
Relationship between Cholesterol-Related Lipids and Severe Acute Pancreatitis: From Bench to Bedside

 , ,
, , 
 , ,
, , 
Abstract
:Highlights
- Higher serum levels of total cholesterol and low-density lipoprotein cholesterol are associated with the severity of AP, while the persistent inflammation of AP is linked with decreased serum levels of cholesterol-related lipids.
- Cholesterol-related lipids should be recommended both as risk factors and early predictors for studying the severity of AP.
- Cholesterol-lowering drugs may play a role in the treatment and prevention of AP with hypercholesterolemia.
Abstract
1. Introduction
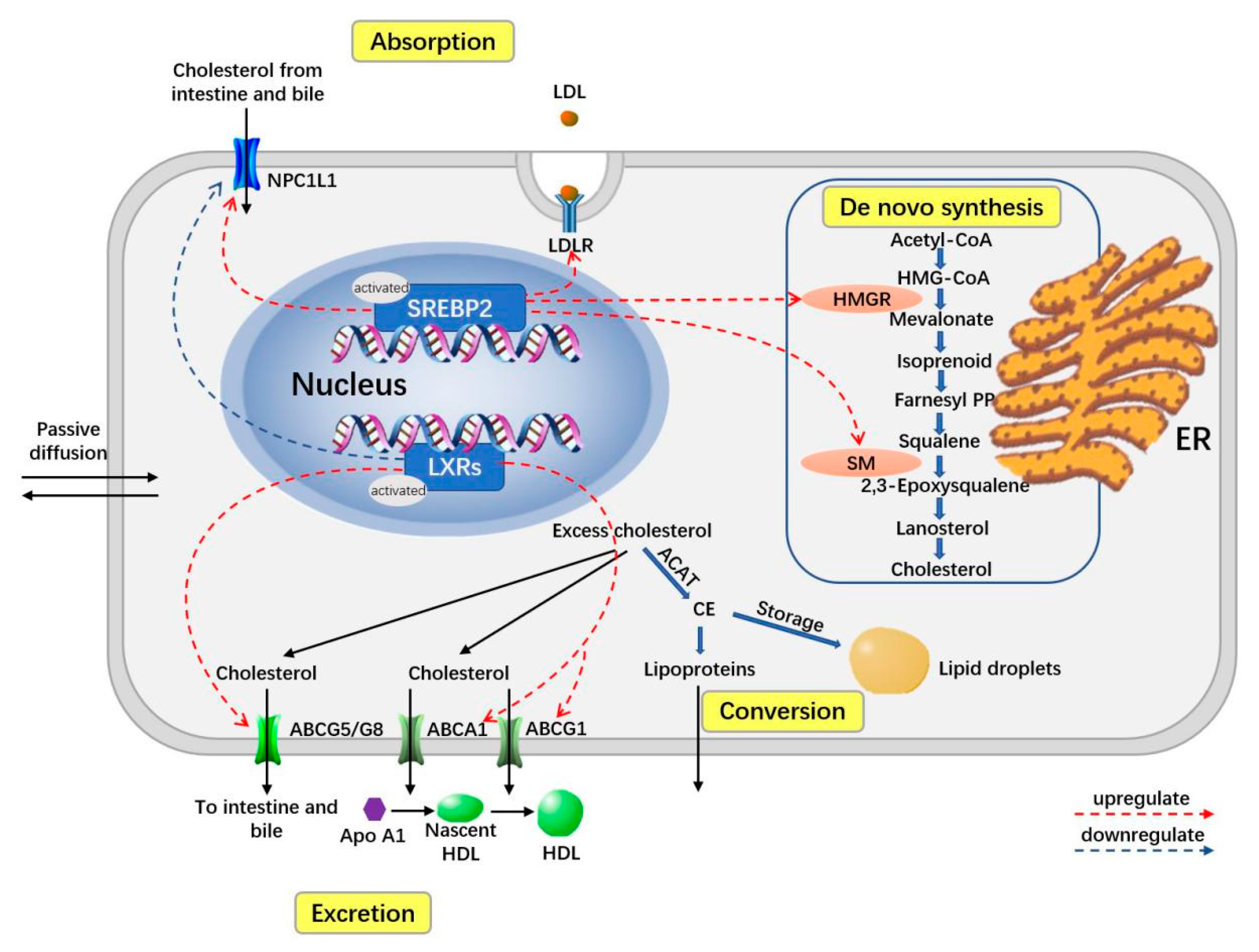
2. Overview of Homeostasis of Cholesterol-Related Body Lipids
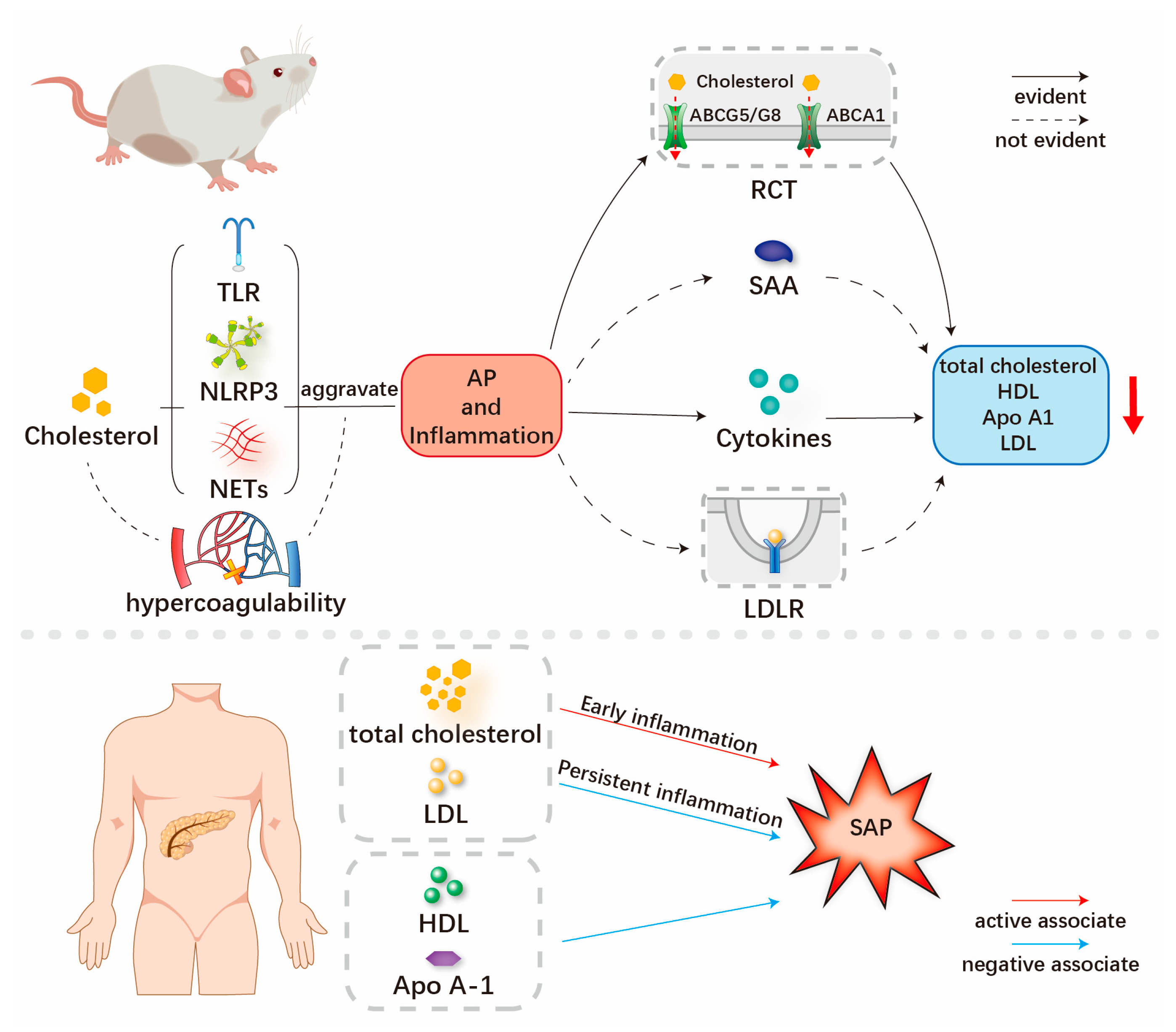
3. Mechanism of Interaction between Cholesterol-Related Lipids, Inflammation and AP
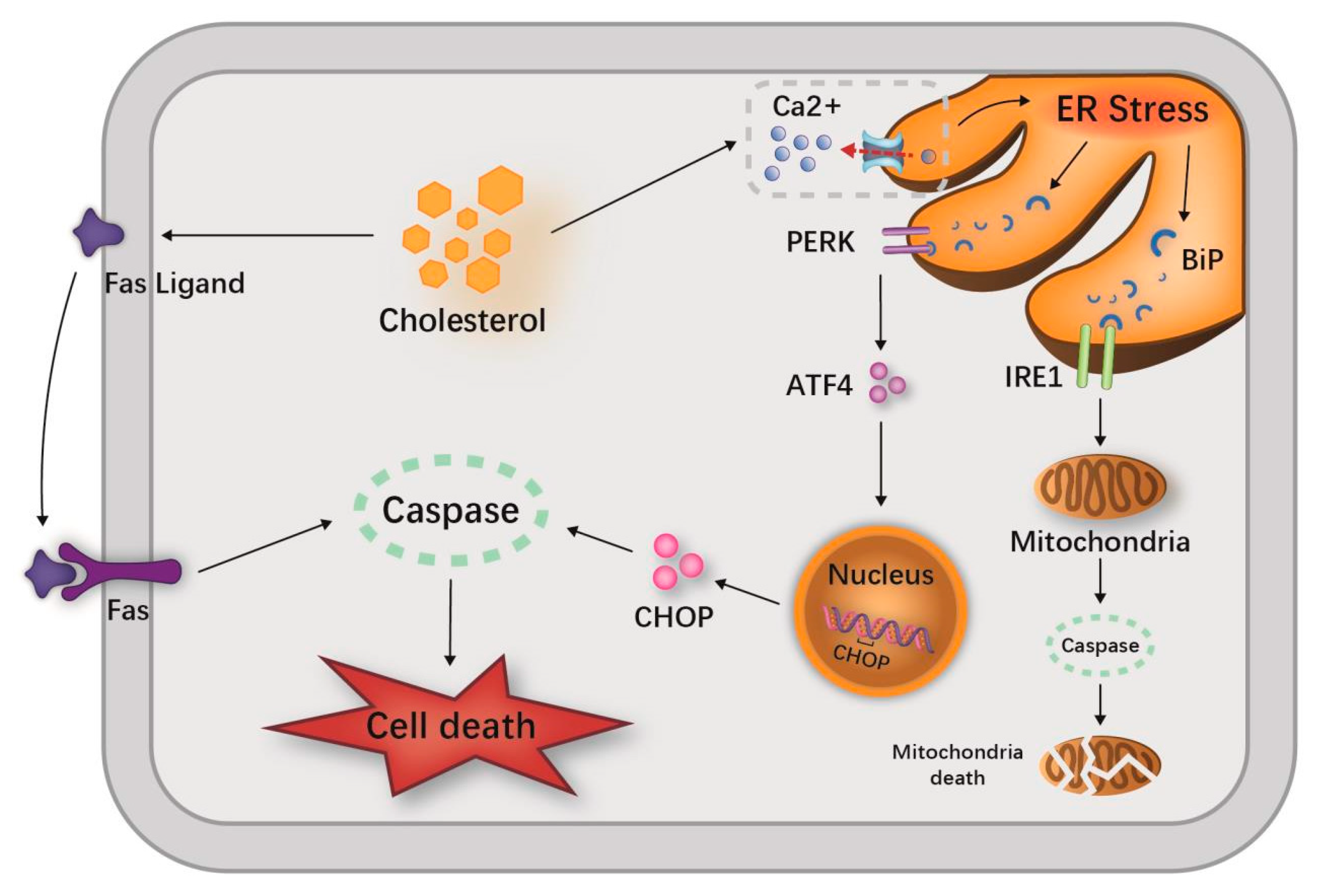
3.1. Hypercholesterolemia, LDL-C May Aggravate the Inflammation and Severity of SAP
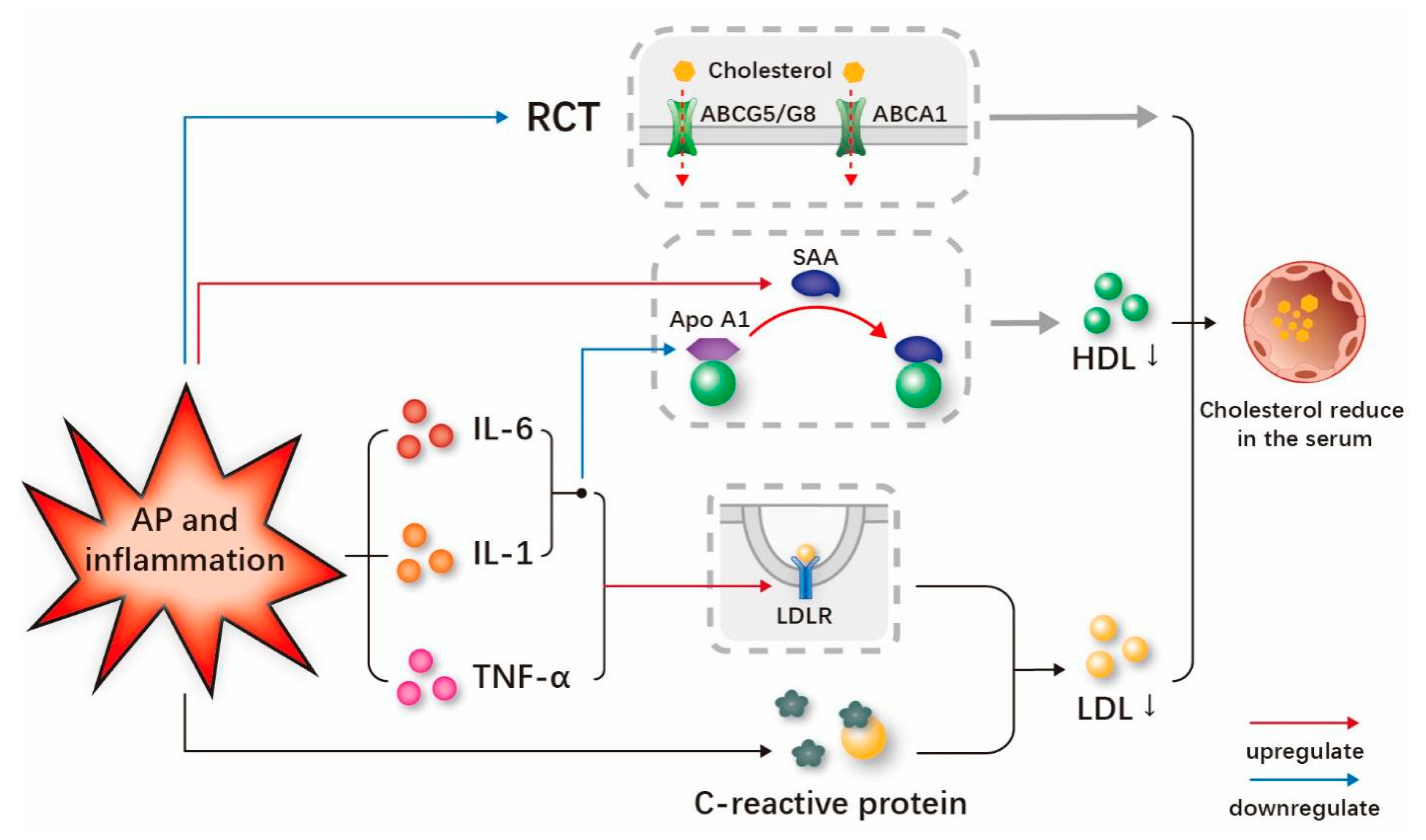
3.2. Persistent Inflammation and AP May Reduce the Serum Cholesterol-Related Lipids
4. Clinical Results of Relationships between Cholesterol-Related Lipids and AP
4.1. Clinical Results on the Relationship between Total Cholesterol and SAP
4.2. Clinical Results of Relationships between LDL-C and SAP
4.3. Clinical Results of Relationships between HDL-C, Apo A1 and SAP
5. Clinical Significance and Future Prospects
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| ACAT | acyl-coenzyme A: cholesterol acyltransferase |
| AP | acute pancreatitis |
| Apo | apolipoprotein |
| ATP | adenosine triphosphate |
| CHOP | CEBP/EBP homologous protein |
| ER | endoplasmic reticulum |
| HDL-C | high-density lipoprotein cholesterol |
| HMG-CoA | acetyl-CoA to 3-hydroxy-3-methylglutaryl-CoA |
| HMGR | HMG-CoA reductase |
| IDL-C | intermediate density lipoprotein cholesterol |
| IL | interleukin |
| LDL-C | low-density lipoprotein cholesterol |
| LPS | lipopolysaccharide |
| LXRs | liver X receptors |
| NETs | neutrophil extracellular traps |
| NF-κB | nuclear factor kappa-B |
| NLRP | NOD-like receptor thermal protein domain associated protein |
| NPC1L1 | Niemann–Pick C1 like 1 |
| RCT | reverse cholesterol transport |
| ROS | reactive oxygen species |
| SAA | Serum amyloid A |
| SAP | severe acute pancreatitis |
| SREBP2 | sterol regulatory element-binding protein-2 |
| TH17 | T-helper cell 17 |
| TLR | Toll-like receptor |
| TNF-α | tumor necrosis factor-α |
| UPR | unfolded protein responses |
| VLDL-C | very low-density lipoprotein cholesterol |
References
- Luo, J.; Yang, H.; Song, B.-L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Ballester, M.; Herrero-Cervera, A.; Vinué, Á.; Martínez-Hervás, S.; González-Navarro, H. Impact of Cholesterol Metabolism in Immune Cell Function and Atherosclerosis. Nutrients 2020, 12, 2021. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, D.; Perucha, E. Cholesterol metabolism: A new molecular switch to control inflammation. Clin. Sci. 2021, 135, 1389–1408. [Google Scholar] [CrossRef]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, J.Y.L. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yvan-Charvet, L.; Bonacina, F.; Guinamard, R.R.; Norata, G.D. Immunometabolic function of cholesterol in cardiovascular disease and beyond. Cardiovasc. Res. 2019, 115, 1393–1407. [Google Scholar] [CrossRef]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.J.; Papachristou, G.I. New insights into acute pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 479–496. [Google Scholar] [CrossRef]
- Li, G.; Wu, X.; Yang, L.; He, Y.; Liu, Y.; Jin, X.; Yuan, H. TLR4-mediated NF-κB signaling pathway mediates HMGB1-induced pancreatic injury in mice with severe acute pancreatitis. Int. J. Mol. Med. 2016, 37, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Abdelmageed, M.E.; Nader, M.A.; Zaghloul, M.S. Targeting HMGB1/TLR4/NF-κB signaling pathway by protocatechuic acid protects against l-arginine induced acute pancreatitis and multiple organs injury in rats. Eur. J. Pharmacol. 2021, 906, 174279. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Chong, E.M.; Pendharkar, S.; Hong, J.; Windsor, J.A.; Ke, L.; Li, W.; Phillips, A. The Effects of NLRP3 Inflammasome Inhibition in Experimental Acute Pancreatitis: A Systematic Review and Meta-Analysis. Pancreas 2022, 51, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Zimmer, V.; Stock, S.; Zippi, M.; Omoshoro-Jones, J.A.; Zhou, M. Relationship between low-density lipoprotein cholesterol and severe acute pancreatitis (“the lipid paradox”). Ther. Clin. Risk Manag. 