Google Scholar] [CrossRef] [PubMed]
- Hyafil, F.; Pelisek, J.; Laitinen, I.; Schottelius, M.; Mohring, M.; Döring, Y.; van der Vorst, E.P.; Kallmayer, M.; Steiger, K.; Poschenrieder, A.; et al. Imaging the Cytokine Receptor CXCR4 in Atherosclerotic Plaques with the Radiotracer 68Ga-Pentixafor for PET. J. Nucl. Med. 2017, 58, 499–506. [Google Scholar] [CrossRef] [Green Version]
- Senders, M.L.; Que, X.; Cho, Y.S.; Yeang, C.; Groenen, H.; Fay, F.; Calcagno, C.; Meerwaldt, A.E.; Green, S.; Miu, P.; et al. PET/MR Imaging of Malondialdehyde-Acetaldehyde Epitopes with a Human Antibody Detects Clinically Relevant Atherothrombosis. J. Am. Coll Cardiol. 2018, 71, 321–335. [Google Scholar] [CrossRef]
- Matter, C.M.; Wyss, M.T.; Meier, P.; Späth, N.; von Lukowicz, T.; Lohmann, C.; Weber, B.; Ramirez de Molina, A.; Lacal, J.C.; Ametamey, S.M.; et al. 18F-choline images murine atherosclerotic plaques ex vivo. Arter. Thromb. Vasc. Biol. 2006, 26, 584–589. [Google Scholar] [CrossRef] [Green Version]
- Simantiris, S.; Antonopoulos, A.S.; Papastamos, C.; Benetos, G.; Koumallos, N.; Tsioufis, K.; Tousoulis, D. Lipoprotein(a) and inflammation- pathophysiological links and clinical implications for cardiovascular disease. J. Clin. Lipidol. 2023, 17, 55–63. [Google Scholar] [CrossRef]
- Hoogeveen, R.C.; Ballantyne, C.M. Residual Cardiovascular Risk at Low LDL: Remnants, Lipoprotein(a), and Inflammation. Clin. Chem. 2021, 67, 143–153. [Google Scholar] [CrossRef]
- Zhang, W.; Speiser, J.L.; Ye, F.; Tsai, M.Y.; Cainzos-Achirica, M.; Nasir, K.; Herrington, D.M.; Shapiro, M.D. High-Sensitivity C-Reactive Protein Modifies the Cardiovascular Risk of Lipoprotein(a): Multi-Ethnic Study of Atherosclerosis. J. Am. Coll Cardiol. 2021, 78, 1083–1094. [Google Scholar] [CrossRef]
- Puri, R.; Nissen, S.E.; Arsenault, B.J.; St John, J.; Riesmeyer, J.S.; Ruotolo, G.; McErlean, E.; Menon, V.; Cho, L.; Wolski, K.; et al. Effect of C-Reactive Protein on Lipopro-tein(a)-Associated Cardiovascular Risk in Optimally Treated Patients with High-Risk Vascular Disease: A Prespecified Secondary Analysis of the ACCELERATE Trial. JAMA Cardiol. 2020, 5, 1136–1143. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Trial | Tested Drug | Mechanism of Action | Study Design | N Patients | Study Population | Follow-Up Duration | Main Findings |
|---|---|---|---|---|---|---|---|
| CANTOS [47] | Canakinumab | IL-1β inhibition | Randomized Controlled Double-blind Phase 3 Trial | 10,061 | Previous MI and high CRP levels | 3.7 years * | Canakinumab reduced cardiovascular events |
| COLCOT [50] | Colchicine | Tubulin polymerization and inflammasome inhibition | Randomized Controlled Double-blind Phase 3 Trial | 4745 | Patients who had had a MI within 30 days before recruitment | 22.6 months * | Significant reduction in ischemic cardiovascular events |
| LoDoCo2 [51] | Colchicine | Tubulin polymerization and inflammasome inhibition | Randomized Controlled Double-blind Phase 3 Trial | 5522 | Chronic coronary disease | 28.6 months * | Colchicine vs. placebo reduced cardiovascular event risk |
| VCUART3 [53] | Anakinra | IL-1 receptor antagonist | Randomized Controlled Double-blind Phase 2 Trial | 99 | ST-segment-elevation myocardial infarction | 12 months | Significant reduction in hsCRP area under the curve during the first 14 days. Lower incidence of HF. |
| RESCUE [54] | Ziltivekimab | IL-6 inhibition | Randomized Controlled Double-blind Phase 2 Trial | 264 | Chronic kidney disease and high CRP levels | 24 weeks | Ziltivekimab (dose dependent) reduced inflammation and thrombosis biomarkers |
| Tocilizumab in NSTEMI patients (ClinicalTrials.gov, NCT01491074) [55] | Tocilizumab | IL-6 receptor inhibition | Randomized Controlled Double-blind Phase 2 trial | 117 | NSTEMI patients scheduled for angiography | 6 months | Tocilizumab reduced hsCRP levels and troponin T release after PCI |
| ASSAIL-MI [56] | Tocilizumab | IL-6 receptor inhibition | Randomized Controlled Double-blind Phase 2 trial | 199 | STEMI patients within 6 h of symptom onset | 6 months | Greater myocardial salvage index (Measured with CMR after 3–6 days) |
| CIRT [38] | Metothrexate | Nucleotide synthesis inhibition leading to suppression of inflammation | Randomized Controlled Double-blind Phase 3 Trial | 4786 | Previous MI or multivessel CAD and Type 2 diabetes or metabolic syndrome | 2.3 years * | Low-dose methotrexate did not result in lower IL-1B/ IL-6 or CRP levels |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Fusco, S.A.; Maggioni, A.P.; Scicchitano, P.; Zuin, M.; D’Elia, E.; Colivicchi, F. Lipoprotein (a), Inflammation, and Atherosclerosis. J. Clin. Med. 2023, 12, 2529. https://doi.org/10.3390/jcm12072529
Di Fusco SA, Maggioni AP, Scicchitano P, Zuin M, D’Elia E, Colivicchi F. Lipoprotein (a), Inflammation, and Atherosclerosis. Journal of Clinical Medicine. 2023; 12(7):2529. https://doi.org/10.3390/jcm12072529
Chicago/Turabian StyleDi Fusco, Stefania Angela, Aldo Pietro Maggioni, Pietro Scicchitano, Marco Zuin, Emilia D’Elia, and Furio Colivicchi. 2023. "Lipoprotein (a), Inflammation, and Atherosclerosis" Journal of Clinical Medicine 12, no. 7: 2529. https://doi.org/10.3390/jcm12072529