
A Novel Heterozygous Desmoplakin Variant Causes Cardiocutaneous Syndrome with Arrhythmogenic Cardiomyopathy and Palmoplantar Keratosis
 , , , and
, , , and 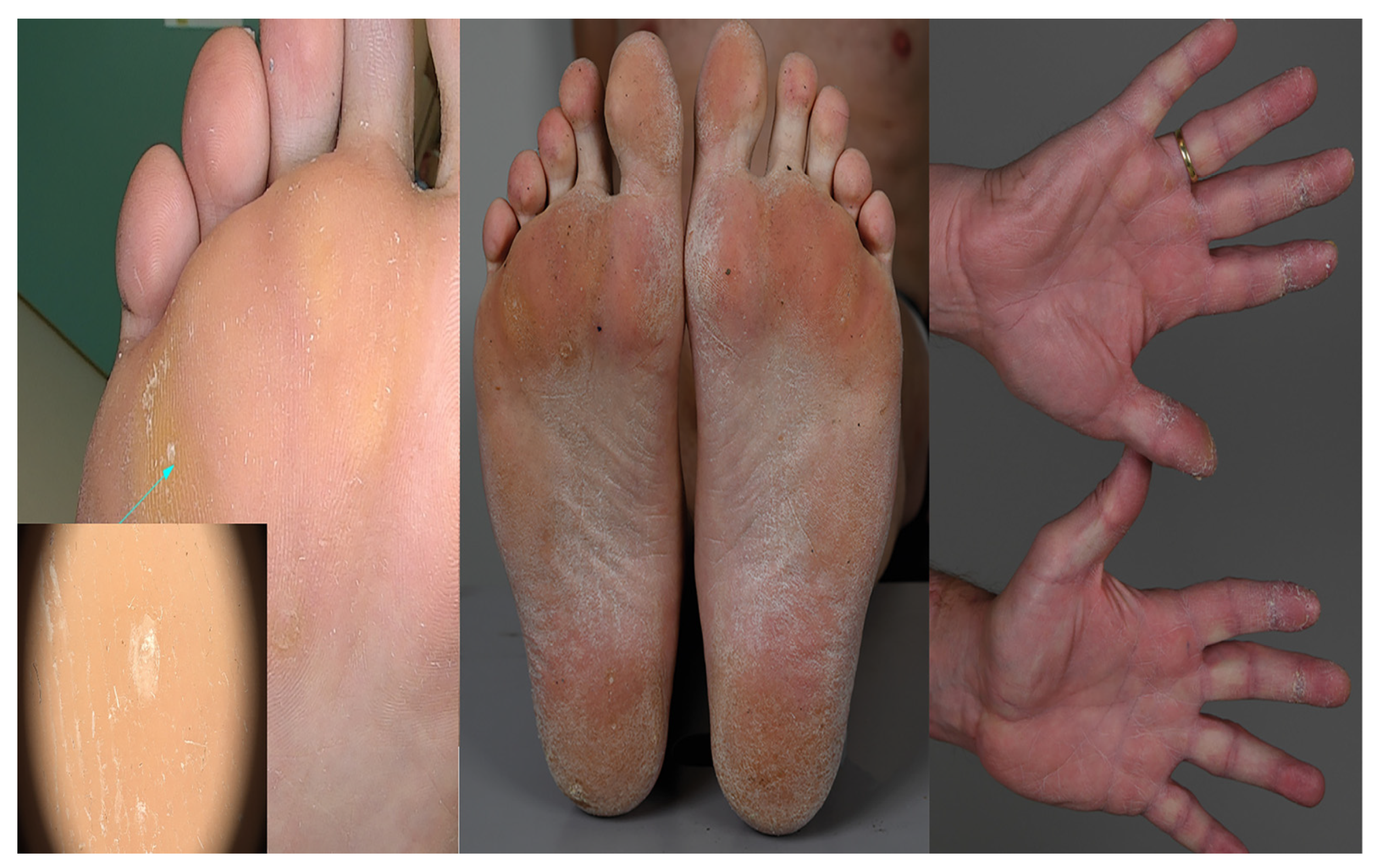{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
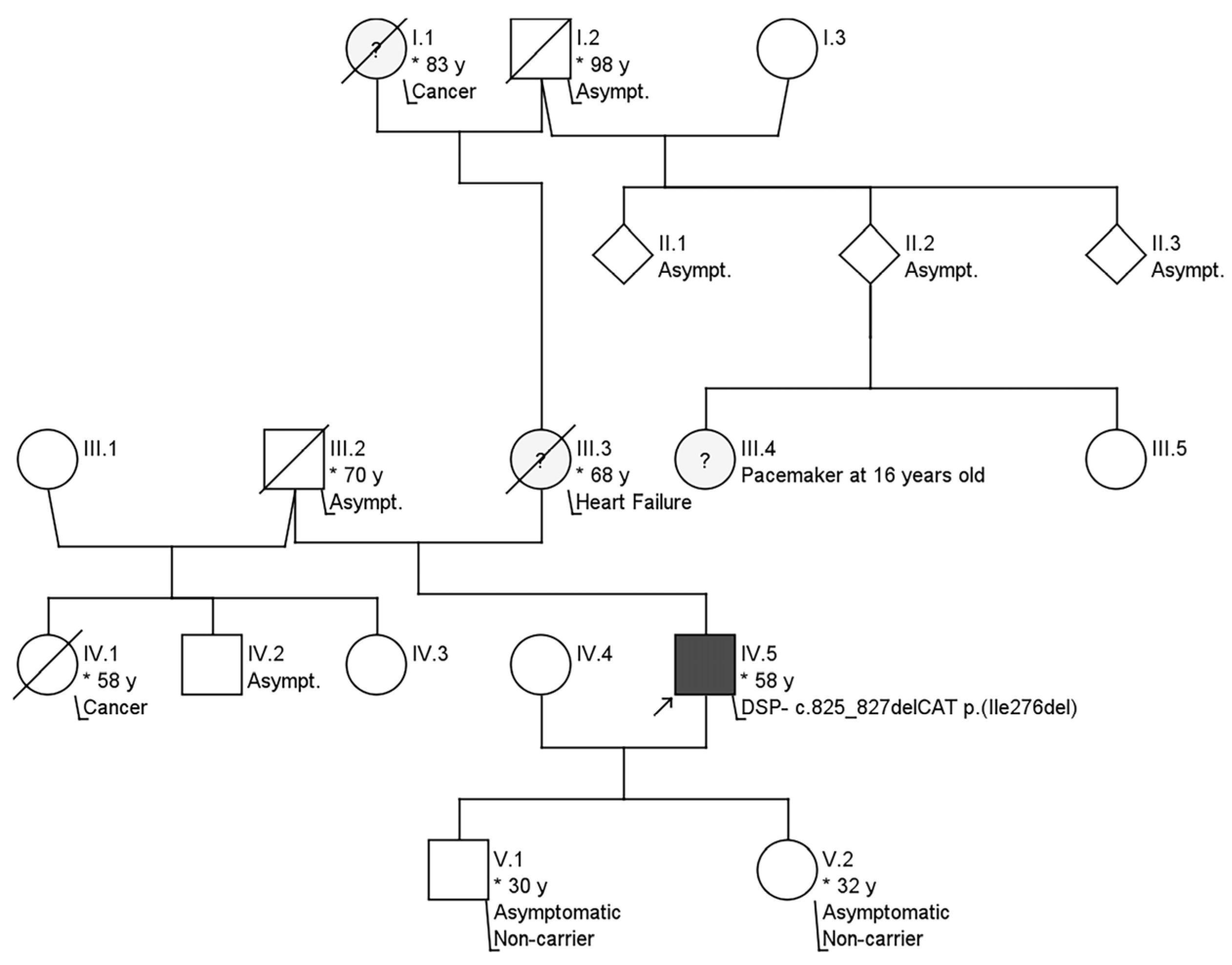
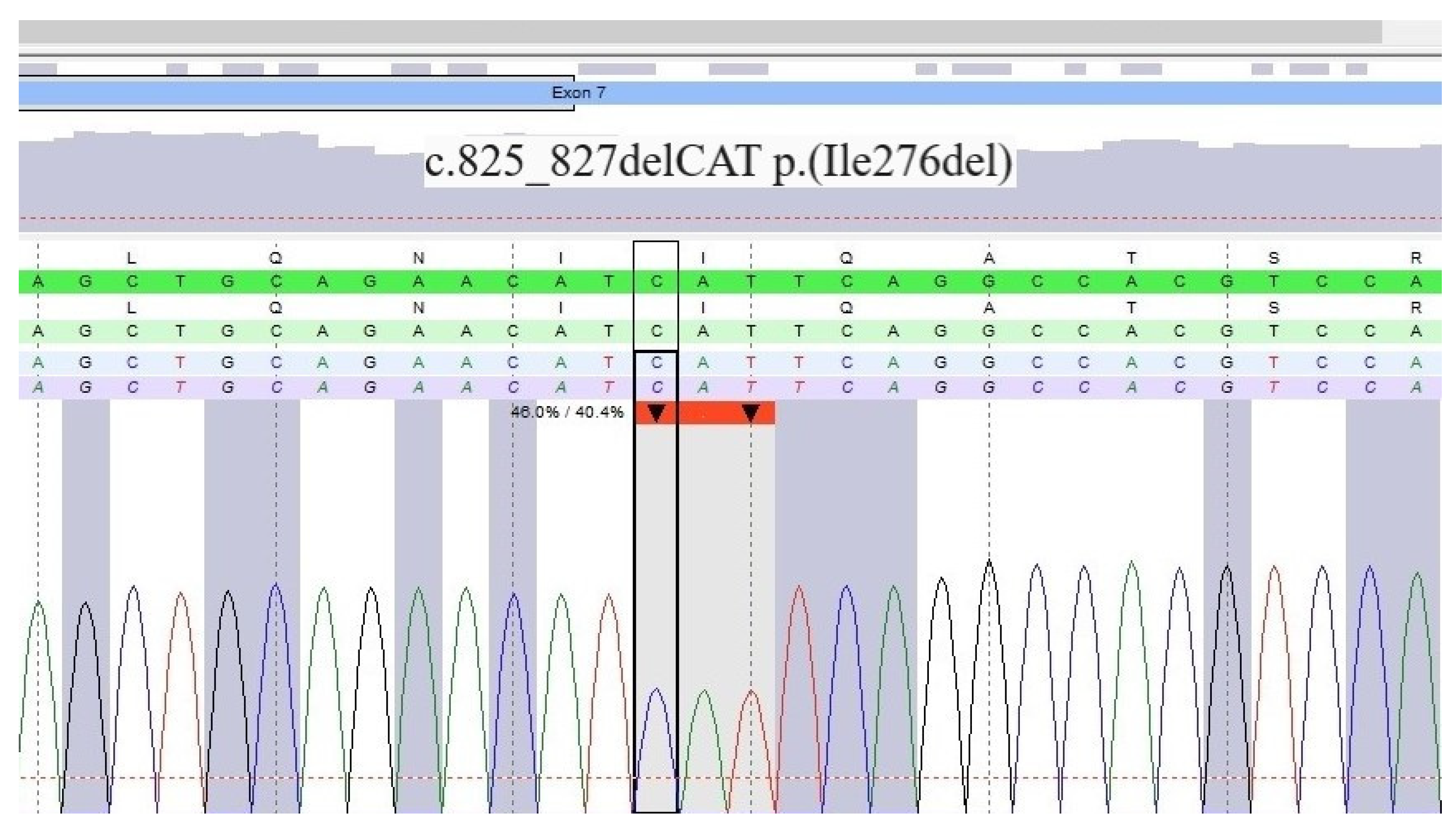
2. Case Description
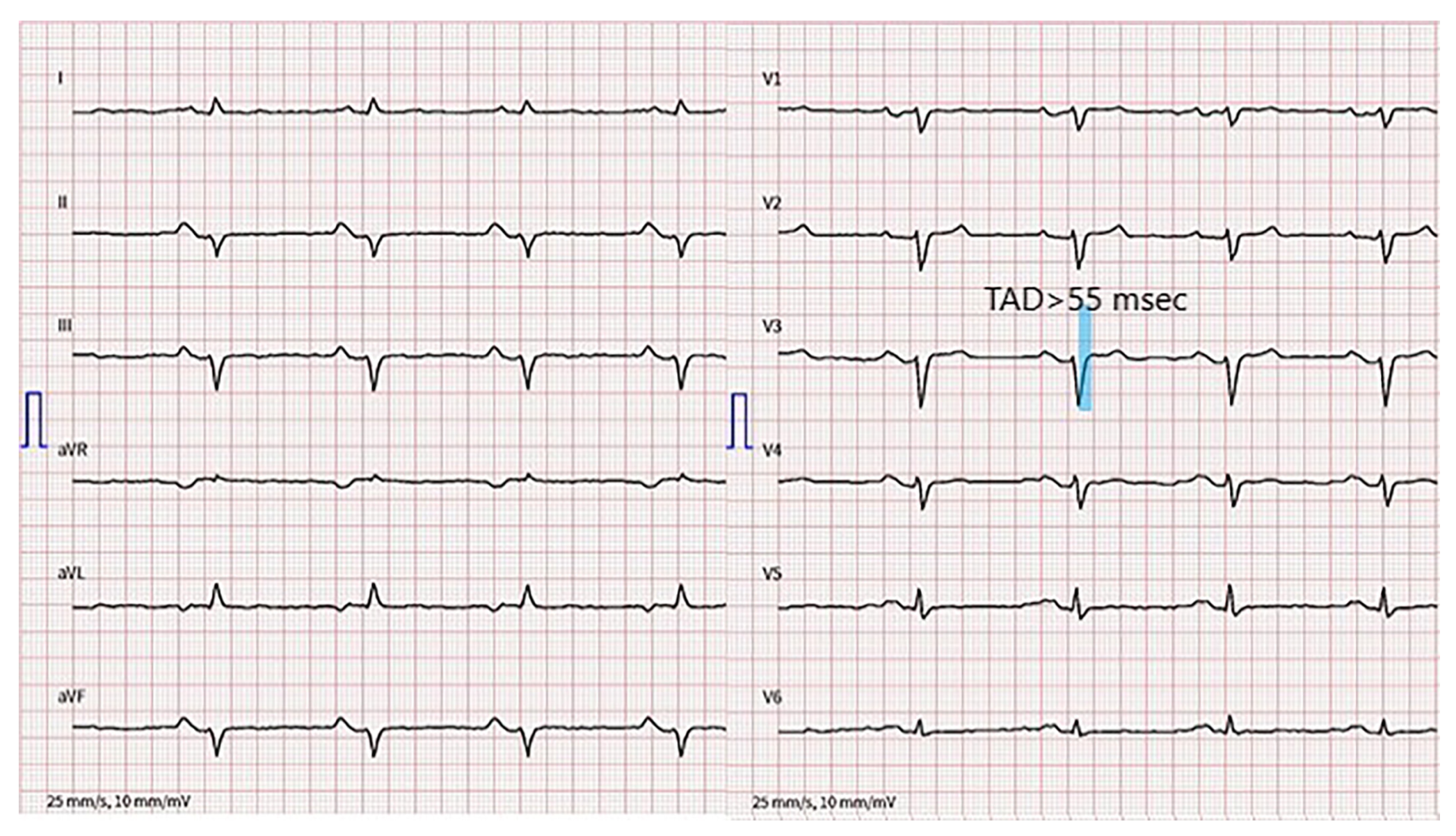
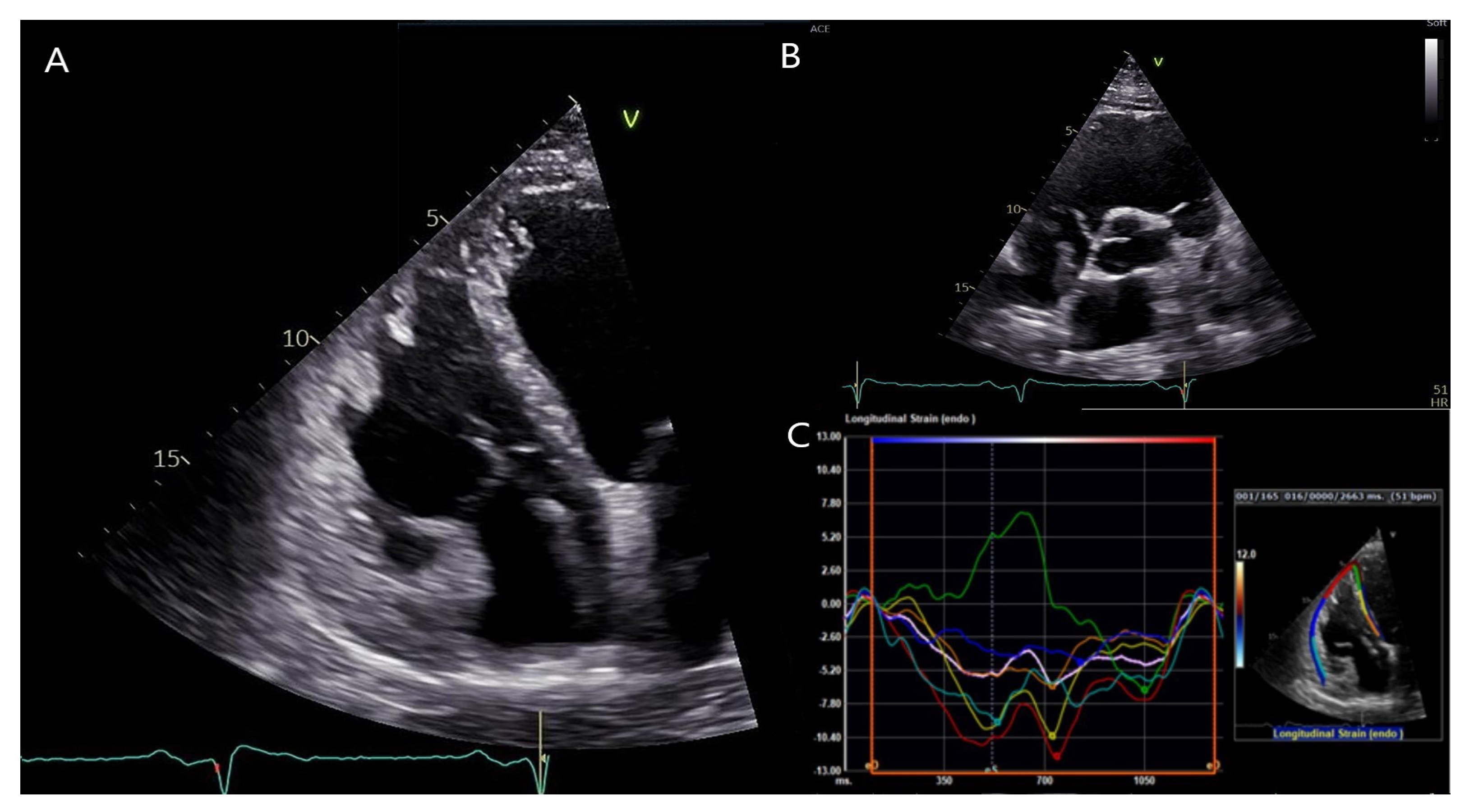
3. Diagnostic Assessment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Smith, E.D.; Lakdawala, N.K.; Papoutsidakis, N.; Aubert, G.; Mazzanti, A.; McCanta, A.C.; Agarwal, P.P.; Arscott, P.; Dellefave-Castillo, L.M.; Vorovich, E.E.; et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct From Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2020, 141, 1872–1884. [Google Scholar] [CrossRef] [PubMed]