2018, 14, 981–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, W.; Zimmer, V.; Basharat, Z.; Zippi, M.; Stock, S.; Geng, W.; Bao, X.; Dong, J.; Pan, J.; Zhou, M. Association of total cholesterol with severe acute pancreatitis: A U-shaped relationship. Clin. Nutr. 2020, 39, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.L.; Zhang, C.-H.; Zhao, X.-Y.; Chen, S.-H.; Liang, H.-J.; Hu, C.-L.; Chen, N.-W. Early prediction of persistent organ failure by serum apolipoprotein A-I and high-density lipoprotein cholesterol in patients with acute pancreatitis. Clin. Chim. Acta 2018, 476, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Innerarity, T.L.; Rall, S.C., Jr.; Weisgraber, K.H. Plasma lipoproteins: Apolipoprotein structure and function. J. Lipid Res. 1984, 25, 1277–1294. [Google Scholar] [CrossRef]
- Sèdes, L.; Thirouard, L.; Maqdasy, S.; Garcia, M.; Caira, F.; Lobaccaro, J.-M.A.; Beaudoin, C.; Volle, D.H. Cholesterol: A Gatekeeper of Male Fertility? Front. Endocrinol. 2018, 9, 369. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Liu, J.; Zhao, K.; Gao, L.; Zhao, J. Cholesterol-induced toxicity: An integrated view of the role of cholesterol in multiple diseases. Cell Metab. 2021, 33, 1911–1925. [Google Scholar] [CrossRef]
- Yu, X.-H.; Qian, K.; Jiang, N.; Zheng, X.-L.; Cayabyab, F.S.; Tang, C.-K. ABCG5/ABCG8 in cholesterol excretion and atherosclerosis. Clin. Chim. Acta 2014, 428, 82–88. [Google Scholar] [CrossRef]
- Garcia-Ruiz, C.; de la Rosa, L.C.; Ribas, V.; Fernandez-Checa, J.C. Mitochondrial Cholesterol and Cancer. Semin. Cancer Biol. 2021, 73, 76–85. [Google Scholar] [CrossRef]
- Repa, J.J.; Mangelsdorf, D.J. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu. Rev. Cell Dev. Biol. 2000, 16, 459–481. [Google Scholar] [CrossRef] [PubMed]
- Škara, L.; Turković, A.H.; Pezelj, I.; Vrtarić, A.; Sinčić, N.; Krušlin, B.; Ulamec, M. Prostate Cancer—Focus on Cholesterol. Cancers 2021, 13, 4696. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhou, Y.; Goldstein, J.L.; Brown, M.S.; Radhakrishnan, A. Cholesterol-induced conformational changes in the sterol-sensing domain of the Scap protein suggest feedback mechanism to control cholesterol synthesis. J. Biol. Chem. 2017, 292, 8729–8737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrero-Andrés, A.; Panisello-Roselló, A.; Roselló-Catafau, J.; Folch-Puy, E. NLRP3 Inflammasome-Mediated Inflammation in Acute Pancreatitis. Int. J. Mol. Sci. 2020, 21, 5386. [Google Scholar] [CrossRef]
- Zheng, H.; Wang, D.; Wang, X.; Lin, Y.; Lu, Z.; Chen, Y.; Feng, G.; Yang, N. Dynamic changes of lipid profile in severe hypertriglyceridemia-induced acute pancreatitis patients under double filtration plasmapheresis: A retrospective observational study. Lipids Health Dis. 2020, 19, 206. [Google Scholar] [CrossRef]
- Triantafilou, M.; Miyake, K.; Golenbock, D.T.; Triantafilou, K. Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J. Cell Sci. 2002, 115, 2603–2611. [Google Scholar] [CrossRef]
- van der Vorst, E.P.C.; Theodorou, K.; Wu, Y.; Hoeksema, M.A.; Goossens, P.; Bursill, C.A.; Aliyev, T.; Huitema, L.F.A.; Tas, S.W.; Wolfs, I.M.J.; et al. High-Density Lipoproteins Exert Pro-inflammatory Effects on Macrophages via Passive Cholesterol Depletion and PKC-NF-κB/STAT1-IRF1 Signaling. Cell Metab. 2017, 25, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Castrillo, A.; Joseph, S.B.; Vaidya, S.A.; Haberland, M.; Fogelman, A.M.; Cheng, G.; Tontonoz, P. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol. Cell 2003, 12, 805–816. [Google Scholar] [CrossRef]
- Dumnicka, P.; Maduzia, D.; Ceranowicz, P.; Olszanecki, R.; Drożdż, R.; Kuśnierz-Cabala, B. The Interplay between Inflammation, Coagulation and Endothelial Injury in the Early Phase of Acute Pancreatitis: Clinical Implications. Int. J. Mol. Sci. 2017, 18, 354. [Google Scholar] [CrossRef] [Green Version]
- Elwakkad, A.S.; Mohamed, S.I.; Fathalla, M. Relation between hypercholesterolaemia and vascular endothelial microinflammation. East. Mediterr. Health J. 2007, 13, 515–521. [Google Scholar]
- Pirro, M.; Bagaglia, F.; Paoletti, L.; Razzi, R.; Mannarino, M.R. Hypercholesterolemia-associated endothelial progenitor cell dysfunction. Ther. Adv. Cardiovasc. Dis. 2008, 2, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.Y.; Protty, M.B.; Davies, I.G.; Lip, G.Y.H. Relationship between lipoproteins, thrombosis, and atrial fibrillation. Cardiovasc. Res. 2022, 118, 716–731. [Google Scholar] [CrossRef] [PubMed]
