
Unraveling the Underlying Molecular Mechanism of ‘Silent Hypoxia’ in COVID-19 Patients Suggests a Central Role for Angiotensin II Modulation of the AT1R-Hypoxia-Inducible Factor Signaling Pathway
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
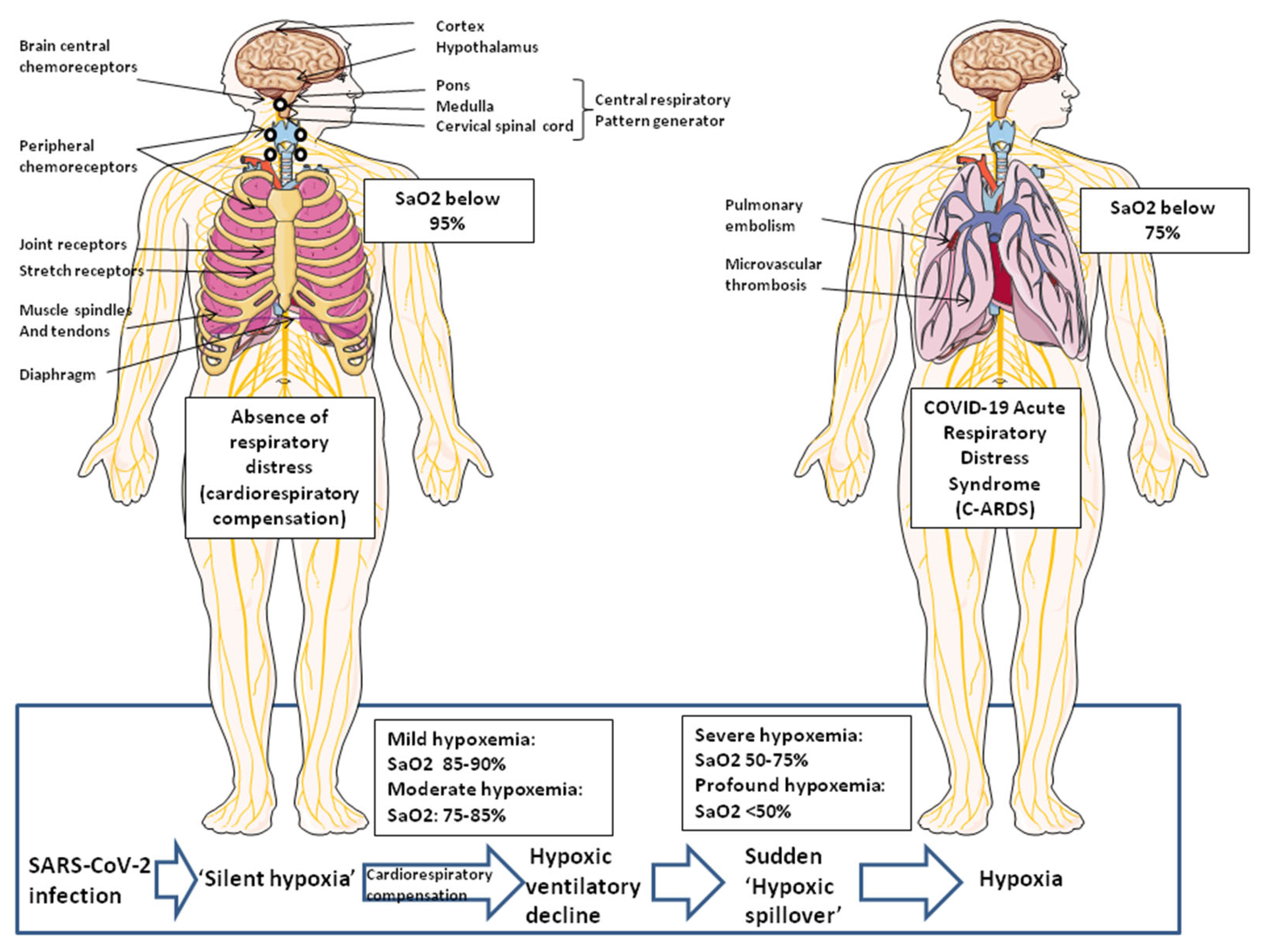
:1. Introduction
2. Clinical Evidence of ‘Silent Hypoxia’
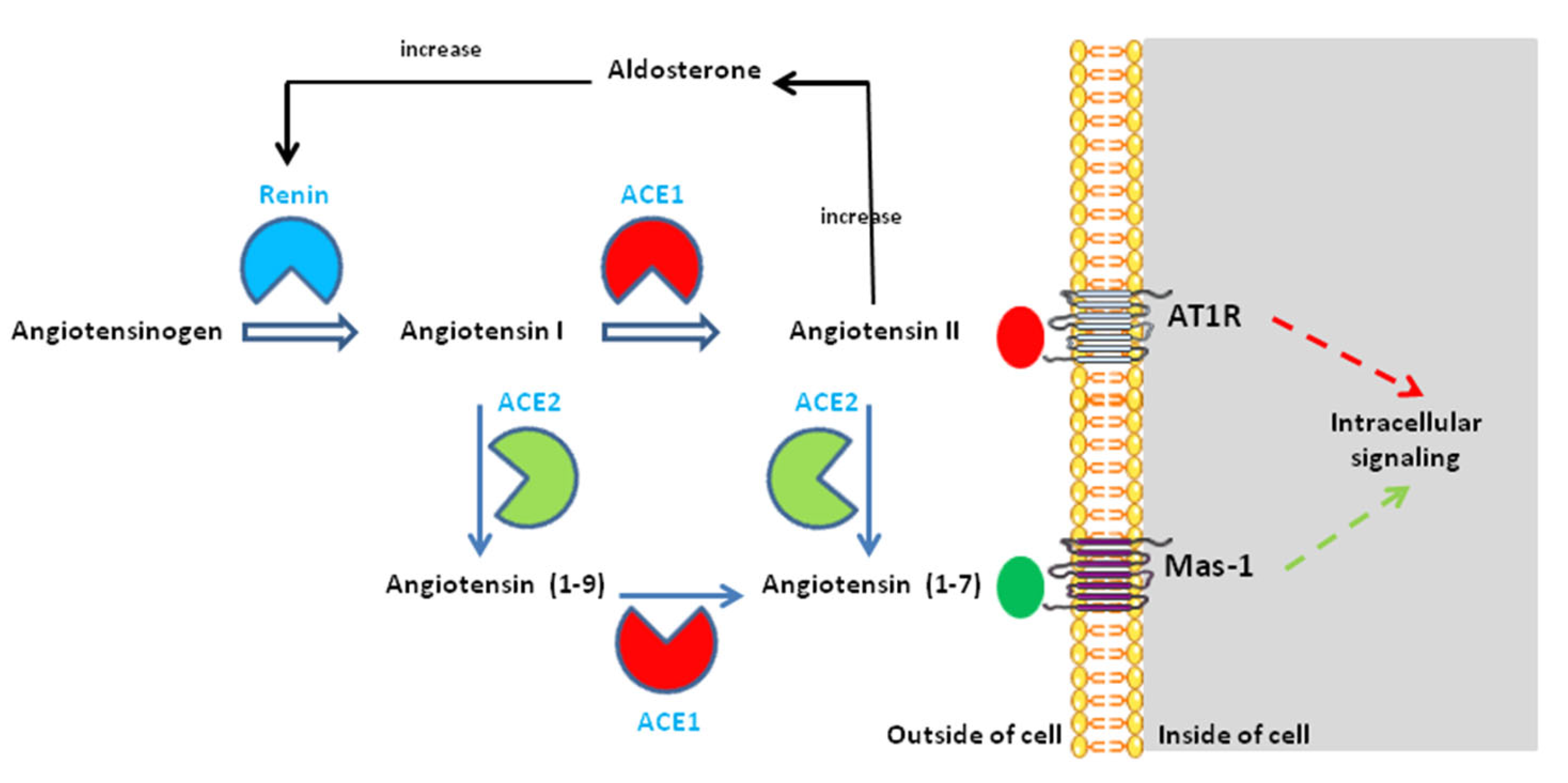
3. Regulation of Blood Pressure Homeostasis via the RAS
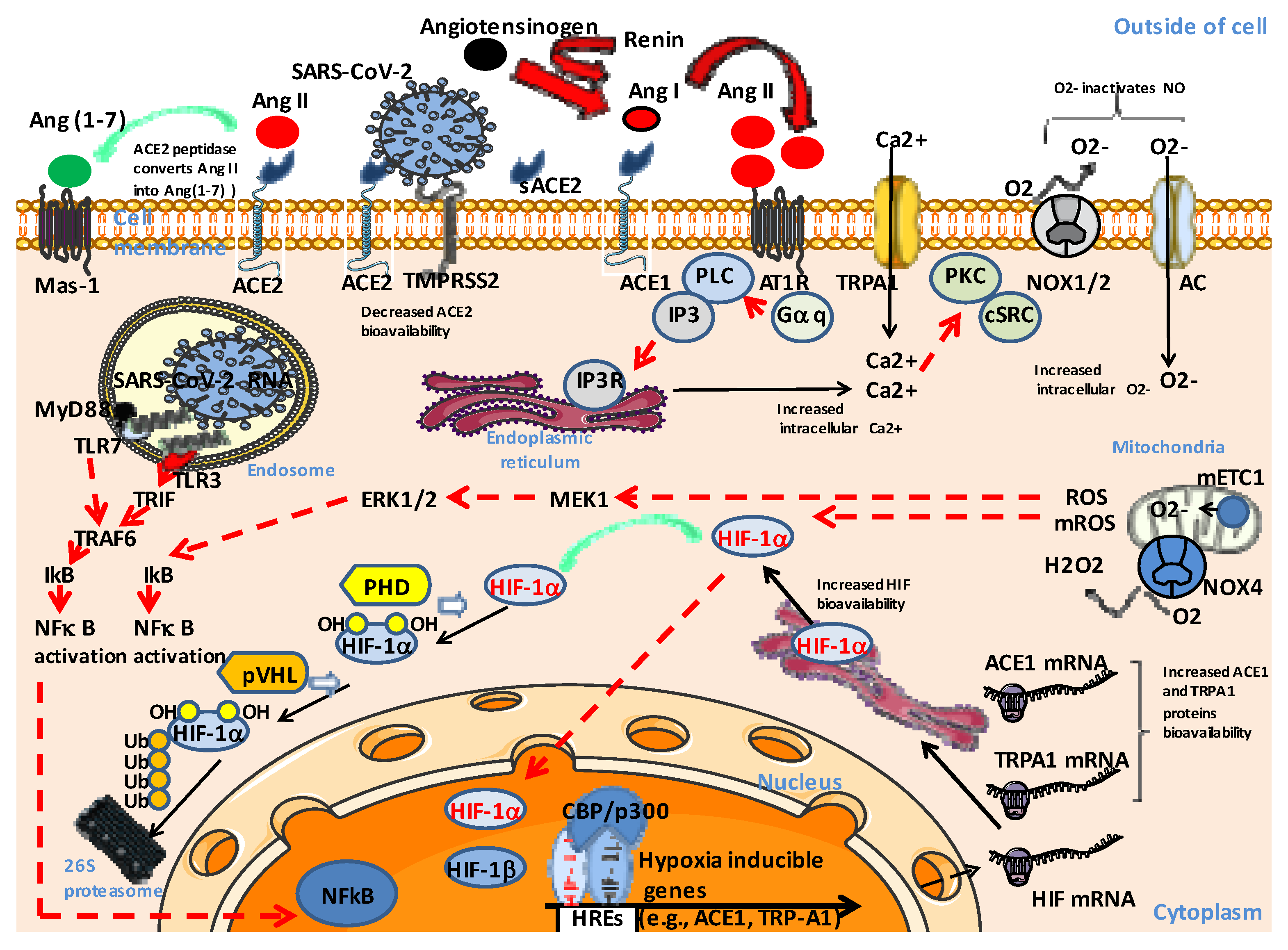
4. The RAS Imbalance in COVID-19
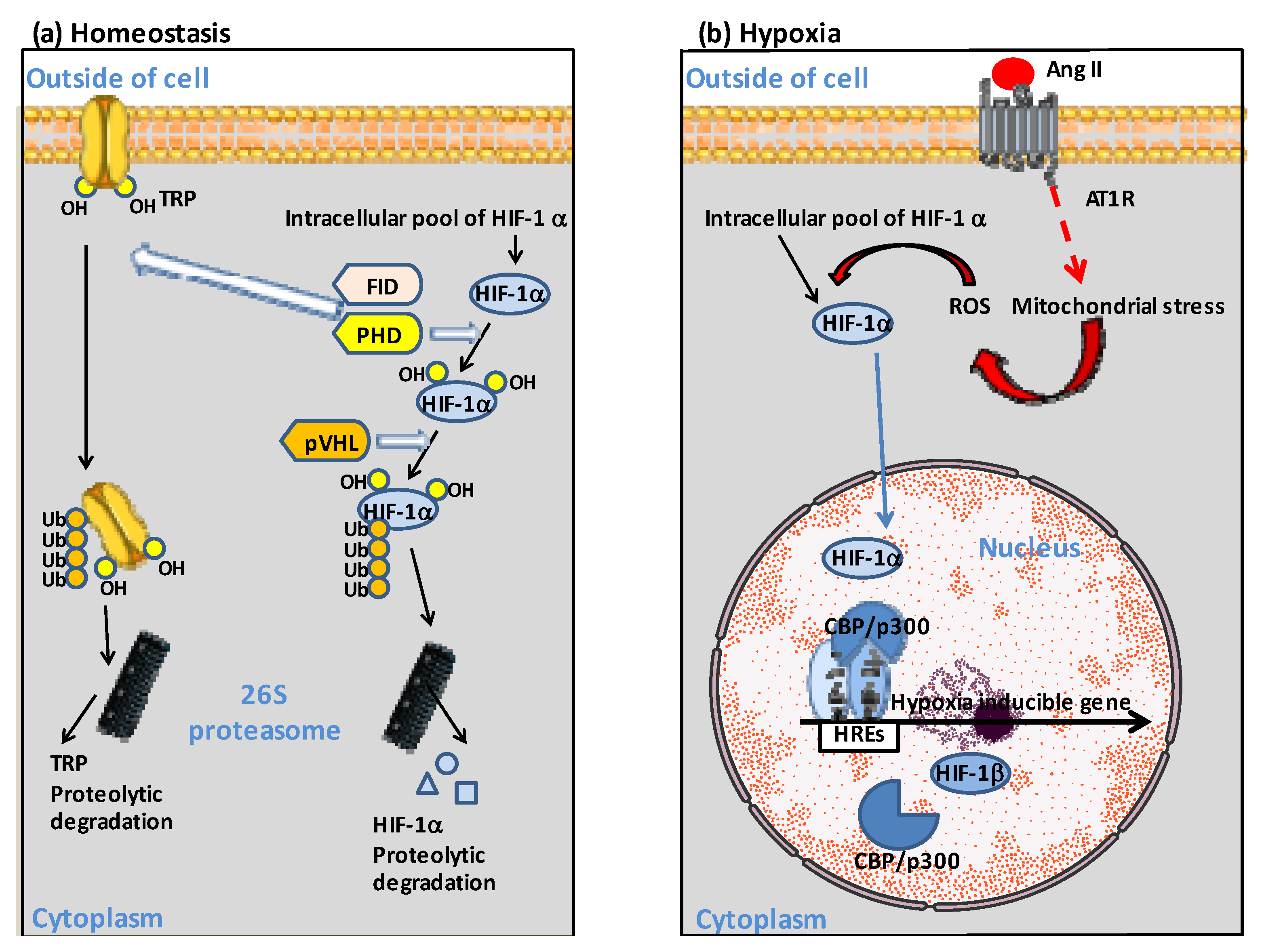
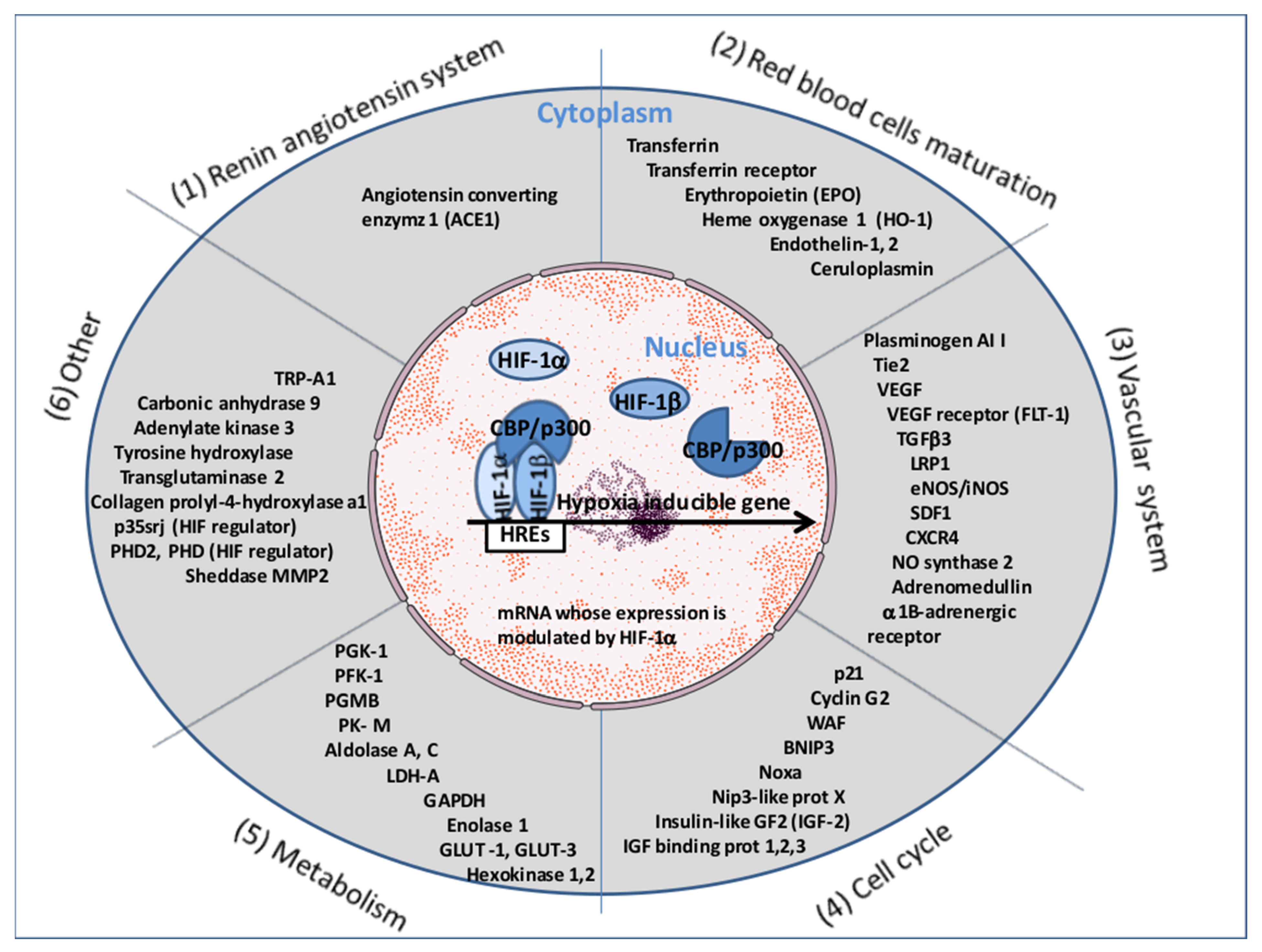
5. The Ang II-AT1R-HIF-1α Axis
6. Pathological Consequences of Ang II-AT1R-HIF-1α Axis Activation in COVID-19
7. The Involvement of Cells, Tissues, and Organs in ‘Silent Hypoxia’
8. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A Pneumonia Outbreak Associated With a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of the SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Zhao, Y.-B.; Wang, Q.; Li, J.-Y.; Zhou, Z.-J.; Liao, C.-H.; Ge, X.-Y. Predicting the angiotensin converting enzyme 2 (ACE2) utilizing capability as the receptor of SARS-CoV-2. Microbes Infect. 2020, 22, 221–225. [Google Scholar] [CrossRef]
- Ortiz, M.E.; Thurman, A.; Pezzulo, A.A.; Leidinger, M.R.; Klesney-Tait, J.A.; Karp, P.H.; Tan, P.; Wohlford-Lenane, C.; McCray, P.B.; Meyerholz, D.K. Heterogeneous expression of the SARS Coronavirus-2 receptor ACE2 in the human respiratory tract. EBioMedicine 2020, 60, 102976. [Google Scholar] [CrossRef]
- Delorey, T.M.; Ziegler, C.G.K.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, A.; Abbondanza, D.; Fleming, S.J.; Subramanian, A.; Montoro, D.T.; et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 2021, 595, 107–113. [Google Scholar] [CrossRef]
- Devaux, C.A.; Rolain, J.M.; Raoult, D. ACE2 receptor polymorphism: Susceptibility to SARS-CoV-2, hypertension, multi-organ failure, and COVID-19 disease outcome. J. Microbiol. Immunol. Inf. 2020, 53, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Fuglebjerg, N.; Jensen, T.; Hoyer, N.; Ryrsø, C.; Madsen, B.; Harboe, Z. Silent hypoxia in patients with SARS CoV-2 infection before hospital discharge. Int. J. Infect. Dis. 2020, 99, 100–101. [Google Scholar] [CrossRef]
- Tobin, M.; Laghi, F.; Jubran, A. Why COVID-19 silent hypoxemia Is baffling to physicians. Am. J. Respir. Crit. Care Med. 2020, 202, 356–360. [Google Scholar] [CrossRef]
- Couzin-Frankel, J. The mystery of the pandemic’s ‘happy hypoxia’. Science 2020, 368, 455–456. [Google Scholar] [CrossRef]
- Brouqui, P.; Amrane, S.; Million, M.; Cortaredona, S.; Parola, P.; Lagier, J.-C.; Raoult, D. Asymptomatic hypoxia in COVID-19 is associated with poor outcome. Int. J. Infect. Dis. 2021, 102, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Gusmao Ramos, S.; da Cruz Rattis, B.A.; Ottaviani, G.; Nunes Celes, M.R.; Pedra Dias, E. ACE2 Down-Regulation May Act as a Transient Molecular Disease Causing RAAS Dysregulation and Tissue Damage in the Microcirculatory Environment Among COVID-19 Patients. Am. J. Pathol. 2021, 191, 1154–1166. [Google Scholar] [CrossRef] [PubMed]
- Osman, I.O.; Garrec, C.; de Souza, G.A.P.; Zarubica, A.; Belhaouari, D.B.; Baudoin, J.-P.; Lepidi, H.; Mege, J.-L.; Malissen, B.; La Scola, B.; et al. Control of CDH1/E-Cadherin Gene Expression and Release of a Soluble Form of E-Cadherin in SARS-CoV-2 Infected Caco-2 Intestinal Cells: Physiopathological Consequences for the Intestinal Forms of COVID-19. Front. Cell. Infect. Microbiol. 2022, 12, 798767. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, J.T.; Shimoda, L.A.; Aaronson, P.I.; Ward, J.P.T. Hypoxic pulmonary vasoconstriction. Physiol. Rev. 2012, 92, 367–520. [Google Scholar] [CrossRef]
- Boerrigter, B.G.; Boonstra, A.; Westerhof, N.; Postmus, P.E.; Vonk-Noordegraaf, A. Cardiac shunt in COPD as a cause of severe hypoxaemia: Probably not so uncommon after all. Eur. Respir. J. 2011, 37, 960–970. [Google Scholar] [CrossRef] [Green Version]
- Vodoz, J.-F.; Cottin, V.; Glérant, J.-C.; Derumeaux, G.; Khouatra, C.; Blanchet, A.-S.; Mastroïanni, B.; Bayle, J.-Y.; Mornex, J.-F.; Cordier, J.-F. Right-to-left shunt with hypoxemia in pulmonary hypertension. BMC Cardiovasc. Disord. 2009, 9, 15. [Google Scholar] [CrossRef]
- Lau, V.I.; Mah, G.D.; Wang, X.; Byker, L.; Robinson, A.; Milovanovic, L.; Alherbish, A.; Odenbach, J.; Vadeanu, C.; Lu, D.; et al. Intra-pulmonary and intra-cardiac shunts in adult COVID-19 versus non-COVID ARDS ICU patients using echocardiography and contrast bubble studies (COVID-Shunt Study): A prospective, observational cohort study. MedRxiv, 2022; preprint. [Google Scholar] [CrossRef]
- Sharaf, M.; Rajaram, M.; Mulji, A. Intracardiac shunt with hypoxemia caused by right ventricular dysfunction following pericardiocentesis. Can. J. Cardiol. 2008, 24, e60–e62. [Google Scholar] [CrossRef] [Green Version]
