
Macrophages, Low-Grade Inflammation, Insulin Resistance and Hyperinsulinemia: A Mutual Ambiguous Relationship in the Development of Metabolic Diseases


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Insulin Signaling and Mechanisms of Insulin Resistance
3. Macrophage Sub-Populations and Their Function
3.1. Adipose Tissue Macrophages
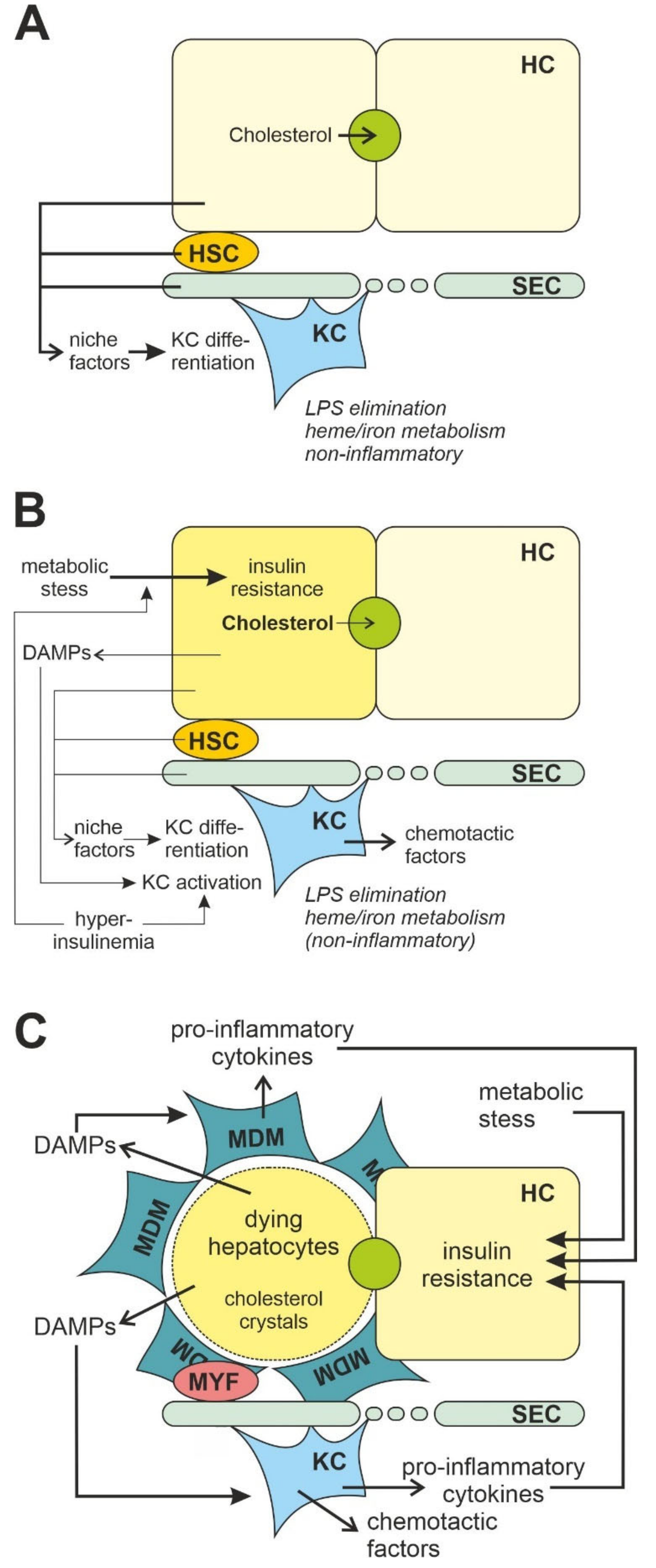
3.2. Liver Macrophages
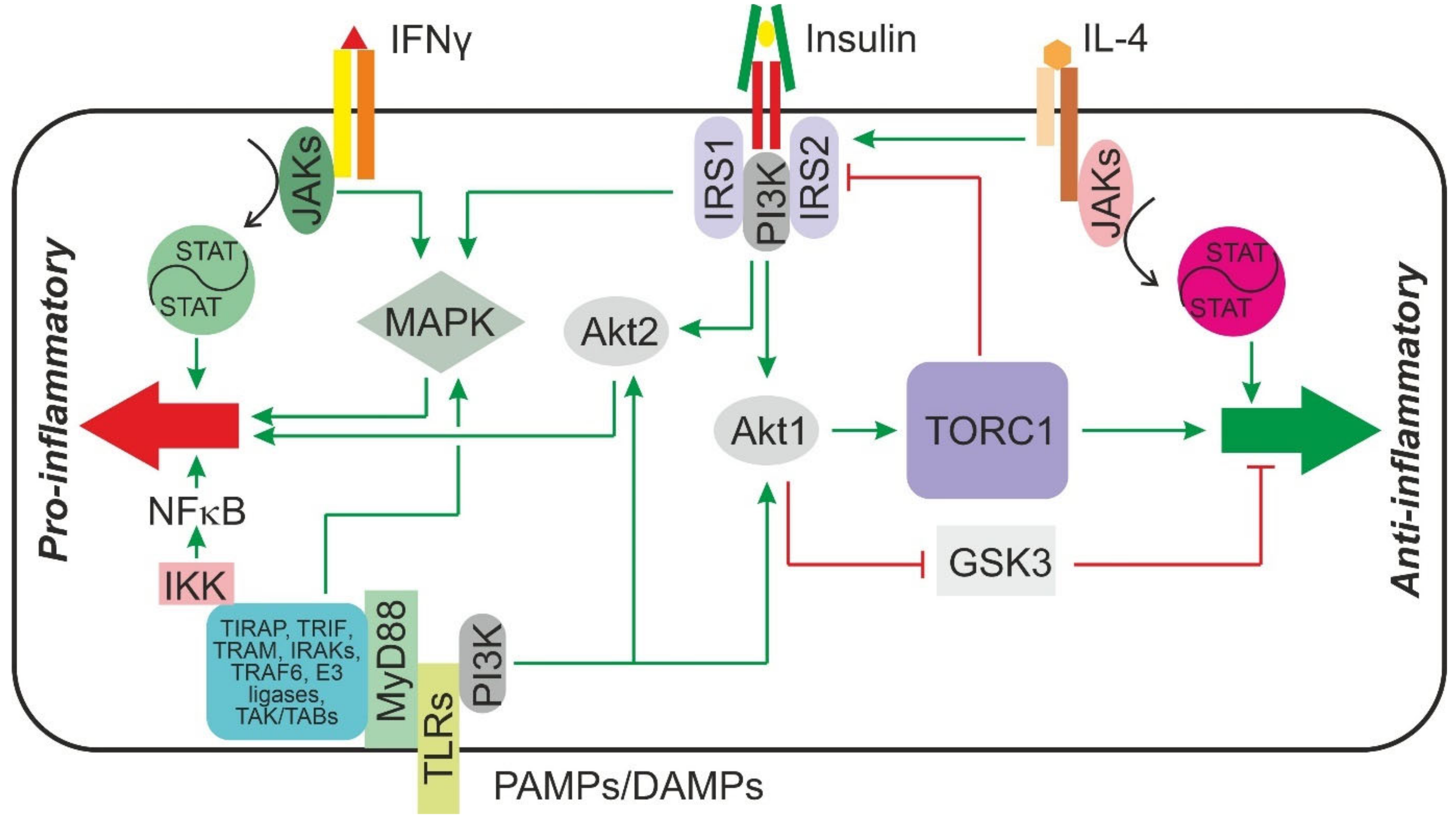
3.3. Signaling Chains Involved in Macrophage Polarization and Activation
4. Role of Macrophages in the Development of Insulin Resistance: Inflammation as a Cause of Insulin Resistance
4.1. Adipose Tissue
4.2. Liver
5. Role of Macrophages in the Development of Insulin Resistance: Insulin Resistance as a Cause of Inflammation
5.1. Adipose Tissue
5.2. Liver
6. Direct Modulation of Macrophage Function by Insulin
6.1. Pro-Inflammatory Actions of Insulin
6.2. Anti-Inflammatory Actions of Insulin
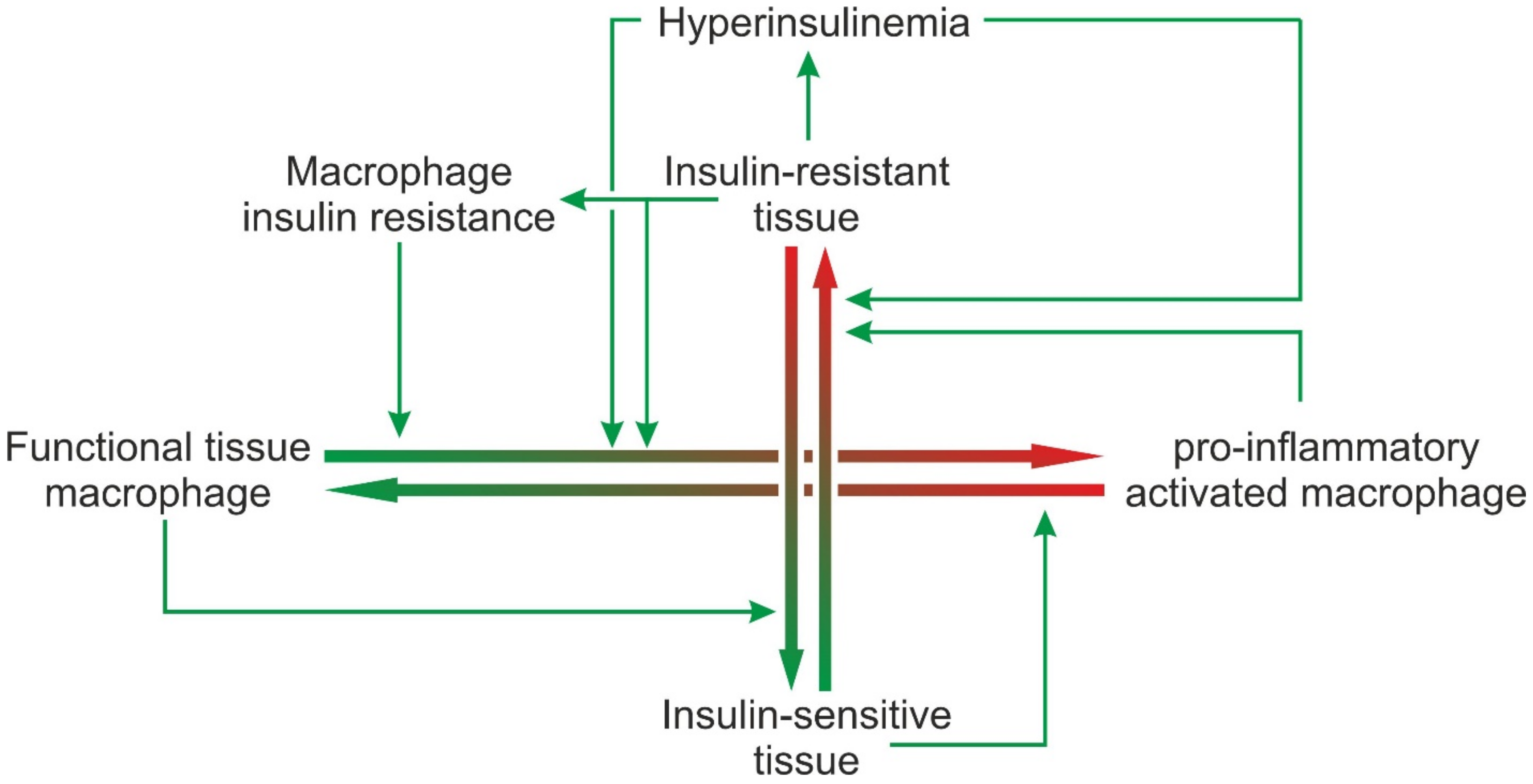
6.3. Impaired Macrophage Function Due to Macrophage Insulin Resistance
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Vettori, A.; Pompucci, G.; Paolini, B.; Del Ciondolo, I.; Bressan, S.; Dundar, M.; Kenanoğlu, S.; Unfer, V.; Bertelli, M. Genetic background, nutrition and obesity: A review. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1751–1761. [Google Scholar] [PubMed]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Cell Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef]
- Roden, M.; Shulman, G.I. The integrative biology of type 2 diabetes. Nature 2019, 576, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.D. On the causal relationships between hyperinsulinaemia, insulin resistance, obesity and dysglycaemia in type 2 diabetes. Diabetologia 2021, 64, 2138–2146. [Google Scholar] [CrossRef]
- Esser, N.; Utzschneider, K.M.; Kahn, S.E. Early beta cell dysfunction vs insulin hypersecretion as the primary event in the pathogenesis of dysglycaemia. Diabetologia 2020, 63, 2007–2021. [Google Scholar] [CrossRef]
- Grosjean, A.; Venteclef, N.; Dalmas, E. Understanding the heterogeneity and functions of metabolic tissue macrophages. Semin. Cell Dev. Biol. 2021, 119, 130–139. [Google Scholar] [CrossRef]
- Kolliniati, O.; Ieronymaki, E.; Vergadi, E.; Tsatsanis, C. Metabolic Regulation of Macrophage Activation. J. Innate Immun. 2022, 14, 51–68. [Google Scholar] [CrossRef]
- White, M.F.; Kahn, C.R. Insulin action at a molecular level—100 years of progress. Mol. Metab. 2021, 52, 101304. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Olefsky, J. Chronic tissue inflammation and metabolic disease. Genes Dev. 2021, 35, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Batista, T.M.; Haider, N.; Kahn, C.R. Defining the underlying defect in insulin action in type 2 diabetes. Diabetologia 2021, 64, 994–1006. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [Green Version]
- James, D.E.; Brown, R.; Navarro, J.; Pilch, P.F. Insulin-regulatable tissues express a unique insulin-sensitive glucose transport protein. Nature 1988, 333, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Merz, K.E.; Thurmond, D.C. Role of Skeletal Muscle in Insulin Resistance and Glucose Uptake. Compr. Physiol. 2020, 10, 785–809. [Google Scholar]
- Rachek, L.I. Free fatty acids and skeletal muscle insulin resistance. Prog. Mol. Biol. Transl. Sci. 2014, 121, 267–292. [Google Scholar]
- Morigny, P.; Boucher, J.; Arner, P.; Langin, D. Lipid and glucose metabolism in white adipocytes: Pathways, dysfunction and therapeutics. Nat. Rev. Endocrinol. 2021, 17, 276–295. [Google Scholar] [CrossRef]
- Krycer, J.R.; Quek, L.-E.; Francis, D.; Zadoorian, A.; Weiss, F.C.; Cooke, K.C.; Nelson, M.E.; Diaz-Vegas, A.; Humphrey, S.J.; Scalzo, R.; et al. Insulin signaling requires glucose to promote lipid anabolism in adipocytes. J. Biol. Chem. 2020, 295, 13250–13266. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Iynedjian, P.B. Molecular physiology of mammalian glucokinase. Cell. Mol. Life Sci. 2009, 66, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.C.; Puigserver, P.; Chen, G.; Donovan, J.; Wu, Z.; Rhee, J.; Adelmant, G.; Stafford, J.; Kahn, C.R.; Granner, D.K.; et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 2001, 413, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Thirone, A.C.P.; Huang, C.; Klip, A. Tissue-specific roles of IRS proteins in insulin signaling and glucose transport. Trends Endocrinol. Metab. 2006, 17, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.R. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 1997, 16, 5572–5581. [Google Scholar] [CrossRef]
- Wu, J.; Tseng, Y.D.; Xu, C.-F.; Neubert, T.A.; White, M.F.; Hubbard, S.R. Structural and biochemical characterization of the KRLB region in insulin receptor substrate-2. Nat. Struct. Mol. Biol. 2008, 15, 251–258. [Google Scholar] [CrossRef]
- Lennicke, C.; Cochemé, H.M. Redox regulation of the insulin signalling pathway. Redox Biol. 2021, 42, 101964. [Google Scholar] [CrossRef]
- Scheid, M.P.; Woodgett, J.R. Unravelling the activation mechanisms of protein kinase B/Akt. FEBS Lett. 2003, 546, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Downward, J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr. Opin. Cell Biol. 1998, 10, 262–267. [Google Scholar] [CrossRef]
- Matheny, R.W.; Adamo, M.L. Current perspectives on Akt Akt-ivation and Akt-ions. Exp. Biol. Med. 2009, 234, 1264–1270. [Google Scholar] [CrossRef]
- Boura-Halfon, S.; Zick, Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am. J. Physiology. Endocrinol. Metab. 2009, 296, E581–E591. [Google Scholar] [CrossRef] [Green Version]
- Gual, P.; Le Marchand-Brustel, Y.; Tanti, J.-F. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie 2005, 87, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Inflammatory mechanisms in the regulation of insulin resistance. Mol. Med. 2008, 14, 222–231. [Google Scholar] [CrossRef]
- Samuel, V.T.; Petersen, K.F.; Shulman, G.I. Lipid-induced insulin resistance: Unravelling the mechanism. Lancet 2010, 375, 2267–2277. [Google Scholar] [CrossRef] [Green Version]