- Hendrikx, T.; Walenbergh, S.M.A.; Hofker, M.H.; Shiri-Sverdlov, R. Lysosomal cholesterol accumulation: Driver on the road to inflammation during atherosclerosis and non-alcoholic steatohepatitis. Obes. Rev. 2014, 15, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Westerterp, M.; Fotakis, P.; Ouimet, M.; Bochem, A.E.; Zhang, H.; Molusky, M.M.; Wang, W.; Abramowicz, S.; la Bastide-van Gemert, S.; Wang, N.; et al. Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis. Circulation 2018, 138, 898–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajamäki, K.; Lappalainen, J.; Öörni, K.; Välimäki, E.; Matikainen, S.; Kovanen, P.T.; Eklund, K.K. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: A novel link between cholesterol metabolism and inflammation. PLoS ONE 2010, 5, e11765. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Chi, Z.; Jiang, D.; Xu, T.; Yu, W.; Wang, Z.; Chen, S.; Zhang, L.; Liu, Q.; Guo, X.; et al. Cholesterol Homeostatic Regulator SCAP-SREBP2 Integrates NLRP3 Inflammasome Activation and Cholesterol Biosynthetic Signaling in Macrophages. Immunity 2018, 49, 842–856.e7. [Google Scholar] [CrossRef] [Green Version]
- Grebe, A.; Latz, E. Cholesterol crystals and inflammation. Curr. Rheumatol. Rep. 2013, 15, 313. [Google Scholar] [CrossRef] [Green Version]
- Catapano, A.L.; Pirillo, A.; Norata, G.D. Vascular inflammation and low-density lipoproteins: Is cholesterol the link? A lesson from the clinical trials. Br. J. Pharmacol. 2017, 174, 3973–3985. [Google Scholar] [CrossRef] [Green Version]
- Latz, E. The inflammasomes: Mechanisms of activation and function. Curr. Opin. Immunol. 2010, 22, 28–33. [Google Scholar] [CrossRef] [Green Version]
- Neumann, A.; Brogden, G.; Jerjomiceva, N.; Brodesser, S.; Naim, H.Y.; von Köckritz-Blickwede, M. Lipid alterations in human blood-derived neutrophils lead to formation of neutrophil extracellular traps. Eur. J. Cell Biol. 2014, 93, 347–354. [Google Scholar] [CrossRef]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerasimenko, J.V.; Gryshchenko, O.; Ferdek, P.E.; Stapleton, E.; Hébert, T.O.G.; Bychkova, S.; Peng, S.; Begg, M.; Gerasimenko, O.V.; Petersen, O.H. Ca2+ release-activated Ca2+ channel blockade as a potential tool in antipancreatitis therapy. Proc. Natl. Acad. Sci. USA 2013, 110, 13186–13191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biczo, G.; Vegh, E.T.; Shalbueva, N.; Mareninova, O.A.; Elperin, J.; Lotshaw, E.; Gretler, S.; Lugea, A.; Malla, S.R.; Dawson, D.; et al. Mitochondrial Dysfunction, Through Impaired Autophagy, Leads to Endoplasmic Reticulum Stress, Deregulated Lipid Metabolism, and Pancreatitis in Animal Models. Gastroenterology 2018, 154, 689–703. [Google Scholar] [CrossRef] [Green Version]
- Aghdassi, A.A.; John, D.S.; Sendler, M.; Weiss, F.U.; Reinheckel, T.; Mayerle, J.; Lerch, M.M. Cathepsin D regulates cathepsin B activation and disease severity predominantly in inflammatory cells during experimental pancreatitis. J. Biol. Chem. 2018, 293, 1018–1029. [Google Scholar] [CrossRef] [Green Version]
- Mutua, V.; Gershwin, L.J. A Review of Neutrophil Extracellular Traps (NETs) in Disease: Potential Anti-NETs Therapeutics. Clin. Rev. Allergy Immunol. 2021, 61, 194–211. [Google Scholar] [CrossRef] [PubMed]
- Hoque, R.; Sohail, M.; Malik, A.; Sarwar, S.; Luo, Y.; Shah, A.; Barrat, F.; Flavell, R.; Gorelick, F.; Husain, S.; et al. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology 2011, 141, 358–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, J.; Ren, Y.; Yang, X.; Li, X.; Xia, L.; Lu, N. The Role of Neutrophils and Neutrophil Extracellular Traps in Acute Pancreatitis. Front. Cell Dev. Biol. 2020, 8, 565758. [Google Scholar] [CrossRef]
- Feng, B.; Yao, P.M.; Li, Y.; Devlin, C.M.; Zhang, D.; Harding, H.P.; Sweeney, M.; Rong, J.X.; Kuriakose, G.; Fisher, E.A.; et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 2003, 5, 781–792. [Google Scholar] [CrossRef]
- Bretscher, M.S.; Munro, S. Cholesterol and the Golgi apparatus. Science 1993, 261, 1280–1281. [Google Scholar] [CrossRef]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Investig. 2002, 110, 1389–1398. [Google Scholar] [CrossRef]
- Kaufman, R.J.; Scheuner, D.; Schröder, M.; Shen, X.; Lee, K.; Liu, C.Y.; Arnold, S.M. The unfolded protein response in nutrient sensing and differentiation. Nat. Rev. Mol. Cell Biol. 2002, 3, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kaufman, R.J. Unfolding the toxicity of cholesterol. Nat. Cell Biol. 2003, 5, 769–770. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, D.; Song, B.; McEwen, E.; Liu, C.; Laybutt, R.; Gillespie, P.; Saunders, T.; Bonner-Weir, S.; Kaufman, R.J. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 2001, 7, 1165–1176. [Google Scholar] [CrossRef]
- Gukovskaya, A.S.; Pandol, S.J.; Gukovsky, I. New insights into the pathways initiating and driving pancreatitis. Curr. Opin. Gastroenterol. 2016, 32, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Yuan, H.-X.; Guan, K.-L. Autophagy regulation by nutrient signaling. Cell Res. 2014, 24, 42–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, H. Regulation of Autophagy by mTOR Signaling Pathway. Adv. Exp. Med. Biol. 2019, 1206, 67–83. [Google Scholar] [CrossRef]