- Ji, J.Y.; Chung, J.H.; Kim, N.S.; Seo, Y.H.; Jung, H.S.; Chun, H.R.; Gong, H.Y.; Kim, W.J.; Ahn, J.M.; Park, Y.J. Causes and treatment of hypoxia during total hip arthroplasty in elderly patients: A case report. Int. J. Environ. Res. Public Health 2021, 18, 12931. [Google Scholar] [CrossRef]
- Machluf, Y.; Rosenfeld, S.; Ben Shlomo, I.; Chaiter, Y.; Dekel, Y. The misattributed and silent causes of poor COVID-19 outcomes among pregnant women. Front. Med. 2021, 8, 745797. [Google Scholar] [CrossRef]
- Bepouka, B.; Odio, O.; Mayasi, N.; Longokolo, M.; Mangala, D.; Mandina, M.; Mbula, M.; Kayembe, J.M.; Situakibanza, H. Prevalence and outcomes of COVID-19 patients with happy hypoxia: A systematic review. Infect. Drug Resist. 2022, 15, 5619–5628. [Google Scholar] [CrossRef] [PubMed]
- Masi, P.; Bagate, F.; d’Humières, T.; Al-Assaad, L.; Abou Chakra, L.; Derumeaux, G.; Mekontso Dessap, A. Is hypoxemia explained by intracardiac or intrapulmonary shunt in COVID-19-related acute respiratory distress syndrome? Ann. Intensive Care 2020, 10, 108. [Google Scholar] [CrossRef]
- Alhusain, F.; Alromaih, A.; Alhajress, G.; Alsaghyir, A.; Alqobaisi, A.; Alaboodi, T.; Alsalamah, M. Predictors and clinical outcomes of silent hypoxia in COVID-19 patients, a single-center retrospective cohort study. J. Inf. Public Health 2021, 14, 1595–1599. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Qu, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Bai, W.Z.; Hashikawa, T. The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J. Med. Virol. 2020, 92, 552–555. [Google Scholar] [CrossRef]
- Hsieh, J.Y.C.; Kan, J.Y.L.; Mattar, S.A.M.; Qin, Y. The clinical implications of sinus tachycardia in mild COVID-19 infection: A retrospective cohort study. SAGE Open Med. 2021, 9, 1–6. [Google Scholar] [CrossRef]
- Recasens, B.B.; Martinez-Llorens, J.M.; Rodriguez-Sevilla, J.J.; Rubio, M.A. Lack of dyspnea in patients with COVID-19: Another neurological conundrum ? Eur. J. Neurol. 2020, 27, e40. [Google Scholar] [CrossRef]
- De Vito, E.L. Possible role of corollary discharge in lack of dyspnea in patients with COVID-19 disease. Front. Physiol. 2021, 12, 719166. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Favaloro, E.J. D-Dimer is associated with severity of coronavirus disease 2019: A pooled analysis. Thromb. Haemostasis 2020, 120, 876–878. [Google Scholar] [CrossRef] [Green Version]
- Sakka, M.; Connors, J.M.; He’kimian, G.; Martin-Toutain, I.; Crichi, B.; Colmegna, I.; Bonnefont-Rousselot, D.; Farge, D.; Frere, C. Association between D-dimer levels and mortality in patients with coronavirus disease 2019 COVID19: A systematic review and pooled analysis. J. Med. Vasc. 2020, 45, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Smadja, D.M.; Mentzer, S.J.; Fontenay, M.; Laffan, M.A.; Ackermann, M.; Helms, J.; Jonigk, D.; Chocron, R.; Pier, G.B.; Gendron, N.; et al. COVID-19 is a systemic vascular hemopathy: Insight for mechanistic and clinical aspects. Angiogenesis 2021, 24, 755–788. [Google Scholar] [CrossRef]
- Stefely, J.A.; Christensen, B.B.; Gogakos, T.; Sullivan, J.K.C.; Montgomery, G.G.; Barranco, J.P.; Van Cott, E.M. Marked factor V activity elevation in severe COVID-19 is associated with venous thromboembolism. Am. J. Hematol. 2020, 95, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- Middeldorp, S.; Coppens, M.; van Haaps, T.F.; Foppen, M.; Vlaar, A.P.; Miller, M.C.A.; Bouman, C.C.S.; Beenen, L.F.M.; Kootte, R.S.; Heijmanset, J. Incidence of venous thromboembolism in hospitalized patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1995–2002. [Google Scholar] [CrossRef] [PubMed]
- Léonard-Lorant, I.; Delabranche, X.; Séverac, F.; Helms, J.; Pauzet, C.; Collange, O.; Schneider, F.; Labani, A.; Bilbault, P.; Molière, S.; et al. Acute pulmonary embolism in COVID-19 patients on CT angiography and relationship to D-dimer levels. Radiology 2020, 296, E189–E191. [Google Scholar] [CrossRef] [Green Version]
- Faggiano, P.; Bonelli, A.; Paris, S.; Milesi, G.; Bisegna, S.; Bernardi, N.; Curnis, A.; Agricola, E.; Maroldi, R. Acute Pulmonary Embolism in COVID-19 Disease: Preliminary Report on Seven Patients. Int. J. Cardiol. 2020, 313, 129–131. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [Green Version]
- Kashani, K.B. Hypoxia in COVID-19: Sign of Severity or Cause for Poor Outcomes. Mayo Clin. Proc. 2020, 95, 1094–1096. [Google Scholar] [CrossRef]
- Xie, J.; Covassin, N.; Fan, Z.; Singh, P.; Gao, W.; Li, G.; Kara, T.; Somers, V.K. Association between hypoxemia and mortality in patients with COVID-19. Mayo Clin. Proc. 2020, 95, 1138–1147. [Google Scholar] [CrossRef]
- Nitsure, M.; Sarangi, B.; Shankar, G.H.; Reddy, V.S.; Walimbe, A.; Sharma, V.; Prayag, S. Mechanisms of Hypoxia in COVID-19 Patients: A Pathophysiologic Reflection. Indian J. Crit. Care Med. 2020, 24, 967–970. [Google Scholar] [CrossRef]