- Karvonen, V.; Harjama, L.; Helio, K.; Kettunen, K.; Elomaa, O.; Koskenvuo, J.W.; Kere, J.; Weckstrom, S.; Holmstrom, M.; Saarela, J.; et al. A novel desmoplakin mutation causes dilated cardiomyopathy with palmoplantar keratoderma as an early clinical sign. J. Eur. Acad. Dermatol. Venereol. 2022, 36, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Robyns, T.; Kuiperi, C.; Breckpot, J.; Devriendt, K.; Souche, E.; Van Cleemput, J.; Willems, R.; Nuyens, D.; Matthijs, G.; Corveleyn, A. Repeat genetic testing with targeted capture sequencing in primary arrhythmia syndrome and cardiomyopathy. Eur. J. Hum. Genet. 2017, 25, 1313–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the Task Force Criteria. Eur. Heart J. 2010, 31, 806–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Gasperetti, A.; Rossi, V.A.; Chiodini, A.; Casella, M.; Costa, S.; Akdis, D.; Buchel, R.; Deliniere, A.; Pruvot, E.; Gruner, C.; et al. Differentiating hereditary arrhythmogenic right ventricular cardiomyopathy from cardiac sarcoidosis fulfilling 2010 ARVC Task Force Criteria. Heart Rhythm. 2021, 18, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Rampazzo, A.; Nava, A.; Malacrida, S.; Beffagna, G.; Bauce, B.; Rossi, V.; Zimbello, R.; Simionati, B.; Basso, C.; Thiene, G.; et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2002, 71, 1200–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauce, B.; Basso, C.; Rampazzo, A.; Beffagna, G.; Daliento, L.; Frigo, G.; Malacrida, S.; Settimo, L.; Danieli, G.; Thiene, G.; et al. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur. Heart J. 2005, 26, 1666–1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruthappu, T.; Posafalvi, A.; Castelletti, S.; Delaney, P.J.; Syrris, P.; O’Toole, E.A.; Green, K.J.; Elliott, P.M.; Lambiase, P.D.; Tinker, A.; et al. Loss-of-function desmoplakin I and II mutations underlie dominant arrhythmogenic cardiomyopathy with a hair and skin phenotype. Br. J. Dermatol. 2019, 180, 1114–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Lint, F.H.M.; Murray, B.; Tichnell, C.; Zwart, R.; Amat, N.; Lekanne Deprez, R.H.; Dittmann, S.; Stallmeyer, B.; Calkins, H.; van der Smagt, J.J.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy-Associated Desmosomal Variants Are Rarely De Novo. Circ. Genom. Precis. Med. 2019, 12, e002467. [Google Scholar] [CrossRef] [PubMed]
- Zorzi, A.; Cipriani, A.; Bariani, R.; Pilichou, K.; Corrado, D.; Bauce, B. Role of Exercise as a Modulating Factor in Arrhythmogenic Cardiomyopathy. Curr. Cardiol. Rep. 2021, 23, 57. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.M.; Semsarian, C.; Marquez, M.F.; Sepehri Shamloo, A.; Ackerman, M.J.; Ashley, E.A.; Sternick Eduardo, B.; Barajas-Martinez, H.; Behr, E.R.; Bezzina, C.R.; et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for cardiac diseases. J. Arrhythm. 2022, 38, 491–553. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Çimen, T.; Medeiros-Domingo, A.; Kolios, A.; Akdiş, D.; Anwer, S.; Tanner, F.C.; Brunckhorst, C.; Duru, F.; Saguner, A.M. A Novel Heterozygous Desmoplakin Variant Causes Cardiocutaneous Syndrome with Arrhythmogenic Cardiomyopathy and Palmoplantar Keratosis. J. Clin. Med. 2023, 12, 913. https://doi.org/10.3390/jcm12030913
Çimen T, Medeiros-Domingo A, Kolios A, Akdiş D, Anwer S, Tanner FC, Brunckhorst C, Duru F, Saguner AM. A Novel Heterozygous Desmoplakin Variant Causes Cardiocutaneous Syndrome with Arrhythmogenic Cardiomyopathy and Palmoplantar Keratosis. Journal of Clinical Medicine. 2023; 12(3):913. https://doi.org/10.3390/jcm12030913
Chicago/Turabian StyleÇimen, Tolga, Argelia Medeiros-Domingo, Antonios Kolios, Deniz Akdiş, Shehab Anwer, Felix C. Tanner, Corinna Brunckhorst, Firat Duru, and Ardan M. Saguner. 2023. "A Novel Heterozygous Desmoplakin Variant Causes Cardiocutaneous Syndrome with Arrhythmogenic Cardiomyopathy and Palmoplantar Keratosis" Journal of Clinical Medicine 12, no. 3: 913. https://doi.org/10.3390/jcm12030913