- Mareninova, O.A.; Vegh, E.T.; Shalbueva, N.; Wightman, C.J.; Dillon, D.L.; Malla, S.; Xie, Y.; Takahashi, T.; Rakonczay, Z., Jr.; French, S.W.; et al. Dysregulation of mannose-6-phosphate-dependent cholesterol homeostasis in acinar cells mediates pancreatitis. J. Clin. Investig. 2021, 131, e146870. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, W.-X.; Li, T. Cholesterol and bile acid-mediated regulation of autophagy in fatty liver diseases and atherosclerosis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 726–733. [Google Scholar] [CrossRef]
- Wouters, K.; van Gorp, P.J.; Bieghs, V.; Gijbels, M.J.; Duimel, H.; Lütjohann, D.; Kerksiek, A.; van Kruchten, R.; Maeda, N.; Staels, B.; et al. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology 2008, 48, 474–486. [Google Scholar] [CrossRef]
- Marí, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; García-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef]
- Hermida, N.; Balligand, J.-L. Low-density lipoprotein-cholesterol-induced endothelial dysfunction and oxidative stress: The role of statins. Antioxid. Redox Signal. 2014, 20, 1216–1237. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Lin, S.; Zippi, M.; Geng, W.; Stock, S.; Zimmer, V.; Xu, C.; Zhou, M. High-Density Lipoprotein Cholesterol, Blood Urea Nitrogen, and Serum Creatinine Can Predict Severe Acute Pancreatitis. Biomed Res. Int. 2017, 2017, 1648385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, T.; Yang, T.; Chen, H.; Fu, D.; Hu, Y.; Wang, J.; Yuan, Q.; Yu, H.; Xu, W.; Xie, X. New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol. 2019, 20, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Sánchez, L.; Madrid-Miller, A.; Chávez-Rueda, K.; Legorreta-Haquet, M.V.; Tesoro-Cruz, E.; Blanco-Favela, F. Activation of TLR2 and TLR4 by minimally modified low-density lipoprotein in human macrophages and monocytes triggers the inflammatory response. Hum. Immunol. 2010, 71, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Al-Shali, K.Z.; Hegele, R.A. Hypertriglyceridemia: Its etiology, effects and treatment. Can. Med Assoc. J. 2007, 176, 1113–1120. [Google Scholar] [CrossRef] [Green Version]
- Khovidhunkit, W.; Kim, M.-S.; Memon, R.A.; Shigenaga, J.K.; Moser, A.H.; Feingold, K.R.; Grunfeld, C. Effects of infection and inflammation on lipid and lipoprotein metabolism: Mechanisms and consequences to the host. J. Lipid Res. 2004, 45, 1169–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Coresh, J.; Eustace, J.A.; Longenecker, J.C.; Jaar, B.; Fink, N.E.; Tracy, R.P.; Powe, N.R.; Klag, M.J. Association between cholesterol level and mortality in dialysis patients: Role of inflammation and malnutrition. JAMA 2004, 291, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Wilson, P.G.; Manji, M.; Neoptolemos, J.P. Acute pancreatitis as a model of sepsis. J. Antimicrob. Chemother. 1998, 41 (Suppl. A), 51–63. [Google Scholar] [CrossRef] [Green Version]
- van Leeuwen, H.J.; Heezius, E.C.J.M.; Dallinga, G.M.; van Strijp, J.A.G.; Verhoef, J.; van Kessel, K.P.M. Lipoprotein metabolism in patients with severe sepsis. Crit. Care Med. 2003, 31, 1359–1366. [Google Scholar] [CrossRef]
- Hong, W.; Lin, S.; Zippi, M.; Geng, W.; Stock, S.; Basharat, Z.; Cheng, B.; Pan, J.; Zhou, M. Serum Albumin Is Independently Associated with Persistent Organ Failure in Acute Pancreatitis. Can. J. Gastroenterol. Hepatol. 2017, 2017, 5297143. [Google Scholar] [CrossRef] [Green Version]
- Kaysen, G.A. Biochemistry and biomarkers of inflamed patients: Why look, what to assess. Clin. J. Am. Soc. Nephrol. 2009, 4 (Suppl. 1), S56–S63. [Google Scholar] [CrossRef] [Green Version]
- van den Kommer, T.N.; Dik, M.G.; Comijs, H.C.; Jonker, C.; Deeg, D.J. Role of lipoproteins and inflammation in cognitive decline: Do they interact? NeuroBiol. Aging 2012, 33, 196.e1–196.e12. [Google Scholar] [CrossRef]
- Nawaz, H.; Koutroumpakis, E.; Easler, J.; Slivka, A.; Whitcomb, D.C.; Singh, V.P.; Yadav, D.; Papachristou, G.I. Elevated serum triglycerides are independently associated with persistent organ failure in acute pancreatitis. Am. J. Gastroenterol. 2015, 110, 1497–1503. [Google Scholar] [CrossRef]
- Su, X.; Zhang, G.; Cheng, Y.; Wang, B. New insights into the emerging effects of inflammatory response on HDL particles structure and function. Mol. Biol. Rep. 2021, 48, 5723–5733. [Google Scholar] [CrossRef]
- Feingold, K.R.; Grunfeld, C. Effect of inflammation on HDL structure and function. Curr. Opin. Lipidol. 2016, 27, 521–530. [Google Scholar] [CrossRef]