- Herrmann, J.; Mori, V.; Bates, J.H.T.; Suki, B. Modeling lung perfusion abnormalities to explain early COVID-19 hypoxemia. Nature Com. 2020, 11, 4883. [Google Scholar] [CrossRef]
- Rahman, A.; Tabassum, T.; Araf, Y.; Al Nahid, A.; Ullah, M.A.; Jakir Hosen, M. Silent hypoxia in COVID-19: Pathomechanism and possible management strategy. Mol. Biol. Rep. 2021, 48, 3863–3869. [Google Scholar] [CrossRef]
- Singh, A.; Kataria, S.; Das, P.; Sharma, A. A proposal to make the pulse oximetry as omnipresent as thermometry in public health care systems. J. Glob. Health 2020, 10, 0203102. [Google Scholar] [CrossRef]
- Huynh, D.N.; Millan, A.; Quijada, E.; John, D.; Khan, S.; Funahashi, T. Description and early results of the Kaiser Permanente Southern California COVID-19 home monitoring program. Perm. J. 2021, 25, 20.281. [Google Scholar] [CrossRef]
- Lee, K.C.; Morgan, A.U.; Chaiyachati, K.H.; Asch, D.A.; Xiong, R.A.; Do, D.; Kilaru, A.S.; Lam, D.; Parambath, A.; Friedman, A.B.; et al. Pulse oximetry for monitoring patients with Covid-19 at home—A pragmatic, randomized trial. N. Engl. J. Med 2022, 386, 19. [Google Scholar] [CrossRef]
- Gallo, G.; Calvez, V.; Savoia, C. Hypertension and COVID-19: Current evidence and perspectives. High Blood Pressure Cardiovasc. Prevent. 2022, 29, 115–123. [Google Scholar] [CrossRef]
- Wakahara, S.; Konoshita, T.; Mizuno, S.; Motomura, M.; Aoyama, C.; Makino, Y.; Kato, N.; Koni, I.; Miyamori, I. Synergistic expression of angiotensin-converting enzyme (ACE) and ACE2 in human renal tissue and confounding effects of hypertension on the ACE to ACE2 Ratio. Endocrinology 2007, 148, 2453–2457. [Google Scholar] [CrossRef] [Green Version]
- Arendse, L.B.; Danser, A.H.J.; Poglitsch, M.; Touyz, R.M.; Burnett, J.C., Jr.; Llorens-Cortes, C.; Ehlers, M.R.; Sturrock, E.D. Novel therapeutic approaches targeting the Renin-Angiotensin System and associated peptides in hypertension and heart failure. Pharmacol. Rev. 2019, 71, 539–570. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, F.; Holstein-Rathlou, N.H. Angiotensin II modulates conducted vasoconstriction to norepinephrine and local electrical stimulation in rat mesenteric arterioles. Cardiovasc. Res. 1999, 44, 176–184. [Google Scholar] [CrossRef] [Green Version]
- Samavati, L.; Uhal, B.D. ACE2, much more than just a receptor for SARS-COV-2. Front. Cell. Infect. Microbiol. 2020, 10, 317. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, M.; Moniwa, N.; Takizawa, H.; Ura, N.; Shimamoto, K. Potential differential effects of renin-angiotensin system inhibitors on SARS-CoV-2 infection and lung injury in COVID-19. Hypertens. Res. 2020, 43, 837–840. [Google Scholar] [CrossRef] [PubMed]
- Sadoshima, J.; Qiu, Z.; Morgan, J.P.; Izumo, S. Angiotensin-II and other hypertrophic stimuli mediated by G-protein-coupled receptors activate tyrosine kinase, mitogen-activated protein-kinase, and 90-Kd S6 kinase in cardiac myocytes—The critical role of Ca2+-dependent signaling. Circ. Res. 1995, 76, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Runge, M.S.; Brasier, A.R. Angiotensin II induces Interleukin-6 transcription in vascular smooth muscle cells through pleiotropic activation of nuclear Factor-kB transcription Factors. Circ. Res. 1999, 84, 695–703. [Google Scholar] [CrossRef]
- Rushworth, C.A.; Guy, J.L.; Turner, A.J. Residues affecting the chloride regulation and substrate selectivity of the angiotensin-converting enzymes (ACE and ACE2) identified by site-directed mutagenesis. FEBS J. 2008, 275, 6033–6042. [Google Scholar] [CrossRef]
- Luther, J.; Gainer, J.V.; Murphey, L.J.; Yu, C.; Vaughan, D.E.; Morrow, J.D.; Brown, N.J. Angiotensin II Induces Interleukin-6 in Humans Through a Mineralocorticoid Receptor–Dependent Mechanism. Hypertension 2006, 48, 1050–1057. [Google Scholar] [CrossRef] [Green Version]
- AbdAlla, S.; Lother, H.; Quitterer, U. AT1-receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature 2000, 407, 94–98. [Google Scholar] [CrossRef]
- Gáborik, Z.; Szaszák, M.; Szidonya, L.; Balla, B.; Paku, S.; Catt, K.J.; Clark, A.J.L.; Hunyady, L. Beta-arrestin- and dynamin-dependent endocytosis of the AT1 angiotensin receptor. Mol. Pharmacol. 2001, 59, 239–247. [Google Scholar] [CrossRef]
- Garrido, A.M.; Griendling, K.K. NADPH Oxidases and Angiotensin II Receptor Signaling. Mol. Cell. Endocrinol. 2009, 302, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Nataraj, C.; Oliverio, M.I.; Mannon, R.B.; Mannon, P.J.; Audoly, L.P.; Amuchastegui, C.S.; Ruiz, P.; Smithies, O.; Coffman, T.M. Angiotensin II regulates cellular immune responses through a calcineurin-dependent pathway. J. Clin. Investig. 1999, 104, 1693–1701. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Ortega, M.; Lorenzo, O.; Suzuki, Y.; Rupérez, M.; Egido, J. Proinflammatory actions of angiotensins. Curr. Opin. Nephrol. Hypertens. 2001, 10, 321–329. [Google Scholar] [CrossRef]