- Fayyaz, S.; Japtok, L.; Kleuser, B. Divergent role of sphingosine 1-phosphate on insulin resistance. Cell. Physiol. Biochem. 2014, 34, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Coppack, S.W. Pro-inflammatory cytokines and adipose tissue. Proc. Nutr. Soc. 2001, 60, 349–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akash, M.S.H.; Rehman, K.; Liaqat, A. Tumor Necrosis Factor-Alpha: Role in Development of Insulin Resistance and Pathogenesis of Type 2 Diabetes Mellitus. J. Cell. Biochem. 2018, 119, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Senn, J.J.; Klover, P.J.; Nowak, I.A.; Zimmers, T.A.; Koniaris, L.G.; Furlanetto, R.W.; Mooney, R.A. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J. Biol. Chem. 2003, 278, 13740–13746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, P.; Lehmann, U.; Sobota, R.M.; Schmitz, J.; Niemand, C.; Linnemann, S.; Haan, S.; Behrmann, I.; Yoshimura, A.; Johnston, J.A.; et al. The role of the inhibitors of interleukin-6 signal transduction SHP2 and SOCS3 for desensitization of interleukin-6 signalling. Biochem. J. 2004, 378, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Hamann, J.; Koning, N.; Pouwels, W.; Ulfman, L.H.; van Eijk, M.; Stacey, M.; Lin, H.-H.; Gordon, S.; Kwakkenbos, M.J. EMR1, the human homolog of F4/80, is an eosinophil-specific receptor. Eur. J. Immunol. 2007, 37, 2797–2802. [Google Scholar] [CrossRef] [PubMed]
- Misharin, A.V.; Morales-Nebreda, L.; Mutlu, G.M.; Budinger, G.R.S.; Perlman, H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am. J. Respir. Cell Mol. Biol. 2013, 49, 503–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kratz, M.; Coats, B.R.; Hisert, K.B.; Hagman, D.; Mutskov, V.; Peris, E.; Schoenfelt, K.Q.; Kuzma, J.N.; Larson, I.; Billing, P.S.; et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014, 20, 614–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Blériot, C.; Chakarov, S.; Ginhoux, F. Determinants of Resident Tissue Macrophage Identity and Function. Immunity 2020, 52, 957–970. [Google Scholar] [CrossRef]
- Gentek, R.; Molawi, K.; Sieweke, M.H. Tissue macrophage identity and self-renewal. Immunol. Rev. 2014, 262, 56–73. [Google Scholar] [CrossRef]
- Van de Laar, L.; Saelens, W.; de Prijck, S.; Martens, L.; Scott, C.L.; van Isterdael, G.; Hoffmann, E.; Beyaert, R.; Saeys, Y.; Lambrecht, B.N.; et al. Yolk Sac Macrophages, Fetal Liver, and Adult Monocytes Can Colonize an Empty Niche and Develop into Functional Tissue-Resident Macrophages. Immunity 2016, 44, 755–768. [Google Scholar] [CrossRef] [Green Version]
- Molawi, K.; Wolf, Y.; Kandalla, P.K.; Favret, J.; Hagemeyer, N.; Frenzel, K.; Pinto, A.R.; Klapproth, K.; Henri, S.; Malissen, B.; et al. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 2014, 211, 2151–2158. [Google Scholar] [CrossRef]
- Oishi, Y.; Manabe, I. Macrophages in age-related chronic inflammatory diseases. NPJ Aging Mech. Dis. 2016, 2, 16018. [Google Scholar] [CrossRef] [Green Version]
- Theurl, I.; Hilgendorf, I.; Nairz, M.; Tymoszuk, P.; Haschka, D.; Asshoff, M.; He, S.; Gerhardt, L.M.S.; Holderried, T.A.W.; Seifert, M.; et al. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat. Med. 2016, 22, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.L.; Zheng, F.; de Baetselier, P.; Martens, L.; Saeys, Y.; de Prijck, S.; Lippens, S.; Abels, C.; Schoonooghe, S.; Raes, G.; et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat. Commun. 2016, 7, 10321. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef] [Green Version]
- Guillot, A.; Tacke, F. Liver Macrophages: Old Dogmas and New Insights. Hepatol. Commun. 2019, 3, 730–743. [Google Scholar] [CrossRef] [Green Version]
- Deczkowska, A.; Weiner, A.; Amit, I. The Physiology, Pathology, and Potential Therapeutic Applications of the TREM2 Signaling Pathway. Cell 2020, 181, 1207–1217. [Google Scholar] [CrossRef]
- Hamerman, J.A.; Jarjoura, J.R.; Humphrey, M.B.; Nakamura, M.C.; Seaman, W.E.; Lanier, L.L. Cutting edge: Inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J. Immunol. 2006, 177, 2051–2055. [Google Scholar] [CrossRef]
- Jaitin, D.A.; Adlung, L.; Thaiss, C.A.; Weiner, A.; Li, B.; Descamps, H.; Lundgren, P.; Bleriot, C.; Liu, Z.; Deczkowska, A.; et al. Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell 2019, 178, 686–698.e14. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef] [Green Version]
- De Luca, C.; Olefsky, J.M. Inflammation and insulin resistance. FEBS Lett. 2008, 582, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Engin, A.B. Adipocyte-Macrophage Cross-Talk in Obesity. Adv. Exp. Med. Biol. 2017, 960, 327–343. [Google Scholar]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef] [PubMed]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Devisscher, L.; Scott, C.L.; Lefere, S.; Raevens, S.; Bogaerts, E.; Paridaens, A.; Verhelst, X.; Geerts, A.; Guilliams, M.; van Vlierberghe, H. Non-alcoholic steatohepatitis induces transient changes within the liver macrophage pool. Cell. Immunol. 2017, 322, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Plüddemann, A. Tissue macrophages: Heterogeneity and functions. BMC Biol. 2017, 15, 53. [Google Scholar] [CrossRef] [PubMed]
- Reid, D.T.; Reyes, J.L.; McDonald, B.A.; Vo, T.; Reimer, R.A.; Eksteen, B. Kupffer Cells Undergo Fundamental Changes during the Development of Experimental NASH and Are Critical in Initiating Liver Damage and Inflammation. PLoS ONE 2016, 11, e0159524. [Google Scholar] [CrossRef] [Green Version]
- Dowman, J.K.; Tomlinson, J.W.; Newsome, P.N. Pathogenesis of non-alcoholic fatty liver disease. QJM Mon. J. Assoc. Physicians 2010, 103, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Brunt, E.M.; Wong, V.W.-S.; Nobili, V.; Day, C.P.; Sookoian, S.; Maher, J.J.; Bugianesi, E.; Sirlin, C.B.; Neuschwander-Tetri, B.A.; Rinella, M.E. Nonalcoholic fatty liver disease. Nat. Rev. Dis. Primers 2015, 1, 15080. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Maimone, D.; Grimaldi, L.M.; Incorpora, G.; Biondi, R.; Sofia, V.; Mancuso, G.R.; Siciliano, L.; Ruscica, M.; Pavone, L. Intrathecal interferon in subacute sclerosing panencephalitis. Acta Neurol. Scand. 1988, 78, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.W.; Adams, L.A. Nonalcoholic fatty liver disease and diabetes mellitus: Pathogenesis and treatment. Nat. Rev. Endocrinol. 2011, 7, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef]
- David, B.A.; Rezende, R.M.; Antunes, M.M.; Santos, M.M.; Freitas Lopes, M.A.; Diniz, A.B.; Sousa Pereira, R.V.; Marchesi, S.C.; Alvarenga, D.M.; Nakagaki, B.N.; et al. Combination of Mass Cytometry and Imaging Analysis Reveals Origin, Location, and Functional Repopulation of Liver Myeloid Cells in Mice. Gastroenterology 2016, 151, 1176–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacParland, S.A.; Liu, J.C.; Ma, X.-Z.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef] [Green Version]