- Navarro, M.A.; Carpintero, R.; Acin, S.; Arbones-Mainar, J.M.; Calleja, L.; Carnicer, R.; Surra, J.C.; Guzmán-García, M.A.; González-Ramón, N.; Iturralde, M.; et al. Immune-regulation of the apolipoprotein A-I/C-III/A-IV gene cluster in experimental inflammation. Cytokine 2005, 31, 52–63. [Google Scholar] [CrossRef]
- Carpentier, Y.A.; Scruel, O. Changes in the concentration and composition of plasma lipoproteins during the acute phase response. Curr. Opin. Clin. Nutr. Metab. Care 2002, 5, 153–158. [Google Scholar] [CrossRef]
- Prüfer, N.; Kleuser, B.; van der Giet, M. The role of serum amyloid A and sphingosine-1-phosphate on high-density lipoprotein functionality. Biol. Chem. 2015, 396, 573–583. [Google Scholar] [CrossRef] [Green Version]
- Rohrer, L.; Hersberger, M.; von Eckardstein, A. High density lipoproteins in the intersection of diabetes mellitus, inflammation and cardiovascular disease. Curr. Opin. Lipidol. 2004, 15, 269–278. [Google Scholar] [CrossRef]
- Khovidhunkit, W.; Moser, A.H.; Shigenaga, J.K.; Grunfeld, C.; Feingold, K.R. Endotoxin down-regulates ABCG5 and ABCG8 in mouse liver and ABCA1 and ABCG1 in J774 murine macrophages: Differential role of LXR. J. Lipid Res. 2003, 44, 1728–1736. [Google Scholar] [CrossRef] [Green Version]
- McGillicuddy, F.C.; de la Llera Moya, M.; Hinkle, C.C.; Joshi, M.R.; Chiquoine, E.H.; Billheimer, J.T.; Rothblat, G.H.; Reilly, M.P. Inflammation impairs reverse cholesterol transport in vivo. Circulation 2009, 119, 1135–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsun, J.G.; Shiu, S.W.; Wong, Y.; Yung, S.; Chan, T.M.; Tan, K.C. Impact of serum amyloid A on cellular cholesterol efflux to serum in type 2 diabetes mellitus. Atherosclerosis 2013, 231, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, R.; Gao, F.; Wang, L.; Feng, S.; Li, J.; Huang, Z. Association between high-density lipoprotein cholesterol and apolipoprotein A-I and severe acute pancreatitis: A case-control study. Eur. J. Gastroenterol. Hepatol. 2021, 33, 1517–1523. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.H.; Jung, S.; Cho, S.K.; Lee, K.J.; Kim, J.W. Predictive value of apolipoprotein B and A-I ratio in severe acute pancreatitis. J. Gastroenterol. Hepatol. 2018, 33, 548–553. [Google Scholar] [CrossRef]
- Georgila, K.; Vyrla, D.; Drakos, E. Apolipoprotein A-I (ApoA-I), Immunity, Inflammation and Cancer. Cancers 2019, 11, 1097. [Google Scholar] [CrossRef] [Green Version]
- Vuilleumier, N.; Dayer, J.M.; von Eckardstein, A.; Roux-Lombard, P. Pro- or anti-inflammatory role of apolipoprotein A-1 in high-density lipoproteins? Swiss Med. Wkly. 2013, 143, w13781. [Google Scholar] [CrossRef]
- Moore, R.E.; Navab, M.; Millar, J.S.; Zimetti, F.; Hama, S.; Rothblat, G.H.; Rader, D.J. Increased atherosclerosis in mice lacking apolipoprotein A-I attributable to both impaired reverse cholesterol transport and increased inflammation. Circ. Res. 2005, 97, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Tietge, U.J.; Maugeais, C.; Lund-Katz, S.; Grass, D.; deBeer, F.C.; Rader, D.J. Human secretory phospholipase A 2 mediates decreased plasma levels of HDL cholesterol and apoA-I in response to inflammation in human apoA-I transgenic mice. Arter. Thromb. Vasc. Biol. 2002, 22, 1213–1218. [Google Scholar] [CrossRef] [Green Version]
- Ettinger, W.H.; Varma, V.K.; Sorci-Thomas, M.; Parks, J.S.; Sigmon, R.C.; Smith, T.K.; Verdery, R.B. Cytokines decrease apolipoprotein accumulation in medium from Hep G2 cells. Arter. Thromb 1994, 14, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.-J.; Lu, H.-M.; Liu, Y.; Li, M.; Hu, W.-M.; Zhou, Z.-G. Development and validation of a prediction model for moderately severe and severe acute pancreatitis in pregnancy. World J. Gastroenterol. 2022, 28, 1588–1600. [Google Scholar] [CrossRef]
- Hong, W.; Lu, Y.; Zhou, X.; Jin, S.; Pan, J.; Lin, Q.; Yang, S.; Basharat, Z.; Zippi, M.; Goyal, H. Usefulness of Random Forest Algorithm in Predicting Severe Acute Pancreatitis. Front. Cell. Infect. MicroBiol. 2022, 12, 893294. [Google Scholar] [CrossRef]
- Haiyan, Z.; Na, P.; Jialin, H.; Qingjian, L.; Jianying, B.; Xiumei, B. Acute Pancreatitis in Pregnancy: A Ten-Year Noninterventional, Retrospective Cohort Experience. Gastroenterol. Res. Pract. 2022, 2022, 3663079. [Google Scholar] [CrossRef]
- Shen, Z.; Wang, X.; Zhen, Z.; Wang, Y.; Sun, P. Metabolic syndrome components and acute pancreatitis: A case-control study in China. BMC Gastroenterol. 2021, 21, 17. [Google Scholar] [CrossRef]
- Düzenci, D.; Yalnız, M.; İspiroğlu, M. Comparison between prognostic indicators in organ insufficiency with acute pancreatitis. Ulus Travma Acil Cerrahi Derg. 2021, 27, 410–420. [Google Scholar] [CrossRef]