- Watanabe, T.; Barker, T.A.; Berk, B.C. Angiotensin II and the endothelium: Diverse signals and effects. Hypertension 2005, 45, 163–169. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Nazarewicz, R.R. Angiotensin II-Induced Production of Mitochondrial Reactive Oxygen Species: Potential Mechanisms and Relevance for Cardiovascular Disease. Antioxid. Redox Signal. 2013, 19, 1085–1094. [Google Scholar] [CrossRef]
- Nisimoto, Y.; Diebold, B.A.; Cosentino-Gomes, D.; Lambeth, J.D. Nox4: A Hydrogen Peroxide-Generating Oxygen Sensor. Biochemistry 2014, 53, 5111–5120. [Google Scholar] [CrossRef] [Green Version]
- Devaux, C.A.; Raoult, D. The impact of COVID-19 on populations living at high altitude: Role of hypoxiainducible factors (HIFs) signaling pathway in SARS-CoV-2 infection and replication. Front. Physiol. 2022, 13, 960308. [Google Scholar] [CrossRef]
- Abu Nabah, Y.N.; Mateo, T.; Estellés, R.; Mata, M.; Zagorski, J.; Sarau, H.; Cortijo, J.; Morcillo, E.J.; Jose, P.J.; Sanz, M.-J.; et al. Angiotensin II induces neutrophil accumulation in vivo through generation and release of CXC chemokines. Circulation 2004, 110, 3581–3586. [Google Scholar] [CrossRef] [Green Version]
- Jamaluddin, M.; Meng, T.; Sun, J.; Boldogh, I.; Han, Y.; Brasier, A.R. Angiotensin II induces nuclear factor (NF)-kappaB1 isoforms to bind the angiotensinogen gene acute-phase response element: A stimulus-specific pathway for NF-kappaB activation. Mol. Endocrinol. 2000, 14, 99–113. [Google Scholar]
- Skurk, T.; van Harmelen, V.; Hauner, H. Angiotensin II stimulates the release of interleukin-6 and interleukin-8 from cultured human adipocytes by activation of NF-kappaB. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1199–1203. [Google Scholar] [CrossRef] [Green Version]
- Gorin, Y.; Ricono, J.M.; Wagner, B.; Kim, N.H.; Bhandari, B.; Choudhury, G.G. Angiotensin II-induced ERK1/ERK2 activation and protein synthesis are redox-dependent in glomerular mesangial cells. Biochem. J. 2004, 381, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Devaux, C.A.; Camoin-Jau, L. An update on ACE2 structure/functions, polymorphism, and duplicitous nature in the pathophysiology of COVID-19: Implications for vascular and coagulation disease associated with SARS-CoV-2 infection. Front. Microbiol. 2022, 13, 1042200. [Google Scholar] [CrossRef]
- Chappell, M.C. The Angiotensin-(1-7) Axis: Formation and Metabolism Pathways. In Angiotensin-(1-7); Santos, R.A.S., Ed.; Springer Nature Switzerland: Cham, Switzerland, 2019; Volume 22, pp. 1–26. [Google Scholar] [CrossRef]
- Santos, R.A.; Simoes e Silva, A.C.; Maric, C.; Silva, D.M.R.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.B.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/angiotensin-(1–7)/MAS axis of the renin-angiotensin system: Focus on angiotensin-(1–7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, M.; Alenina, N.; Young, D.; Santos, R.A.S.; Touyz, R.M.; Savergnini, S.Q.; Beiman, M.; Lautner, R.Q.; de Paula-Carvalho, V.; Allahdadi, K.; et al. The meaning of Mas. Hypertension 2018, 72, 1072–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rysz, S.; Al-Saadi, J.; Sjöström, A.; Farm, M.; Jalde, F.C.; Plattén, M.; Eriksson, H.; Klein, M.; Vargas-Paris, R.; Nyrén, S.; et al. COVID-19 pathophysiology may be driven by an imbalance in the renin-angiotensin-aldosterone system. Nat. Com. 2021, 12, 2417. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Qiao, Y.; Wang, X.-M.; Mannan, R.; Pitchiaya, S.; Zhang, Y.; Wotring, J.W.; Xiao, L.; Robinson, D.R.; Wu, Y.-M.; Tien, J.C.-Y.; et al. Targeting transcriptional regulation of SARS-CoV-2 entry factors ACE2 and TMPRSS2. Proc. Natl. Acad. Sci. USA 2021, 118, e2021450118. [Google Scholar] [CrossRef]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef] [Green Version]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–33019. [Google Scholar] [CrossRef] [Green Version]
- Oarhe, C.I.; Dang, V.; Dang, M.; Nguyen, H.; Gopallawa, I.; Gewolb, I.H.; Uhal, B.D. Hyperoxia downregulates angiotensin-converting enzyme-2 in human fetal lung fibroblasts. Pediatr. Res. 2015, 77, 656–662. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Osman, I.O.; Melenotte, C.; Brouqui, P.; Million, M.; Lagier, J.-C.; Parola, P.; Stein, A.; La Scola, B.; Meddeb, L.; Mege, J.-L.; et al. Expression of ACE2, Soluble ACE2, Angiotensin I, Angiotensin II and Angiotensin-(1-7) Is Modulated in COVID-19 Patients. Front. Immunol. 2021, 12, 625732. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Hu, R.; Zhang, C.; Ren, W.; Yu, A.; Zhou, X. Elevation of plasma angiotensin II level is a potential pathogenesis for the critically ill COVID-19 patients. Crit. Care 2020, 24, 290. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef] [Green Version]