- Roth, K.; Rockwell, C.E.; Copple, B.L. Differential Sensitivity of Kupffer Cells and Hepatic Monocyte-Derived Macrophages to Bacterial Lipopolysaccharide. Clin. Exp. Gastroenterol. Hepatol. 2019, 1, 106. [Google Scholar]
- Bonnardel, J.; T’Jonck, W.; Gaublomme, D.; Browaeys, R.; Scott, C.L.; Martens, L.; Vanneste, B.; de Prijck, S.; Nedospasov, S.A.; Kremer, A.; et al. Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche. Immunity 2019, 51, 638–654.e9. [Google Scholar] [CrossRef] [Green Version]
- Tran, S.; Baba, I.; Poupel, L.; Dussaud, S.; Moreau, M.; Gélineau, A.; Marcelin, G.; Magréau-Davy, E.; Ouhachi, M.; Lesnik, P.; et al. Impaired Kupffer Cell Self-Renewal Alters the Liver Response to Lipid Overload during Non-alcoholic Steatohepatitis. Immunity 2020, 53, 627–640.e5. [Google Scholar] [CrossRef]
- Wen, Y.; Lambrecht, J.; Ju, C.; Tacke, F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell. Mol. Immunol. 2021, 18, 45–56. [Google Scholar] [CrossRef]
- Remmerie, A.; Martens, L.; Thoné, T.; Castoldi, A.; Seurinck, R.; Pavie, B.; Roels, J.; Vanneste, B.; de Prijck, S.; Vanhockerhout, M.; et al. Osteopontin Expression Identifies a Subset of Recruited Macrophages Distinct from Kupffer Cells in the Fatty Liver. Immunity 2020, 53, 641–657.e14. [Google Scholar] [CrossRef]
- Sakai, M.; Troutman, T.D.; Seidman, J.S.; Ouyang, Z.; Spann, N.J.; Abe, Y.; Ego, K.M.; Bruni, C.M.; Deng, Z.; Schlachetzki, J.C.M.; et al. Liver-Derived Signals Sequentially Reprogram Myeloid Enhancers to Initiate and Maintain Kupffer Cell Identity. Immunity 2019, 51, 655–670.e8. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, M.; Hanyu, N.; Wako, T.; Wada, S.; Yanagisawa, N. A case of portal-systemic encephalopathy, showing synchronous sharp wave on the electroencephalogram. Nihon Naika Gakkai Zasshi J. Jpn. Soc. Intern. Med. 1988, 77, 1257–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.; Kuang, H.; Ansari, S.; Liu, T.; Gong, J.; Wang, S.; Zhao, X.-Y.; Ji, Y.; Li, C.; Guo, L.; et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol. Cell 2019, 75, 644–660.e5. [Google Scholar] [CrossRef] [PubMed]
- Seidman, J.S.; Troutman, T.D.; Sakai, M.; Gola, A.; Spann, N.J.; Bennett, H.; Bruni, C.M.; Ouyang, Z.; Li, R.Z.; Sun, X.; et al. Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis. Immunity 2020, 52, 1057–1074.e7. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Bar, R.S.; Kahn, C.R.; Koren, H.S. Insulin inhibition of antibody-dependent cytoxicity and insulin receptors in macrophages. Nature 1977, 265, 632–635. [Google Scholar] [CrossRef]
- Manowsky, J.; Camargo, R.G.; Kipp, A.P.; Henkel, J.; Püschel, G.P. Insulin-induced cytokine production in macrophages causes insulin resistance in hepatocytes. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E938–E946. [Google Scholar] [CrossRef] [Green Version]
- Toda, G.; Soeda, K.; Okazaki, Y.; Kobayashi, N.; Masuda, Y.; Arakawa, N.; Suwanai, H.; Masamoto, Y.; Izumida, Y.; Kamei, N.; et al. Insulin- and Lipopolysaccharide-Mediated Signaling in Adipose Tissue Macrophages Regulates Postprandial Glycemia through Akt-mTOR Activation. Mol. Cell 2020, 79, 43–53.e4. [Google Scholar] [CrossRef]
- Brown, J.; Wang, H.; Hajishengallis, G.N.; Martin, M. TLR-signaling networks: An integration of adaptor molecules, kinases, and cross-talk. J. Dent. Res. 2011, 90, 417–427. [Google Scholar] [CrossRef]
- Chen, T.-L.; Chang, C.-C.; Lin, Y.-L.; Ueng, Y.-F.; Chen, R.-M. Signal-transducing mechanisms of ketamine-caused inhibition of interleukin-1 beta gene expression in lipopolysaccharide-stimulated murine macrophage-like Raw 264.7 cells. Toxicol. Appl. Pharmacol. 2009, 240, 15–25. [Google Scholar] [CrossRef]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.; Chen, Z.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Laird, M.H.W.; Rhee, S.H.; Perkins, D.J.; Medvedev, A.E.; Piao, W.; Fenton, M.J.; Vogel, S.N. TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J. Leukoc. Biol. 2009, 85, 966–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Sierra, S.; Deshmukh, S.D.; Kalnitski, J.; Küenzi, P.; Wymann, M.P.; Golenbock, D.T.; Henneke, P. Mal connects TLR2 to PI3Kinase activation and phagocyte polarization. EMBO J. 2009, 28, 2018–2027. [Google Scholar] [CrossRef]
- Arranz, A.; Doxaki, C.; Vergadi, E.; La Martinez de Torre, Y.; Vaporidi, K.; Lagoudaki, E.D.; Ieronymaki, E.; Androulidaki, A.; Venihaki, M.; Margioris, A.N.; et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc. Natl. Acad. Sci. USA 2012, 109, 9517–9522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J. Immunol. 2017, 198, 1006–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keegan, A.D.; Zamorano, J.; Keselman, A.; Heller, N.M. IL-4 and IL-13 Receptor Signaling From 4PS to Insulin Receptor Substrate 2: There and Back Again, a Historical View. Front. Immunol. 2018, 9, 1037. [Google Scholar] [CrossRef]
- Lee, A.J.; Ashkar, A.A. The Dual Nature of Type I and Type II Interferons. Front. Immunol. 2018, 9, 2061. [Google Scholar] [CrossRef] [Green Version]
- Orliaguet, L.; Ejlalmanesh, T.; Alzaid, F. Metabolic and Molecular Mechanisms of Macrophage Polarisation and Adipose Tissue Insulin Resistance. Int. J. Mol. Sci. 2020, 21, 5731. [Google Scholar] [CrossRef]
- Saha, S.; Shalova, I.N.; Biswas, S.K. Metabolic regulation of macrophage phenotype and function. Immunol. Rev. 2017, 280, 102–111. [Google Scholar] [CrossRef]
- Van Diepen, J.A.; Robben, J.H.; Hooiveld, G.J.; Carmone, C.; Alsady, M.; Boutens, L.; Bekkenkamp-Grovenstein, M.; Hijmans, A.; Engelke, U.F.H.; Wevers, R.A.; et al. SUCNR1-mediated chemotaxis of macrophages aggravates obesity-induced inflammation and diabetes. Diabetologia 2017, 60, 1304–1313. [Google Scholar] [CrossRef] [Green Version]