- Song, K.; Guo, C.; Li, C.; Ding, N. Risk Factors of Recurrence of Acute Pancreatitis: A Retrospective Research. Turk. J. Gastroenterol. 2021, 32, 971–978. [Google Scholar] [CrossRef]
- Khan, J.; Nordback, I.; Sand, J. Serum lipid levels are associated with the severity of acute pancreatitis. Digestion 2013, 87, 223–228. [Google Scholar] [CrossRef]
- Peng, Y.-S.; Chen, Y.-C.; Tian, Y.-C.; Yang, C.-W.; Lien, J.-M.; Fang, J.-T.; Wu, C.-S.; Hung, C.-F.; Hwang, T.-L.; Tsai, Y.-H.; et al. Serum levels of apolipoprotein A-I and high-density lipoprotein can predict organ failure in acute pancreatitis. Crit. Care 2015, 19, 88. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cui, Z.; Li, H.; Saleen, A.F.; Zhang, D.; Miao, B.; Cui, Y.; Zhao, E.; Li, Z.; Cui, N. Nosocomial mortality and early prediction of patients with severe acute pancreatitis. J. Gastroenterol. Hepatol. 2010, 25, 1386–1393. [Google Scholar] [CrossRef]
- Liao, W.; Niu, X.; Zhang, W.; Liu, X. Comparison of the Development and Prognosis in Patients of Hypertriglyceridemic Pancreatitis with and without Diabetes. Gastroenterol. Res. Pract. 2021, 2021, 8895268. [Google Scholar] [CrossRef]
- Hong, W.; Geng, W.; Chen, B.; Basharat, Z.; Wu, Q.; Zimmer, V.; Zhou, M. Predictors of acute pancreatitis with low elevation of serum amylase. Ther. Clin. Risk Manag. 2017, 13, 1577–1584. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wang, Y.; Li, H.; Tan, W.; Chen, X.; Ye, S. Serum apolipoprotein B-to-apolipoprotein A1 ratio is independently associated with disease severity in patients with acute pancreatitis. Sci. Rep. 2019, 9, 7764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, P.A.; Bollen, T.L.; Dervenis, C.; Gooszen, H.G.; Johnson, C.D.; Sarr, M.G.; Tsiotos, G.G.; Vege, S.S.; Acute Pancreatitis Classification Working Group. Classification of acute pancreatitis—2012: Revision of the Atlanta classification and definitions by international consensus. Gut 2013, 62, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Bradley, E.L., 3rd. A clinically based classification system for acute pancreatitis. Ann. Chir. 1993, 47, 537–541. [Google Scholar] [CrossRef]
- Kröner, P.T.; Wallace, M.B.; Raimondo, M.; Antwi, S.O.; Ma, Y.; Li, Z.; Ji, B.; Bi, Y. Systemic anticoagulation is associated with decreased mortality and morbidity in acute pancreatitis. Pancreatology 2021, 21, 1428–1433. [Google Scholar] [CrossRef]
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001, 285, 2486–2497. [Google Scholar] [CrossRef]
- Kim, D.-G.; Bae, G.-S.; Choi, S.-B.; Jo, I.-J.; Shin, J.-Y.; Lee, S.-K.; Kim, M.-J.; Kim, M.-J.; Jeong, H.-W.; Choi, C.-M.; et al. Guggulsterone attenuates cerulein-induced acute pancreatitis via inhibition of ERK and JNK activation. Int. Immunopharmacol. 2015, 26, 194–202. [Google Scholar] [CrossRef]
- Osadnik, T.; Goławski, M.; Lewandowski, P.; Morze, J.; Osadnik, K.; Pawlas, N.; Lejawa, M.; Jakubiak, G.K.; Mazur, A.; Schwingschackl, L.; et al. A network meta-analysis on the comparative effect of nutraceuticals on lipid profile in adults. Pharmacol. Res. 2022, 183, 106402. [Google Scholar] [CrossRef]
- Matalka, I.I.; Mhaidat, N.M.; Fatlawi, L.A. Antioxidant activity of simvastatin prevents L-arginine-induced acute toxicity of pancreas. Int. J. Physiol. Pathophysiol. Pharmacol. 2013, 5, 102–108. [Google Scholar]
- Gornik, I.; Gašparović, V.; Vrdoljak, N.G.; Haxiu, A.; Vucelić, B. Prior statin therapy is associated with milder course and better outcome in acute pancreatitis—A cohort study. Pancreatology 2013, 13, 196–200. [Google Scholar] [CrossRef]
- Lee, P.J.; Modha, K.; Chua, T.; Chak, A.; Jang, D.; Lopez, R.; Gougol, A.; Papachristou, G.I.; Stevens, T. Association of Statins With Decreased Acute Pancreatitis Severity: A Propensity Score Analysis. J. Clin. Gastroenterol. 2018, 52, 742–746. [Google Scholar] [CrossRef] [PubMed]
- Thiruvengadam, N.R.; Schaubel, D.E.; Forde, K.; Lee, P.; Saumoy, M.; Kochman, M.L. Association of Statin Usage and the Development of Diabetes Mellitus after Acute Pancreatitis. Clin. Gastroenterol. Hepatol. 2022. [Google Scholar] [CrossRef]
- Ruiz-Rebollo, M.L.; Muñoz-Moreno, M.F.; Mayo-Iscar, A.; Udaondo-Cascante, M.A.; Nistal, R.B. Statin intake can decrease acute pancreatitis severit. Pancreatology 2019, 19, 807–812. [Google Scholar] [CrossRef]
- de-Madaria, E. Statins for the Prevention of Acute Pancreatitis. Am. J. Gastroenterol. 2017, 112, 1765–1767. [Google Scholar] [CrossRef]