- Reindl-Schwaighofer, R.; Hödlmoser, S.; Eskandary, F.; Poglitsch, M.; Bonderman, D.; Strassl, R.; Aberle, J.H.; Oberbauer, R.; Zoufaly, A.; Hecking, M. ACE2 Elevation in Severe COVID-19. Am. J. Resp. Critic. Care Med. 2021, 203, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Zoufaly, A.; Poglitsch, M.; Aberle, J.H.; Hoepler, W.; Seitz, T.; Traugott, M.; Grieb, A.; Pawelka, E.; Laferl, H.; Wenisch, C.; et al. Human Recombinant Soluble ACE2 in Severe COVID-19. Lancet Resp. Med. 2020, 8, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Minieri, C.A.; Ollerenshaw, J.D.; Alexander, R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994, 74, 1141–1148. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, S.; Kurz, S.; Münzel, T.; Tarpey, M.; Freeman, B.A.; Griendling, K.K.; Harrison, D.G. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J. Clin. Investig. 1996, 97, 1916–1923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dandona, P.; Kumar, V.; Aljada, A.; Ghanim, H.; Syed, T.; Hofmayer, D.; Mohanty, P.; Tripathy, D.; Garg, R. Angiotensin II receptor blocker valsartan suppresses reactive oxygen species generation in leukocytes, nuclear factor-kappa B, in mononuclear cells of normal subjects: Evidence of an antiinflammatory action. J. Clin. Endocrinol. Metab. 2003, 88, 4496–4501. [Google Scholar] [CrossRef] [Green Version]
- Duprez, D.A. Role of the renin-angiotensin-aldosterone system in vascular remodeling and inflammation: A clinical review. J. Hypertens. 2006, 24, 983–991. [Google Scholar] [CrossRef]
- Ratcliffe, P.J. Oxygen sensing and hypoxia signalling pathways in animals: The implications of physiology for cancer. J. Physiol. 2013, 591, 2027–2042. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.-W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxiainducible factor 1 is a basic-helix-loop-helixPAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [Green Version]
- Kallio, P.J.; Okamoto, K.; O’Brien, S.; Carrero, P.; Makino, Y.; Tanaka, H.; Poellinger, L. Signal transduction in hypoxic cells: Inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1α. EMBO J. 1998, 17, 6573–6586. [Google Scholar] [CrossRef]
- Dames, S.A.; Martinez-Yamout, M.; De Guzman, R.N.; Dyson, H.J.; Wright, P.E. Structural basis for Hif-1α/CBP recognition in the cellular hypoxic response. Proc. Natl. Acad. Sci. USA 2002, 99, 5271–5276. [Google Scholar] [CrossRef] [Green Version]
- Gesang, L.; Liu, G.; Cen, W.; Qiu, C.; Zhuoma, C.; Zhuang, L.; Ren, D.; Pincuo, Z.; Chan, Y. Angiotensinconverting enzyme gene polymorphism and its association with essential hypertension in a Tibetan population. Hypertens. Res. 2002, 25, 481–485. [Google Scholar] [CrossRef] [Green Version]
- Beall, C.M.; Cavalleri, G.L.; Deng, L.; Elston, R.C.; Gao, Y.; Knight, J.; Li, C.; Li, J.C.; Liang, Y.; McCormack, M.; et al. Natural selection on EPAS1 (HIF2α) associated with low hemoglobin concentration in Tibetan highlanders. Proc. Natl. Acad. Sci. USA 2010, 107, 11459–11464. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. HIF-1: Mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol. 2000, 88, 1474–1480. [Google Scholar] [CrossRef] [Green Version]
- Seta, K.A.; Yuan, Y.; Spicer, Z.; Lu, G.; Bedard, J.; Ferguson, T.K.; Pathrose, P.; Cole-Strauss, A.; Kaufhold, A.; Millhorn, D.E. The role of calcium in hypoxia-induced signal transduction and gene expression. Cell Calcium 2004, 36, 331–340. [Google Scholar] [CrossRef]
- Hatano, N.; Itoh, Y.; Suzuki, H.; Muraki, Y.; Hayashi, H.; Onozaki, K.; Wood, I.C.; Beech, D.J.; Muraki, K. Hypoxia-inducible factor-1α (HIF1α) switches on transient receptor potential ankyrin repeat 1 (TRPA1) gene expression via a hypoxia response element-like motif to modulate cytokine release. J. Biol. Chem. 2012, 287, 31962–31972. [Google Scholar] [CrossRef] [Green Version]
- Danta, C.C. SARS-CoV-2, Hypoxia, and Calcium Signaling: The Consequences and therapeutic options. ACS Pharmacol. Trans. Sci. 2021, 4, 400–402. [Google Scholar] [CrossRef]
- Mori, Y.; Takahashi, N.; Kurokawa, T.; Kiyonaka, S. TRP channels in oxygen physiology: Distinctive functional properties and roles of TRPA1 in O2 sensing. Proc. Jpn. Acad. 2017, 93, 464–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Arif, G.; Khazaal, S.; Farhat, A.; Harb, J.; Annweiler, C.; Wu, Y.; Cao, Z.; Kovacic, H.; Khattar, Z.A.; Fajloun, Z.; et al. Angiotensin II Type I Receptor (AT1R): The gate towards COVID-19-associated diseases. Molecules 2022, 27, 2048. [Google Scholar] [CrossRef] [PubMed]
- Attiq, A.; Yao, L.J.; Afzal, S.; Khan, M.A. The triumvirate of NF-κB, inflammation and cytokine storm in COVID-19. Int. Immunopharmacol. 2021, 101, 108255. [Google Scholar] [CrossRef] [PubMed]
- Cron, R.Q.; Caricchio, R.; Chatham, W.W. Calming the cytokine storm in COVID-19. Nat. Med. 2021, 27, 1672–1678. [Google Scholar] [CrossRef]
- Zanza, C.; Romenskaya, T.; Manetti, A.C.; Franceschi, F.; La Russa, R.; Bertozzi, G.; Maiese, A.; Savioli, G.; Volonnino, G.; Longhitano, Y. Cytokine storm in COVID-19: Immunopathogenesis and theory. Medicina 2022, 58, 144. [Google Scholar] [CrossRef]