- Serena, C.; Ceperuelo-Mallafré, V.; Keiran, N.; Queipo-Ortuño, M.I.; Bernal, R.; Gomez-Huelgas, R.; Urpi-Sarda, M.; Sabater, M.; Pérez-Brocal, V.; Andrés-Lacueva, C.; et al. Elevated circulating levels of succinate in human obesity are linked to specific gut microbiota. ISME J. 2018, 12, 1642–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Kietzmann, T.; Mennerich, D.; Dimova, E.Y. Hypoxia-Inducible Factors (HIFs) and Phosphorylation: Impact on Stability, Localization, and Transactivity. Front. Cell Dev. Biol. 2016, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, V.A.; Carvalho, D.; Freitas, P. Obesity, Adipose Tissue, and Inflammation Answered in Questions. J. Obes. 2022, 2022, 2252516. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Arkan, M.C.; Hevener, A.L.; Greten, F.R.; Maeda, S.; Li, Z.-W.; Long, J.M.; Wynshaw-Boris, A.; Poli, G.; Olefsky, J.; Karin, M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat. Med. 2005, 11, 191–198. [Google Scholar] [CrossRef]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Dalmas, E.; Venteclef, N.; Caer, C.; Poitou, C.; Cremer, I.; Aron-Wisnewsky, J.; Lacroix-Desmazes, S.; Bayry, J.; Kaveri, S.V.; Clément, K.; et al. T cell-derived IL-22 amplifies IL-1β-driven inflammation in human adipose tissue: Relevance to obesity and type 2 diabetes. Diabetes 2014, 63, 1966–1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amano, S.U.; Cohen, J.L.; Vangala, P.; Tencerova, M.; Nicoloro, S.M.; Yawe, J.C.; Shen, Y.; Czech, M.P.; Aouadi, M. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab. 2014, 19, 162–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, C.; Yang, Q.; Cao, J.; Xie, N.; Liu, K.; Shou, P.; Qian, F.; Wang, Y.; Shi, Y. Local proliferation initiates macrophage accumulation in adipose tissue during obesity. Cell Death Dis. 2016, 7, e2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.; Zhang, H.; Zhu, H. Blocking CXCR7-mediated adipose tissue macrophages chemotaxis attenuates insulin resistance and inflammation in obesity. Biochem. Biophys. Res. Commun. 2016, 479, 649–655. [Google Scholar] [CrossRef]
- Spite, M.; Hellmann, J.; Tang, Y.; Mathis, S.P.; Kosuri, M.; Bhatnagar, A.; Jala, V.R.; Haribabu, B. Deficiency of the leukotriene B4 receptor, BLT-1, protects against systemic insulin resistance in diet-induced obesity. J. Immunol. 2011, 187, 1942–1949. [Google Scholar] [CrossRef] [Green Version]
- Patsouris, D.; Li, P.-P.; Thapar, D.; Chapman, J.; Olefsky, J.M.; Neels, J.G. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. 2008, 8, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Kamei, N.; Tobe, K.; Suzuki, R.; Ohsugi, M.; Watanabe, T.; Kubota, N.; Ohtsuka-Kowatari, N.; Kumagai, K.; Sakamoto, K.; Kobayashi, M.; et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J. Biol. Chem. 2006, 281, 26602–26614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finucane, O.M.; Reynolds, C.M.; McGillicuddy, F.C.; Harford, K.A.; Morrison, M.; Baugh, J.; Roche, H.M. Macrophage migration inhibitory factor deficiency ameliorates high-fat diet induced insulin resistance in mice with reduced adipose inflammation and hepatic steatosis. PLoS ONE 2014, 9, e113369. [Google Scholar] [CrossRef] [PubMed]
- McGillicuddy, F.C.; Harford, K.A.; Reynolds, C.M.; Oliver, E.; Claessens, M.; Mills, K.H.G.; Roche, H.M. Lack of interleukin-1 receptor I (IL-1RI) protects mice from high-fat diet-induced adipose tissue inflammation coincident with improved glucose homeostasis. Diabetes 2011, 60, 1688–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.; Iyer, A.; Suen, J.Y.; Seow, V.; Reid, R.C.; Brown, L.; Fairlie, D.P. C5aR and C3aR antagonists each inhibit diet-induced obesity, metabolic dysfunction, and adipocyte and macrophage signaling. FASEB J. 2013, 27, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Phieler, J.; Chung, K.-J.; Chatzigeorgiou, A.; Klotzsche-von Ameln, A.; Garcia-Martin, R.; Sprott, D.; Moisidou, M.; Tzanavari, T.; Ludwig, B.; Baraban, E.; et al. The complement anaphylatoxin C5a receptor contributes to obese adipose tissue inflammation and insulin resistance. J. Immunol. 2013, 191, 4367–4374. [Google Scholar] [CrossRef] [Green Version]
- Mamane, Y.; Chung Chan, C.; Lavallee, G.; Morin, N.; Xu, L.-J.; Huang, J.; Gordon, R.; Thomas, W.; Lamb, J.; Schadt, E.E.; et al. The C3a anaphylatoxin receptor is a key mediator of insulin resistance and functions by modulating adipose tissue macrophage infiltration and activation. Diabetes 2009, 58, 2006–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramkhelawon, B.; Hennessy, E.J.; Ménager, M.; Ray, T.D.; Sheedy, F.J.; Hutchison, S.; Wanschel, A.; Oldebeken, S.; Geoffrion, M.; Spiro, W.; et al. Netrin-1 promotes adipose tissue macrophage retention and insulin resistance in obesity. Nat. Med. 2014, 20, 377–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.; Schlegel, M.; Brown, E.J.; Sansbury, B.E.; Weinstock, A.; Afonso, M.S.; Corr, E.M.; van Solingen, C.; Shanley, L.C.; Peled, D.; et al. Netrin-1 Alters Adipose Tissue Macrophage Fate and Function in Obesity. Immunometabolism 2019, 1, e190010. [Google Scholar]
- Bu, L.; Gao, M.; Qu, S.; Liu, D. Intraperitoneal injection of clodronate liposomes eliminates visceral adipose macrophages and blocks high-fat diet-induced weight gain and development of insulin resistance. AAPS J. 2013, 15, 1001–1011. [Google Scholar] [CrossRef] [Green Version]
- Feng, B.; Jiao, P.; Nie, Y.; Kim, T.; Jun, D.; van Rooijen, N.; Yang, Z.; Xu, H. Clodronate liposomes improve metabolic profile and reduce visceral adipose macrophage content in diet-induced obese mice. PLoS ONE 2011, 6, e24358. [Google Scholar] [CrossRef] [PubMed]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef]
- Lanthier, N.; Molendi-Coste, O.; Horsmans, Y.; van Rooijen, N.; Cani, P.D.; Leclercq, I.A. Kupffer cell activation is a causal factor for hepatic insulin resistance. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G107–G116. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Metlakunta, A.; Dedousis, N.; Zhang, P.; Sipula, I.; Dube, J.J.; Scott, D.K.; O’Doherty, R.M. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes 2010, 59, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Neyrinck, A.M.; Cani, P.D.; Dewulf, E.M.; de Backer, F.; Bindels, L.B.; Delzenne, N.M. Critical role of Kupffer cells in the management of diet-induced diabetes and obesity. Biochem. Biophys. Res. Commun. 2009, 385, 351–356. [Google Scholar] [CrossRef]