- Kim, B.-K.; Hong, S.-J.; Lee, Y.-J.; Hong, S.J.; Yun, K.H.; Hong, B.-K.; Heo, J.H.; Rha, S.-W.; Cho, Y.-H.; Lee, S.-J.; et al. Long-term efficacy and safety of moderate-intensity statin with ezetimibe combination therapy versus high-intensity statin monotherapy in patients with atherosclerotic cardiovascular disease (RACING): A randomised, open-label, non-inferiority trial. Lancet 2022, 400, 380–390. [Google Scholar] [CrossRef]
- Lin, C.-M.; Liao, K.-F.; Lin, C.-L.; Lai, S.-W. Use of Simvastatin and Risk of Acute Pancreatitis: A Nationwide Case-Control Study in Taiwan. J. Clin. Pharmacol. 2017, 57, 918–923. [Google Scholar] [CrossRef]
- Kuoppala, J.; Pulkkinen, J.; Kastarinen, H.; Kiviniemi, V.; Jyrkkä, J.; Enlund, H.; Happonen, P.; Paajanen, H. Use of statins and the risk of acute pancreatitis: A population-based case-control study. Pharmacoepidemiol. Drug Saf. 2015, 24, 1085–1092. [Google Scholar] [CrossRef]
- Machicado, J.D.; Papachristou, G.I. Pharmacologic management and prevention of acute pancreatitis. Curr. Opin. Gastroenterol. 2019, 35, 460–467. [Google Scholar] [CrossRef]
- Prüller, F.; Raggam, R.B.; Posch, V.; Almer, G.; Truschnig-Wilders, M.; Horejsi, R.; Möller, R.; Weghuber, D.; Ille, R.; Schnedl, W.; et al. Trunk weighted obesity, cholesterol levels and low grade inflammation are main determinants for enhanced thrombin generation. Atherosclerosis 2012, 220, 215–218. [Google Scholar] [CrossRef]
- de Laat-Kremers, R.; Di Castelnuovo, A.; van der Vorm, L.; Costanzo, S.; Ninivaggi, M.; Cerletti, C.; Huskens, D.; De Curtis, A.; Gialluisi, A.; Bai, C.; et al. Increased BMI and Blood Lipids Are Associated With a Hypercoagulable State in the Moli-sani Cohort. Front. Cardiovasc. Med. 2022, 9, 897733. [Google Scholar] [CrossRef]
- Undas, A. Statins in prevention of thromboembolic events: From seminal studies to recent advances. Pol. Arch. Intern. Med. 2022, 132. [Google Scholar] [CrossRef]
- Siudut, J.; Pudło, J.; Konieczyńska, M.; Polak, M.; Jawień, J.; Undas, A. Therapy with high-dose statins reduces soluble P-selectin: The impact on plasma fibrin clot properties. Int. J. Cardiol. 2023, 373, 110–117. [Google Scholar] [CrossRef]
- Patil, B.; Meena, L.N.; Sharma, D.C.; Agarwal, G.; Dadhich, Y.; Gupta, G. Impact of low-molecular-weight heparin in the treatment of moderately severe and severe acute pancreatitis; a randomized, single blind, phase 3 control trial. Int. J. Surg. 2022, 101, 106621. [Google Scholar] [CrossRef]
- Hong, W.; Lillemoe, K.D.; Pan, S.; Zimmer, V.; Kontopantelis, E.; Stock, S.; Zippi, M.; Wang, C.; Zhou, M. Development and validation of a risk prediction score for severe acute pancreatitis. J. Transl. Med. 2019, 17, 146. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author and Year of Publication | Type of Study | Sample Size | Lipids | Result/Conclusion |
|---|---|---|---|---|
| Yang et. al, 2022 [90] | Retrospective study | APIP patients (n = 190) | Triglyceride, total cholesterol | Total cholesterol is an independent predictor of moderately severe and severe AP in pregnancy. |
| Zhao et.al, 2022 [92] | Cohort study | antenatal mothers with AP (n = 45) | Triglycerides, total cholesterol | Total cholesterol levels were significantly higher in patients staying in hospital for >13 days compared with those staying for less than 13 days. |
| Shen et.al, 2021 [93] | Case-control study | Non-AP patients (n = 356) and AP patients (n = 349) | Total cholesterol, triglyceride, Apo A1 | The occurrence of AP was significantly associated with total cholesterol. |
| D. Düzenci et.al, 2021 [94] | Retrospective study | AP patients (n = 153) | Total cholesterol, triglycerides, CRP | A significant correlation was observed between total cholesterol and mortality. |
| Song et.al, 2021 [95] | Retrospective study | non-recurrent AP patients (n = 671) and recurrent AP patient | Triglyceride, HDL-C, total cholesterol | The total cholesterol level was significantly higher in recurrent acute pancreatitis compared to AP at first onset. |
| Hong et.al, 2020 [14] | Retrospective study | AP patients (n = 648) | HDL-C, LDL-C, triglyceride | Both low total cholesterol (<160 mg/dL) and high total cholesterol (>240 mg/dL) levels within 24 h of admission are independently associated with an increased risk of SAP. |
| Peng et.al, 2015 [97] | Prospective study | patients predicted SAP (n = 66) | Total cholesterol, HDL-C, LDL-C, triglyceride | The total cholesterol level was significantly lower in AP patients with organ failure than without organ failure. |
| Khan et.al, 2013 [96] | Retrospective study | AP patients (n = 233) | Total cholesterol, HDL-C, LDL-C | Levels of serum total cholesterol are significantly lower in patients with SAP and are associated with longer hospitalization. |
| Wang et.al, 2010 [98] | Retrospective study | SAP patients (n = 338) | Total Cholesterol | Within 24 h after admission, the serum total cholesterol was a mortality-reduced factor when it is between 4.37 mmol/L and 5.23 mmol/L. |