- Liu, F.; Li, L.; Xu, M.; Wu, J.; Luo, D.; Zhu, Y.; Li, B.; Song, X.; Zhou, X. Prognostic value of interleukin-6, C-reactive protein, and procalcitonin in patients with COVID-19. J. Clin. Virol. 2020, 127, 104370. [Google Scholar] [CrossRef]
- William, C.; Koehne, P.; Jürgensen, J.S.; Gräfe, M.; Wagner, K.D.; Bachmann, S.; Frei, U.; Eckardt, K.W. Tie2 receptor expression ss stimulated by hypoxia and proinflammatory cytokines in human endothelial cells. Circ. Res. 2000, 87, 370–377. [Google Scholar] [CrossRef]
- Melhorn, J.; Alamoudi, A.; Mentzer, A.J.; Fraser, E.; Fries, A.; Cassar, M.P.; Kwok, A.; Knight, J.C.; Raman, B.; Talbot, N.P.; et al. Persistence of inflammatory and vascular mediators five months after hospitalisation with COVID-19 infection. Front. Med. 2023, 10, 1056506. [Google Scholar] [CrossRef]
- Eguchi, S.; Kawai, T.; Scalia, R.; Rizzo, V. Understanding angiotensin II type 1 receptor signaling in vascular pathophysiology. Hypertension 2018, 71, 804–810. [Google Scholar] [CrossRef]
- Lam, S.Y.; Fung, M.-L.; Leung, P.S. Regulation of the angiotensinconverting enzyme activity by a time-course hypoxia in the carotid body. J. Appl. Physiol. 2004, 96, 809–813. [Google Scholar] [CrossRef]
- Morrell, N.W.; A Higham, M.; Phillips, P.G.; Shakur, B.H.; Robinson, P.J.; Beddoes, R.J. Pilot study of losartan for pulmonary hypertension in chronic obstructive pulmonary disease. Respir. Res. 2005, 6, 88. [Google Scholar] [CrossRef] [Green Version]
- Morrell, N.W.; Morris, K.G.; Stenmark, K.R. Role of angiotensin converting enzyme and angiotensin II in development of hypoxic pulmonary hypertension. Am. J. Physiol. 1995, 269, H1186–H1194. [Google Scholar] [CrossRef]
- Koka, V.; Huang, X.R.; Chung, A.C.; Wang, W.; Truong, L.D.; Lan, H.Y. Angiotensin II Up-Regulates Angiotensin I Converting Enzyme (ACE), but Down-Regulates ACE2 via the AT1-ERK/p38 MAP Kinase Pathway. Am. J. Pathol. 2008, 172, 1174–1183. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Wu, Y.; Zhao, M.; Liu, C.; Zhou, L.; Shen, S.; Liao, S.; Yang, K.; Li, Q.; Wan, H. Role of HIF-1 in the regulation ACE and ACE2 expression in hypoxic human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L631–L640. [Google Scholar] [CrossRef]
- Fazeli, G.; Stopper, H.; Schinzel, R.; Ni, C.-W.; Jo, H.; Schupp, N. Angiotensin II induces DNA damage via AT1 receptor and NADPH oxidase isoform Nox4. Mutagenesis 2012, 27, 673–681. [Google Scholar] [CrossRef]
- Park, J.-M.; Do, V.Q.; Seo, Y.-S.; Kim, H.J.; Nam, J.H.; Yin, M.Z.; Kim, H.J.; Kim, S.J.; Griendling, K.K.; Lee, M.-Y. NADPH Oxidase 1 Mediates Acute Blood Pressure Response to Angiotensin II by Contributing to Calcium Influx in Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2022, 42, e117–e130. [Google Scholar] [CrossRef]
- Richard, D.E.; Berra, E.; Gothié, E.; Pouysségur, J.R. p42/p44 Mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1a (HIF-1a) and enhance the transcriptional Activity of HIF-1. J. Biol. Chem. 1999, 274, 32631–32637. [Google Scholar] [CrossRef] [Green Version]
- McClendon, J.; Jansing, N.L.; Redente, E.F.; Gandjeva, A.; Ito, Y.; Colgan, S.P.; Ahmad, A.; Riches, D.W.; Chapman, H.A.; Mason, R.J.; et al. Hypoxia-inducible factor 1α signaling promotes repair of the alveolar epithelium after acute lung injury. Am. J. Pathol. 2017, 187, 1772–1786. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Wang, Z.; Xia, M.; Li, P.-L.; Van Tassell, B.W.; Abbate, A.; Dhaduk, R.; Li, N. Silencing of hypoxia-inducible factor-1α gene attenuated angiotensin II-induced renal injury in Sprague-Dawley rats. Hypertension 2011, 58, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Imperio, G.E.; Lye, P.; Mughis, H.; Hamada, H.; Bloise, E.; Lye, S.J.; Matthews, S.G. Hypoxia alters the expression of ACE2 and TMPRSS2 SARS-CoV-2 cell entry mediators in hCMEC/D3 brain endothelial cells. Microvasc. Res. 2021, 138, 104232. [Google Scholar] [CrossRef]
- Zakheim, R.M.; Molteni, A.; Mattioli, L.; Park, M. Plasma angiotensin II levels in hypoxic and hypovolemic stress in unanesthetized rabbits. J. Appl. Physiol. 1976, 41, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. The impact of the hypoxia-VEGF-vascular permeability on COVID-19-infected patients. Exploration 2021, 1, 20210051. [Google Scholar] [CrossRef] [PubMed]
- Afsar, B.; Kanbay, M.; Afsar, R.E. Hypoxia inducible factor-1 protects against COVID-19: A hypothesis. Med. Hypothesis 2020, 143, 109857. [Google Scholar] [CrossRef] [PubMed]
- Foster, G.E.; Hanly, P.J.; Ahmed, S.B.; Beaudin, A.E.; Pialoux, V.; Poulin, M.J. Intermittent Hypoxia Increases Arterial Blood Pressure in Humans Through a Renin-Angiotensin System–Dependent Mechanism. Hypertension 2010, 56, 369–377. [Google Scholar] [CrossRef]
- Takahashi, S.; Nakamura, Y.; Nishijima, T.; Sakurai, S.; Inoue, H. Essential roles of angiotensin II in vascular endothelial growth factor expression in sleep apnea syndrome. Respir. Med. 2005, 99, 1125–1131. [Google Scholar] [CrossRef] [Green Version]