- Clementi, A.H.; Gaudy, A.M.; van Rooijen, N.; Pierce, R.H.; Mooney, R.A. Loss of Kupffer cells in diet-induced obesity is associated with increased hepatic steatosis, STAT3 signaling, and further decreases in insulin signaling. Biochim. Biophys. Acta 2009, 1792, 1062–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinoshita, M.; Uchida, T.; Sato, A.; Nakashima, M.; Nakashima, H.; Shono, S.; Habu, Y.; Miyazaki, H.; Hiroi, S.; Seki, S. Characterization of two F4/80-positive Kupffer cell subsets by their function and phenotype in mice. J. Hepatol. 2010, 53, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Movita, D.; Kreefft, K.; Biesta, P.; van Oudenaren, A.; Leenen, P.J.M.; Janssen, H.L.A.; Boonstra, A. Kupffer cells express a unique combination of phenotypic and functional characteristics compared with splenic and peritoneal macrophages. J. Leukoc. Biol. 2012, 92, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef]
- Baeck, C.; Wehr, A.; Karlmark, K.R.; Heymann, F.; Vucur, M.; Gassler, N.; Huss, S.; Klussmann, S.; Eulberg, D.; Luedde, T.; et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2012, 61, 416–426. [Google Scholar] [CrossRef]
- Roh, Y.S.; Zhang, B.; Loomba, R.; Seki, E. TLR2 and TLR9 contribute to alcohol-mediated liver injury through induction of CXCL1 and neutrophil infiltration. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G30–G41. [Google Scholar] [CrossRef] [Green Version]
- Karlmark, K.R.; Weiskirchen, R.; Zimmermann, H.W.; Gassler, N.; Ginhoux, F.; Weber, C.; Merad, M.; Luedde, T.; Trautwein, C.; Tacke, F. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009, 50, 261–274. [Google Scholar] [CrossRef]
- Sharifnia, T.; Antoun, J.; Verriere, T.G.C.; Suarez, G.; Wattacheril, J.; Wilson, K.T.; Peek, R.M.; Abumrad, N.N.; Flynn, C.R. Hepatic TLR4 signaling in obese NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G270–G278. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, H.; Nakashima, M.; Kinoshita, M.; Ikarashi, M.; Miyazaki, H.; Hanaka, H.; Imaki, J.; Seki, S. Activation and increase of radio-sensitive CD11b+ recruited Kupffer cells/macrophages in diet-induced steatohepatitis in FGF5 deficient mice. Sci. Rep. 2016, 6, 34466. [Google Scholar] [CrossRef]
- Miura, K.; Yang, L.; van Rooijen, N.; Ohnishi, H.; Seki, E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1310–G1321. [Google Scholar] [CrossRef] [Green Version]
- Krenkel, O.; Puengel, T.; Govaere, O.; Abdallah, A.T.; Mossanen, J.C.; Kohlhepp, M.; Liepelt, A.; Lefebvre, E.; Luedde, T.; Hellerbrand, C.; et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018, 67, 1270–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negrin, K.A.; Roth Flach, R.J.; DiStefano, M.T.; Matevossian, A.; Friedline, R.H.; Jung, D.; Kim, J.K.; Czech, M.P. IL-1 signaling in obesity-induced hepatic lipogenesis and steatosis. PLoS ONE 2014, 9, e107265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tencerova, M.; Aouadi, M.; Vangala, P.; Nicoloro, S.M.; Yawe, J.C.; Cohen, J.L.; Shen, Y.; Garcia-Menendez, L.; Pedersen, D.J.; Gallagher-Dorval, K.; et al. Activated Kupffer cells inhibit insulin sensitivity in obese mice. FASEB J. 2015, 29, 2959–2969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jourdan, T.; Nicoloro, S.M.; Zhou, Z.; Shen, Y.; Liu, J.; Coffey, N.J.; Cinar, R.; Godlewski, G.; Gao, B.; Aouadi, M.; et al. Decreasing CB1 receptor signaling in Kupffer cells improves insulin sensitivity in obese mice. Mol. Metab. 2017, 6, 1517–1528. [Google Scholar] [CrossRef]
- Pardo, V.; González-Rodríguez, Á.; Guijas, C.; Balsinde, J.; Valverde, Á.M. Opposite cross-talk by oleate and palmitate on insulin signaling in hepatocytes through macrophage activation. J. Biol. Chem. 2015, 290, 11663–11677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henkel, J.; Coleman, C.D.; Schraplau, A.; Jöhrens, K.; Weiss, T.S.; Jonas, W.; Schürmann, A.; Püschel, G.P. Augmented liver inflammation in a microsomal prostaglandin E synthase 1 (mPGES-1)-deficient diet-induced mouse NASH model. Sci. Rep. 2018, 8, 16127. [Google Scholar] [CrossRef] [Green Version]
- Henkel, J.; Gärtner, D.; Dorn, C.; Hellerbrand, C.; Schanze, N.; Elz, S.R.; Püschel, G.P. Oncostatin M produced in Kupffer cells in response to PGE2: Possible contributor to hepatic insulin resistance and steatosis. Lab. Investig. 2011, 91, 1107–1117. [Google Scholar] [CrossRef]
- Gierej, P.; Gierej, B.; Kalinowski, P.; Wróblewski, T.; Paluszkiewicz, R.; Kobryń, K.; Ziarkiewicz-Wróblewska, B. Expression of resistin in the liver of patients with non-alcoholic fatty liver disease. Pol. J. Pathol. 2017, 68, 225–233. [Google Scholar] [CrossRef]
- Li, P.; Da Oh, Y.; Bandyopadhyay, G.; Lagakos, W.S.; Talukdar, S.; Osborn, O.; Johnson, A.; Chung, H.; Maris, M.; Ofrecio, J.M.; et al. LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat. Med. 2015, 21, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Sakagami, Y.; Mizoguchi, Y.; Seki, S.; Kobayashi, K.; Morisawa, S.; Yamamoto, S. Release of leukotriene B4 from rat Kupffer cells. Prostaglandins Leukot. Essent. Fat. Acids 1989, 36, 125–128. [Google Scholar] [CrossRef]
- Klauder, J.; Henkel, J.; Vahrenbrink, M.; Wohlenberg, A.-S.; Camargo, R.G.; Püschel, G.P. Direct and indirect modulation of LPS-induced cytokine production by insulin in human macrophages. Cytokine 2020, 136, 155241. [Google Scholar] [CrossRef]
- Henkel, J.; Klauder, J.; Statz, M.; Wohlenberg, A.-S.; Kuipers, S.; Vahrenbrink, M.; Püschel, G.P. Enhanced Palmitate-Induced Interleukin-8 Formation in Human Macrophages by Insulin or Prostaglandin E2. Biomedicines 2021, 9, 449. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.; Neuschäfer-Rube, F.; Pathe-Neuschäfer-Rube, A.; Püschel, G.P. Aggravation by prostaglandin E2 of interleukin-6-dependent insulin resistance in hepatocytes. Hepatology 2009, 50, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Fennekohl, A.; Sugimoto, Y.; Segi, E.; Maruyama, T.; Ichikawa, A.; Püschel, G.P. Contribution of the two Gs-coupled PGE2-receptors EP2-receptor and EP4-receptor to the inhibition by PGE2 of the LPS-induced TNFalpha-formation in Kupffer cells from EP2-or EP4-receptor-deficient mice. Pivotal role for the EP4-receptor in wild type Kupffer cells. J. Hepatol. 2002, 36, 328–334. [Google Scholar] [PubMed]
- Shimobayashi, M.; Albert, V.; Woelnerhanssen, B.; Frei, I.C.; Weissenberger, D.; Meyer-Gerspach, A.C.; Clement, N.; Moes, S.; Colombi, M.; Meier, J.A.; et al. Insulin resistance causes inflammation in adipose tissue. J. Clin. Investig. 2018, 128, 1538–1550. [Google Scholar] [CrossRef] [PubMed]