| Author and Year of Publication | Type of Study | Sample Size | Lipids | Result/Conclusion |
|---|---|---|---|---|
| Hong et.al, 2022 [91] | Retrospective study | Non-SAP patient (n = 438) and SAP patient (n = 49) | HDL-C, LDL-C | Based on a variable importance analysis of the RF model, LDL-C is an important predictor of SAP. |
| Liao et.al, 2021 [99] | Retrospective study | hypertriglyceridemic pancreatitis patients (n = 157) | Triglyceride, LDL-C | Higher LDL-C was associated with HTGP recurrence. |
| Wu et. al, 2019 [101] | Retrospective study | Non-SAP patient (n = 310) and SAP patient (n = 65) | Serum Apo B/A1 ratio | Apo B is the main structure of LDL-C. The serum Apo B/A1 ratio at admission is closely correlated with disease severity in patients with AP and can serve as a reliable indicator for SAP in the clinical setting. |
| Hong et. al, 2018 [13] | Retrospective study | AP patient (n = 647) | HDL-C, LDL-C | LDL-C (90 mg/dL) levels within 24 h of admission are independently associated with an increased risk of SAP. |
| Hong et.al, 2017 [100] | Case-control study | AP patients with amylase ≥300 U/L (n = 108) and AP patients with amylase <300 U/L (n = 108). | LDL-C, triglyceride | Higher LDL-C levels are independently associated with the development of AP along with low serum amylase. |
| Peng et.al, 2015 [97] | Prospective study | patients predicted SAP (n = 66) | Total cholesterol, HDL-C, LDL-C, triglyceride | The serum LDL-C level of patients with persistent organ failure was significantly lower than that of patients without persistent organ failure within 24 h of admission, and persistent organ failure is closely related to the high mortality rate of SAP. |
| Khan et. al, 2013 [96] | Retrospective study | Non-SAP patient (n = 30) and SAP patient (n = 203) | Serum total cholesterol, HDL-C, LDL-C | The level of LDL-C is significantly lower in patients with SAP and is associated with longer hospitalization. |
| Author and Year of Publication | Type of Study | Sample Size | Lipids | Result/Conclusion |
|---|---|---|---|---|
| Hong et al., 2022 [91] | Retrospective study | Non-SAP patient (n = 438) and SAP patient (n = 49) | HDL-C, LDL-C | Based on a variable importance analysis of the RF model, HDL-C is the important predictor of SAP. |
| Li et al., 2021 [83] | Retrospective study | Non-SAP patient (n = 600) and SAP patient (n = 78) | Apo A1, HDL-C | Apo A1 and HDL-C levels were negatively correlated to the occurrence of SAP. |
| Zhou et al., 2018 [15] | Retrospective study | 102 adult AP patients with OF, worsening of previous comorbidities or local complications during hospitalization | Apo A1, HDL-C | Apo A1 and HDL-C levels are negatively associated with some scoring systems and poor clinical outcomes in AP patients. The concentrations of Apo A1 and HDL-C have high predictive value to forecast persistent OF. |
| Hong et al., 2017 [62] | Retrospective study | Non-SAP patient (n = 589) and SAP patient (n = 58) | HDL-C | HDL-C at admission may predict development of SAP. |
| Peng et al., 2015 [97] | Retrospective study | 66 patients with predicted SAP admitted to ICU | Apo A1, HDL-C | Low levels of Apo A1 and HDL-C are associated with high levels of inflammatory cytokines, persistent OF, infected necrosis and hospital mortality. Apo A1 and HDL-C can serve as biomarkers to differentiate persistent OF from transient OF. |
| Khan et al., 2013 [96] | Retrospective study | Non-SAP patient (n = 30) and SAP patient (n = 203) | Serum total cholesterol, HDL-C, LDL-C | The levels of HDL-C are significantly lower in patients with SAP and are associated with longer hospitalization. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Jin, S.; Pan, J.; Lin, Q.; Yang, S.; Lu, Y.; Qiu, M.; Ambe, P.C.; Basharat, Z.; Zimmer, V.; et al. Relationship between Cholesterol-Related Lipids and Severe Acute Pancreatitis: From Bench to Bedside. J. Clin. Med. 2023, 12, 1729. https://doi.org/10.3390/jcm12051729
Zhou X, Jin S, Pan J, Lin Q, Yang S, Lu Y, Qiu M, Ambe PC, Basharat Z, Zimmer V, et al. Relationship between Cholesterol-Related Lipids and Severe Acute Pancreatitis: From Bench to Bedside. Journal of Clinical Medicine. 2023; 12(5):1729. https://doi.org/10.3390/jcm12051729
Chicago/Turabian StyleZhou, Xiaoying, Shengchun Jin, Jingyi Pan, Qingyi Lin, Shaopeng Yang, Yajing Lu, Minhao Qiu, Peter C. Ambe, Zarrin Basharat, Vincent Zimmer, and et al. 2023. "Relationship between Cholesterol-Related Lipids and Severe Acute Pancreatitis: From Bench to Bedside" Journal of Clinical Medicine 12, no. 5: 1729. https://doi.org/10.3390/jcm12051729