- Wright, J.M. Choosing a first-line drug in the management of elevated blood pressure: What is the evidence? 3: Angiotensin-converting-enzyme inhibitors. CMAJ 2000, 163, 293–296. [Google Scholar]
- Mentz, R.J.; Bakris, G.L.; Waeber, B.; McMurray, J.J.; Gheorghiade, M.; Ruilope, L.M.; Maggioni, A.P.; Swedberg, K.; Piña, I.L.; Fiuzat, M.; et al. The past, present and future of renin-angiotensin aldosterone system inhibition. Int. J. Cardiol. 2013, 167, 1677–1687. [Google Scholar] [CrossRef] [Green Version]
- Schiffrin, E.L.; Flack, J.M.; Ito, S.; Muntner, P.; Webb, R.C. Hypertension and COVID-19. Am. J. Hypertens. 2020, 33, 373–374. [Google Scholar] [CrossRef] [Green Version]
- Devaux, C.A. Are ACE inhibitors and ARBs more beneficial than harmful in the treatment of severe COVID-19 disease? J. Cardiovasc. Med. Cardiol. 2020, 7, 101–103. [Google Scholar] [CrossRef]
- de Souza, G.A.P.; Osman, I.O.; Le Bideau, M.; Baudoin, J.-P.; Jaafar, R.; Devaux, C.; La Scola, B. Angiotensin II receptor blockers (ARBs antihypertensive agents) increase replication of SARS-CoV-2 in Vero E6 Cells. Front. Cell. Infect. Microbiol. 2021, 11, 639177. [Google Scholar] [CrossRef]
- Shen, L.; Mo, H.; Cai, L.; Kong, T.; Zheng, W.; Ye, J.; Qi, J.; Xiao, Z. Losartan prevents sepsis-induced acute lung injury and decreases activation of nuclear factor kappaB and mitogen-activated protein kinases. Shock 2009, 31, 500–506. [Google Scholar] [CrossRef]
- Meng, J.; Xiao, G.; Zhang, J.; He, X.; Ou, M.; Bi, J.; Yang, R.; Di, W.; Wang, Z.; Li, Z.; et al. Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg. Microbes Infect. 2020, 9, 757–760. [Google Scholar] [CrossRef]
- Bean, D.M.; Kraljevic, Z.; Searle, T.; Bendayan, R.; Kevin, O.; Pickles, A.; Folarin, A.; Roguski, L.; Noor, K.; Shek, A.; et al. Angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers are not associated with severe COVID-19 infection in a multi-site UK acute hospital trust. Eur. J. Heart Fail. 2020, 22, 967–974. [Google Scholar] [CrossRef]
- Zhang, P.; Zhu, L.; Cai, J.; Lei, F.; Qin, J.-J.; Xie, J.; Yuan, Y.; Loomba, R.; Liu, P.P.; Li, H.; et al. Association of inpatient use of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers with mortality among patients with hypertension hospitalized with COVID-19. Circ. Res. 2020, 126, 1671–1681. [Google Scholar] [CrossRef] [Green Version]
- Abbasi, J. Choose ARBs Over ACE Inhibitors for First-line Hypertension Treatment, Large New Analysis Suggests. JAMA. 2021, 326, 1244–1245. [Google Scholar] [CrossRef]
- Lopes, R.D.; Macedo, A.V.S.; de Barros, E.S.P.G.M.; Moll-Bernardes, R.J.; Dos Santos, T.M.; Mazza, L.; Feldman, A.; D’Andrea Saba Arruda, G.; de Albuquerque, D.C.; Camiletti, A.S.; et al. Continuing angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers on days alive and out of the hospital in patients admitted with COVID-19. A randomized clinical trial. JAMA 2021, 325, 254–264. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captoprilinsensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef] [Green Version]
- Descamps, G.; Verset, L.; Trelcat, A.; Hopkins, C.; Lechien, J.R.; Journe, F.; Saussez, S. ACE2 protein landscape in the head and neck region: The conundrum of SARS-CoV-2 infection. Biology 2020, 9, 235. [Google Scholar] [CrossRef]
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.D.; et al. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J. Pathol. 2020, 251, 228–248. [Google Scholar] [CrossRef]
- Henry, B.M.; Vikse, J.; Benoit, S.; Favaloro, E.J.; Lippi, G. Hyperinflammation and derangement of renin-angiotensin-aldosterone system in COVID-19: A novel hypothesis for clinically suspected hypercoagulopathy and microvascular immunothrombosis. Clin. Chim. Acta 2020, 507, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.J.; Vaughan, D.E. Prothrombotic effects of angiotensin. Adv. Intern. Med. 2000, 45, 419–429. [Google Scholar] [PubMed]
- Larsson, P.T.; Schwieler, J.H.; Wallen, N.H. Platelet activation during angiotensin II infusion in healthy volunteers. Blood Coag. Fibrinolysis 2000, 11, 61–69. [Google Scholar] [CrossRef]
- Fletcher-Sandersjöö, A.; Bellander, B.M. Is COVID-19 associated thrombosis caused by overactivation of the complement cascade? A literature review. Thromb. Res. 2020, 194, 36–41. [Google Scholar] [CrossRef]
- Gando, S.; Wada, T. Thromboplasminflammation in COVID-19 Coagulopathy: Three Viewpoints for Diagnostic and Therapeutic Strategies. Front. Immunol. 2021, 12, 649122. [Google Scholar] [CrossRef]
- Mogielnicki, A.; Kramkowski, K.; Hermanowicz, J.M.; Leszczynska, A.; Przyborowski, K.; Buczko, W. Angiotensin-(1-9) enhances stasisinduced venous thrombosis in the rat because of the impairment of fibrinolysis. J. Renin Angiotensin Aldosterone Syst. 2014, 15, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120. [Google Scholar] [CrossRef]
- Kallet, R.H.; Branson, R.D.; Lipnick, M.S. Respiratory drive, dyspnea, and silent hypoxemia: A physiological review in the context of COVID-19. Resp. Care 2022, 67, 1343–1360. [Google Scholar] [CrossRef]