- Espinosa De Ycaza, A.E.; Søndergaard, E.; Morgan-Bathke, M.; Lytle, K.; Delivanis, D.A.; Ramos, P.; Carranza Leon, B.G.; Jensen, M.D. Adipose Tissue Inflammation Is Not Related to Adipose Insulin Resistance in Humans. Diabetes 2022, 71, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Villarreal-Calderon, J.R.; Cuellar-Tamez, R.; Castillo, E.C.; Luna-Ceron, E.; García-Rivas, G.; Elizondo-Montemayor, L. Metabolic shift precedes the resolution of inflammation in a cohort of patients undergoing bariatric and metabolic surgery. Sci. Rep. 2021, 11, 12127. [Google Scholar] [CrossRef]
- Lee, Y.S.; Li, P.; Huh, J.Y.; Hwang, I.J.; Lu, M.; Kim, J.; Ham, M.; Talukdar, S.; Chen, A.; Lu, W.J.; et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 2011, 60, 2474–2483. [Google Scholar] [CrossRef] [Green Version]
- Holland, W.L.; Brozinick, J.T.; Wang, L.-P.; Hawkins, E.D.; Sargent, K.M.; Liu, Y.; Narra, K.; Hoehn, K.L.; Knotts, T.A.; Siesky, A.; et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007, 5, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Frangioudakis, G.; Cooney, G.J. Acute elevation of circulating fatty acids impairs downstream insulin signalling in rat skeletal muscle in vivo independent of effects on stress signalling. J. Endocrinol. 2008, 197, 277–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roden, M.; Price, T.B.; Perseghin, G.; Petersen, K.F.; Rothman, D.L.; Cline, G.W.; Shulman, G.I. Mechanism of free fatty acid-induced insulin resistance in humans. J. Clin. Investig. 1996, 97, 2859–2865. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.K.; Kim, Y.J.; Fillmore, J.J.; Chen, Y.; Moore, I.; Lee, J.; Yuan, M.; Li, Z.W.; Karin, M.; Perret, P.; et al. Prevention of fat-induced insulin resistance by salicylate. J. Clin. Investig. 2001, 108, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Fillmore, J.J.; Sunshine, M.J.; Albrecht, B.; Higashimori, T.; Kim, D.-W.; Liu, Z.-X.; Soos, T.J.; Cline, G.W.; O’Brien, W.R.; et al. PKC-theta knockout mice are protected from fat-induced insulin resistance. J. Clin. Investig. 2004, 114, 823–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, K.; Zhang, D.; Song, J.; Li, X.; Perry, R.J.; Samuel, V.T.; Shulman, G.I. Short-term overnutrition induces white adipose tissue insulin resistance through sn-1,2-diacylglycerol/PKCε/insulin receptor Thr1160 phosphorylation. JCI Insight 2021, 6, e139946. [Google Scholar]
- Wang, L.; Pydi, S.P.; Zhu, L.; Barella, L.F.; Cui, Y.; Gavrilova, O.; Bence, K.K.; Vernochet, C.; Wess, J. Adipocyte Gi signaling is essential for maintaining whole-body glucose homeostasis and insulin sensitivity. Nat. Commun. 2020, 11, 2995. [Google Scholar] [CrossRef]
- Tirosh, O. Hypoxic Signaling and Cholesterol Lipotoxicity in Fatty Liver Disease Progression. Oxidative Med. Cell. Longev. 2018, 2018, 2548154. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Subramanian, S.; Chait, A.; Haigh, W.G.; Yeh, M.M.; Farrell, G.C.; Lee, S.P.; Savard, C. Cholesterol crystallization within hepatocyte lipid droplets and its role in murine NASH. J. Lipid Res. 2017, 58, 1067–1079. [Google Scholar] [CrossRef] [Green Version]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Legry, V.; van Rooyen, D.M.; Lambert, B.; Sempoux, C.; Poekes, L.; Español-Suñer, R.; Molendi-Coste, O.; Horsmans, Y.; Farrell, G.C.; Leclercq, I.A. Endoplasmic reticulum stress does not contribute to steatohepatitis in obese and insulin-resistant high-fat-diet-fed foz/foz mice. Clin. Sci. 2014, 127, 507–518. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, G.N.; Haigh, W.G.; Thorning, D.; Savard, C. Hepatic cholesterol crystals and crown-like structures distinguish NASH from simple steatosis. J. Lipid Res. 2013, 54, 1326–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannou, G.N.; Landis, C.S.; Jin, G.-Y.; Haigh, W.G.; Farrell, G.C.; Kuver, R.; Lee, S.P.; Savard, C. Cholesterol Crystals in Hepatocyte Lipid Droplets Are Strongly Associated with Human Nonalcoholic Steatohepatitis. Hepatol. Commun. 2019, 3, 776–791. [Google Scholar] [CrossRef]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marí, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; García-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef]
- Schrauwen, P.; Schrauwen-Hinderling, V.; Hoeks, J.; Hesselink, M.K.C. Mitochondrial dysfunction and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Burgos-Morón, E.; Abad-Jiménez, Z.; de Marañón, A.M.; Iannantuoni, F.; Escribano-López, I.; López-Domènech, S.; Salom, C.; Jover, A.; Mora, V.; Roldan, I.; et al. Relationship Between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues. J. Clin. Med. 2019, 8, 1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vejux, A.; Guyot, S.; Montange, T.; Riedinger, J.-M.; Kahn, E.; Lizard, G. Phospholipidosis and down-regulation of the PI3-K/PDK-1/Akt signalling pathway are vitamin E inhibitable events associated with 7-ketocholesterol-induced apoptosis. J. Nutr. Biochem. 2009, 20, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Drake, W.P.; Pendergrast, W.J.; Kramer, R.E.; Mardiney, M.R. The age-dependent efficacy of polyadenylic-polyuridylic acid therapy upon the development of spontaneous leukemia in AKR mice. Cancer Res. 1976, 36, 1172–1175. [Google Scholar]
- Song, S.; Andrikopoulos, S.; Filippis, C.; Thorburn, A.W.; Khan, D.; Proietto, J. Mechanism of fat-induced hepatic gluconeogenesis: Effect of metformin. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E275–E282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, G.F.; Vranic, M.; Harley, P.; Giacca, A. Fatty acids mediate the acute extrahepatic effects of insulin on hepatic glucose production in humans. Diabetes 1997, 46, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Roden, M.; Stingl, H.; Chandramouli, V.; Schumann, W.C.; Hofer, A.; Landau, B.R.; Nowotny, P.; Waldhäusl, W.; Shulman, G.I. Effects of free fatty acid elevation on postabsorptive endogenous glucose production and gluconeogenesis in humans. Diabetes 2000, 49, 701–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.-H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalobos-Labra, R.; Subiabre, M.; Toledo, F.; Pardo, F.; Sobrevia, L. Endoplasmic reticulum stress and development of insulin resistance in adipose, skeletal, liver, and foetoplacental tissue in diabesity. Mol. Asp. Med. 2019, 66, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, S.-W.; Wang, Z.-M.; Sun, S.-M.; Su, Y.; Li, Z.-H.; Shao, J.-J.; Tan, S.-Z.; Chen, A.-P.; Wang, S.-J.; Zhang, Z.-L.; et al. Endoplasmic reticulum stress and protein degradation in chronic liver disease. Pharmacol. Res. 2020, 161, 105218. [Google Scholar] [CrossRef] [PubMed]
- Birkenfeld, A.L.; Lee, H.-Y.; Majumdar, S.; Jurczak, M.J.; Camporez, J.P.; Jornayvaz, F.R.; Frederick, D.W.; Guigni, B.; Kahn, M.; Zhang, D.; et al. Influence of the hepatic eukaryotic initiation factor 2alpha (eIF2alpha) endoplasmic reticulum (ER) stress response pathway on insulin-mediated ER stress and hepatic and peripheral glucose metabolism. J. Biol. Chem. 2011, 286, 36163–36170. [Google Scholar] [CrossRef] [Green Version]
- Williams, L.M.; Campbell, F.M.; Drew, J.E.; Koch, C.; Hoggard, N.; Rees, W.D.; Kamolrat, T.; Thi Ngo, H.; Steffensen, I.-L.; Gray, S.R.; et al. The development of diet-induced obesity and glucose intolerance in C57BL/6 mice on a high-fat diet consists of distinct phases. PLoS ONE 2014, 9, e106159. [Google Scholar] [CrossRef] [Green Version]
- Šiklová, M.; Krauzová, E.; Svobodová, B.; Kračmerová, J.; Štěpán, M.; Koc, M.; Štich, V.; Rossmeislová, L. Circulating Monocyte and Lymphocyte Populations in Healthy First-Degree Relatives of Type 2 Diabetic Patients at Fasting and during Short-Term Hyperinsulinemia. Mediat. Inflamm. 2019, 2019, 1491083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferris, S.T.; Zakharov, P.N.; Wan, X.; Calderon, B.; Artyomov, M.N.; Unanue, E.R.; Carrero, J.A. The islet-resident macrophage is in an inflammatory state and senses microbial products in blood. J. Exp. Med. 2017, 214, 2369–2385. [Google Scholar] [CrossRef]
- Dror, E.; Dalmas, E.; Meier, D.T.; Wueest, S.; Thévenet, J.; Thienel, C.; Timper, K.; Nordmann, T.M.; Traub, S.; Schulze, F.; et al. Postprandial macrophage-derived IL-1β stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol. 2017, 18, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Costa Rosa, L.F.; Safi, D.A.; Cury, Y.; Curi, R. The effect of insulin on macrophage metabolism and function. Cell Biochem. Funct. 1996, 14, 33–42. [Google Scholar] [CrossRef]
- Sun, C.; Sun, L.; Ma, H.; Peng, J.; Zhen, Y.; Duan, K.; Liu, G.; Ding, W.; Zhao, Y. The phenotype and functional alterations of macrophages in mice with hyperglycemia for long term. J. Cell. Physiol. 2012, 227, 1670–1679. [Google Scholar] [CrossRef]
- Jansen, H.J.; Stienstra, R.; van Diepen, J.A.; Hijmans, A.; van der Laak, J.A.; Vervoort, G.M.M.; Tack, C.J. Start of insulin therapy in patients with type 2 diabetes mellitus promotes the influx of macrophages into subcutaneous adipose tissue. Diabetologia 2013, 56, 2573–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krogh-Madsen, R.; Plomgaard, P.; Keller, P.; Keller, C.; Pedersen, B.K. Insulin stimulates interleukin-6 and tumor necrosis factor-alpha gene expression in human subcutaneous adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E234–E238. [Google Scholar] [CrossRef]
- Krogh-Madsen, R.; Møller, K.; Dela, F.; Kronborg, G.; Jauffred, S.; Pedersen, B.K. Effect of hyperglycemia and hyperinsulinemia on the response of IL-6, TNF-alpha, and FFAs to low-dose endotoxemia in humans. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E766–E772. [Google Scholar] [CrossRef] [Green Version]
- Siklova-Vitkova, M.; Polak, J.; Klimcakova, E.; Vrzalova, J.; Hejnova, J.; Kovacikova, M.; Kovacova, Z.; Bajzova, M.; Rossmeislova, L.; Hnevkovska, Z.; et al. Effect of hyperinsulinemia and very-low-calorie diet on interstitial cytokine levels in subcutaneous adipose tissue of obese women. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1154–E1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruge, T.; Lockton, J.A.; Renstrom, F.; Lystig, T.; Sukonina, V.; Svensson, M.K.; Eriksson, J.W. Acute hyperinsulinemia raises plasma interleukin-6 in both nondiabetic and type 2 diabetes mellitus subjects, and this effect is inversely associated with body mass index. Metab. Clin. Exp. 2009, 58, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, D.J.; Guilherme, A.; Danai, L.V.; Heyda, L.; Matevossian, A.; Cohen, J.; Nicoloro, S.M.; Straubhaar, J.; Noh, H.L.; Jung, D.; et al. A major role of insulin in promoting obesity-associated adipose tissue inflammation. Mol. Metab. 2015, 4, 507–518. [Google Scholar] [CrossRef]
- Fain, J.N. Release of interleukins and other inflammatory cytokines by human adipose tissue is enhanced in obesity and primarily due to the nonfat cells. Vitam. Horm. 2006, 74, 443–477. [Google Scholar]
- Li, G.; Barrett, E.J.; Ko, S.-H.; Cao, W.; Liu, Z. Insulin and insulin-like growth factor-I receptors differentially mediate insulin-stimulated adhesion molecule production by endothelial cells. Endocrinology 2009, 150, 3475–3482. [Google Scholar] [CrossRef] [Green Version]
- Madonna, R.; Pandolfi, A.; Massaro, M.; Consoli, A.; de Caterina, R. Insulin enhances vascular cell adhesion molecule-1 expression in human cultured endothelial cells through a pro-atherogenic pathway mediated by p38 mitogen-activated protein-kinase. Diabetologia 2004, 47, 532–536. [Google Scholar] [CrossRef] [Green Version]
- Soop, M.; Duxbury, H.; Agwunobi, A.O.; Gibson, J.M.; Hopkins, S.J.; Childs, C.; Cooper, R.G.; Maycock, P.; Little, R.A.; Carlson, G.L. Euglycemic hyperinsulinemia augments the cytokine and endocrine responses to endotoxin in humans. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E1276–E1285. [Google Scholar] [CrossRef] [Green Version]
- Iida, K.T.; Shimano, H.; Kawakami, Y.; Sone, H.; Toyoshima, H.; Suzuki, S.; Asano, T.; Okuda, Y.; Yamada, N. Insulin up-regulates tumor necrosis factor-alpha production in macrophages through an extracellular-regulated kinase-dependent pathway. J. Biol. Chem. 2001, 276, 32531–32537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerville, M.; Leroy, A.; Sinquin, A.; Laugerette, F.; Michalski, M.-C.; Boudry, G. Western-diet consumption induces alteration of barrier function mechanisms in the ileum that correlates with metabolic endotoxemia in rats. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E107–E120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [Green Version]
- Moreira, A.P.B.; Texeira, T.F.S.; Ferreira, A.B.; Peluzio, M.d.C.G.; Alfenas, R.d.C.G. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br. J. Nutr. 2012, 108, 801–809. [Google Scholar] [CrossRef]