- Carsana, L.; Sanzagni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: A two-Centre descriptive study. Lancet 2020, 20, 1135–1140. [Google Scholar] [CrossRef]
- Hofman, P.; Copin, M.C.; Tauziede-Espariat, A.; Adle-Biassette, H.; Fortarezza, F.; Passeron, T.; Salmon, I.; Calabrese, F. Histopathological features due to the SARS-CoV-2. Ann. Pathol. 2021, 41, 9–22. [Google Scholar] [CrossRef]
- Zubieta-Calleja, G.R.; Zubieta-DeUrioste, N.; Montelongo, F.D.J.; Sanchez, M.G.R.; Campoverdi, A.F.; Rocco, P.R.M.; Battaglini, D.; Ball, L.; Pelosi, P. Morphological and functional findings in COVID-19 lung disease as compared to Pneumonia, ARDS, and High-Altitude Pulmonary Edema. Respir. Physiol. Neurobiol. 2023, 309, 104000. [Google Scholar] [CrossRef]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.-H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Heamatol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Gustafsson, L.; Leone, A.; Persson, M.; Wiklund, N.; Moncada, S. Endogenous Nitric Oxide is present in the exaled air of rabbits, guinea pigs and humans. Biochem. Biophys. Res. Commun. 1991, 181, 852–857. [Google Scholar] [CrossRef]
- Scherrer, U.; Vollenweider, L.; Delabays, A.; Savcic, M.; Eichenberger, U.; Kleger, G.-R.; Fikrle, A.; Ballmer, P.E.; Nicod, P.; Bärtsch, P. Inhaled nitric oxide for high-altitude pulmonary edema. N. Engl. J. Med. 1996, 334, 624–629. [Google Scholar] [CrossRef] [Green Version]
- Brito-Azevedo, A.; Costa Pinto, E.; de Cata Preta Corrêa, G.A.; Bouskela, E. SARS-CoV-2 infection causes pulmonary shunt by vasodilatation. J. Med. Virol. 2021, 93, 573–575. [Google Scholar] [CrossRef]
- Alamé, K.; Lemaitre, E.L.; Vuillaume, L.A.; Noizet, M.; Gottwalles, Y.; Chouihed, T.; Lavoignet, C.-E.; Bérard, L.; Molter, L.; Gennai, S.; et al. Silent hypoxemia in the emergency department: A retrospective cohort of two clinical phenotypes in critical COVID-19. J. Clin. Med. 2022, 11, 5034. [Google Scholar] [CrossRef]
- Ssentongo, A.E.; Ssentongo, P.; Heilbrunn, E.S.; Lekoubou, A.; Du, P.; Liao, D.; Oh, J.S.; Chinchilli, V.M. Renin–angiotensin–aldosterone system inhibitors and the risk of mortality in patients with hypertension hospitalised for COVID-19: Systematic review and meta-analysis. Open Heart 2020, 7, e001353. [Google Scholar] [CrossRef]
- Wing, P.A.C.; Keeley, T.P.; Zhuang, X.; Lee, J.Y.; Prange-Barczynska, M.; Tsukuda, S.; Morgan, S.B.; Harding, A.C.; Argles, I.L.; Kurlekar, S.; et al. Hypoxic and pharmacological activation of HIF inhibits SARS-CoV-2 infection of lung epithelial cells. Cell Rep. 2021, 35, 109020. [Google Scholar] [CrossRef]
- Wing, P.A.C.; Prange-Barczynska, M.; Cross, A.; Crotta, S.; Rubio, C.O.; Cheng, X.; Harris, J.M.; Zhuang, X.; Johnson, R.L.; Ryan, K.A.; et al. Hypoxia inducible factors regulate infectious SARS-CoV-2, epithelial damage and respiratory symptoms in a hamster COVID-19 model. PLoS Pathog. 2022, 18, e1010807. [Google Scholar] [CrossRef]
- Tian, M.; Liu, W.; Li, X.; Zhao, P.; Shereen, M.A.; Zhu, C.; Huang, S.; Liu, S.; Yu, X.; Yue, M.; et al. HIF-1α promotes SARS-CoV-2 infection and aggravates inflammatory responses to COVID-19. Signal Transduct. Target. Therapy 2021, 6, 308. [Google Scholar] [CrossRef]
- Zalpoor, H.; Akbari, A.; Nabi-Afjadi, M.; Forghaniesfidvajani, R.; Tavakol, C.; Barzegar, Z.; Iravanpour, F.; Hosseini, M.; Mousavi, S.R.; Farrokhi, M.R. Hypoxia-inducible factor 1 alpha (HIF-1α) stimulated and P2X7 receptor activated by COVID-19, as a potential therapeutic target and risk factor for epilepsy. Hum. Cell 2022, 35, 1338–1345. [Google Scholar] [CrossRef] [PubMed]
- Kotagiri, P.; Mescia, F.; Hanson, A.L.; Turner, L.; Bergamaschi, L.; Peñalver, A.; Richoz, N.; Moore, S.D.; Ortmann, B.M.; Dunmore, B.J.; et al. The impact of hypoxia on B cells in COVID-19. eBioMedicine 2022, 77, 103878. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Devaux, C.A.; Lagier, J.-C. Unraveling the Underlying Molecular Mechanism of ‘Silent Hypoxia’ in COVID-19 Patients Suggests a Central Role for Angiotensin II Modulation of the AT1R-Hypoxia-Inducible Factor Signaling Pathway. J. Clin. Med. 2023, 12, 2445. https://doi.org/10.3390/jcm12062445
Devaux CA, Lagier J-C. Unraveling the Underlying Molecular Mechanism of ‘Silent Hypoxia’ in COVID-19 Patients Suggests a Central Role for Angiotensin II Modulation of the AT1R-Hypoxia-Inducible Factor Signaling Pathway. Journal of Clinical Medicine. 2023; 12(6):2445. https://doi.org/10.3390/jcm12062445
Chicago/Turabian StyleDevaux, Christian Albert, and Jean-Christophe Lagier. 2023. "Unraveling the Underlying Molecular Mechanism of ‘Silent Hypoxia’ in COVID-19 Patients Suggests a Central Role for Angiotensin II Modulation of the AT1R-Hypoxia-Inducible Factor Signaling Pathway" Journal of Clinical Medicine 12, no. 6: 2445. https://doi.org/10.3390/jcm12062445