- Mauer, J.; Chaurasia, B.; Plum, L.; Quast, T.; Hampel, B.; Blüher, M.; Kolanus, W.; Kahn, C.R.; Brüning, J.C. Myeloid cell-restricted insulin receptor deficiency protects against obesity-induced inflammation and systemic insulin resistance. PLoS Genet. 2010, 6, e1000938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgartl, J.; Baudler, S.; Scherner, M.; Babaev, V.; Makowski, L.; Suttles, J.; McDuffie, M.; Tobe, K.; Kadowaki, T.; Fazio, S.; et al. Myeloid lineage cell-restricted insulin resistance protects apolipoproteinE-deficient mice against atherosclerosis. Cell Metab. 2006, 3, 247–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rached, M.-T.; Millership, S.J.; Pedroni, S.M.A.; Choudhury, A.I.; Costa, A.S.H.; Hardy, D.G.; Glegola, J.A.; Irvine, E.E.; Selman, C.; Woodberry, M.C.; et al. Deletion of myeloid IRS2 enhances adipose tissue sympathetic nerve function and limits obesity. Mol. Metab. 2019, 20, 38–50. [Google Scholar] [CrossRef]
- Pal, S.; Nath, P.; Das, D.; Hajra, S.; Maitra, S. Cross-talk between insulin signalling and LPS responses in mouse macrophages. Mol. Cell. Endocrinol. 2018, 476, 57–69. [Google Scholar] [CrossRef]
- Tachado, S.D.; Li, X.; Swan, K.; Patel, N.; Koziel, H. Constitutive activation of phosphatidylinositol 3-kinase signaling pathway down-regulates TLR4-mediated tumor necrosis factor-alpha release in alveolar macrophages from asymptomatic HIV-positive persons in vitro. J. Biol. Chem. 2008, 283, 33191–33198. [Google Scholar] [CrossRef] [Green Version]
- Fallah, M.P.; Chelvarajan, R.L.; Garvy, B.A.; Bondada, S. Role of phosphoinositide 3-kinase-Akt signaling pathway in the age-related cytokine dysregulation in splenic macrophages stimulated via TLR-2 or TLR-4 receptors. Mech. Ageing Dev. 2011, 132, 274–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuschieri, J.; Bulger, E.; Grinsell, R.; Garcia, I.; Maier, R.V. Insulin regulates macrophage activation through activin A. Shock 2008, 29, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Pengal, R.A.; Cao, X.; Ganesan, L.P.; Wewers, M.D.; Marsh, C.B.; Tridandapani, S. Lipopolysaccharide-induced macrophage inflammatory response is regulated by SHIP. J. Immunol. 2004, 173, 360–366. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Ma, Y.; Li, Y.; Zheng, X.; Lv, P.; Zhang, Y.; Li, J.; Ma, M.; Zhang, L.; Li, C.; et al. Insulin inhibits inflammation and promotes atherosclerotic plaque stability via PI3K-Akt pathway activation. Immunol. Lett. 2016, 170, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Liang, C.-P.; DeVries-Seimon, T.; Ranalletta, M.; Welch, C.L.; Collins-Fletcher, K.; Accili, D.; Tabas, I.; Tall, A.R. Macrophage insulin receptor deficiency increases ER stress-induced apoptosis and necrotic core formation in advanced atherosclerotic lesions. Cell Metab. 2006, 3, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Leffler, M.; Hrach, T.; Stuerzl, M.; Horch, R.E.; Herndon, D.N.; Jeschke, M.G. Insulin attenuates apoptosis and exerts anti-inflammatory effects in endotoxemic human macrophages. J. Surg. Res. 2007, 143, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.O.; Ferracini, M.; Ravanelli, N.; Landgraf, R.G.; Jancar, S. Insulin inhibits LPS-induced signaling pathways in alveolar macrophages. Cell. Physiol. Biochem. 2008, 21, 297–304. [Google Scholar] [CrossRef]
- Tessaro, F.H.G.; Ayala, T.S.; Nolasco, E.L.; Bella, L.M.; Martins, J.O. Insulin Influences LPS-Induced TNF-α and IL-6 Release Through Distinct Pathways in Mouse Macrophages from Different Compartments. Cell. Physiol. Biochem. 2017, 42, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Ferracini, M.; Martins, J.O.; Campos, M.R.M.; Anger, D.B.C.; Jancar, S. Impaired phagocytosis by alveolar macrophages from diabetic rats is related to the deficient coupling of LTs to the Fc gamma R signaling cascade. Mol. Immunol. 2010, 47, 1974–1980. [Google Scholar] [CrossRef]
- Yu, T.; Gao, M.; Yang, P.; Liu, D.; Wang, D.; Song, F.; Zhang, X.; Liu, Y. Insulin promotes macrophage phenotype transition through PI3K/Akt and PPAR-γ signaling during diabetic wound healing. J. Cell. Physiol. 2019, 234, 4217–4231. [Google Scholar] [CrossRef]
- Su, D.; Coudriet, G.M.; Hyun Kim, D.; Lu, Y.; Perdomo, G.; Qu, S.; Slusher, S.; Tse, H.M.; Piganelli, J.; Giannoukakis, N.; et al. FoxO1 links insulin resistance to proinflammatory cytokine IL-1beta production in macrophages. Diabetes 2009, 58, 2624–2633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, E.A.; Devitt, A.; Johnson, J.R. Macrophages: The Good, the Bad, and the Gluttony. Front. Immunol. 2021, 12, 708186. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Inoue, M.; Kubota, N.; Takamoto, I.; Mineyama, T.; Iwayama, K.; Tokuyama, K.; Moroi, M.; Ueki, K.; Yamauchi, T.; et al. Downregulation of macrophage Irs2 by hyperinsulinemia impairs IL-4-indeuced M2a-subtype macrophage activation in obesity. Nat. Commun. 2018, 9, 4863. [Google Scholar] [CrossRef] [PubMed]
- Androulidaki, A.; Iliopoulos, D.; Arranz, A.; Doxaki, C.; Schworer, S.; Zacharioudaki, V.; Margioris, A.N.; Tsichlis, P.N.; Tsatsanis, C. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity 2009, 31, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, E.M.; Wang, J.; Redmond, H.P. The emerging role of microRNA in regulation of endotoxin tolerance. J. Leukoc. Biol. 2012, 91, 721–727. [Google Scholar] [CrossRef]
- Huang, C.; Li, G.; Dong, H.; Sun, S.; Chen, H.; Luo, D.; Sun, L.; Li, X.; Chen, Z.; Yang, H.; et al. Arg⁹⁷2 insulin receptor substrate-1 inhibits endothelial nitric oxide synthase expression in human endothelial cells by upregulating microRNA-155. Int. J. Mol. Med. 2015, 36, 239–248. [Google Scholar] [CrossRef]
- Virtue, A.; Johnson, C.; Lopez-Pastraña, J.; Shao, Y.; Fu, H.; Li, X.; Li, Y.-F.; Yin, Y.; Mai, J.; Rizzo, V.; et al. MicroRNA-155 Deficiency Leads to Decreased Atherosclerosis, Increased White Adipose Tissue Obesity, and Non-alcoholic Fatty Liver Disease: A Novel Mouse Model of Obesity Paradox. J. Biol. Chem. 2017, 292, 1267–1287. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Püschel, G.P.; Klauder, J.; Henkel, J. Macrophages, Low-Grade Inflammation, Insulin Resistance and Hyperinsulinemia: A Mutual Ambiguous Relationship in the Development of Metabolic Diseases. J. Clin. Med. 2022, 11, 4358. https://doi.org/10.3390/jcm11154358
Püschel GP, Klauder J, Henkel J. Macrophages, Low-Grade Inflammation, Insulin Resistance and Hyperinsulinemia: A Mutual Ambiguous Relationship in the Development of Metabolic Diseases. Journal of Clinical Medicine. 2022; 11(15):4358. https://doi.org/10.3390/jcm11154358
Chicago/Turabian StylePüschel, Gerhard Paul, Julia Klauder, and Janin Henkel. 2022. "Macrophages, Low-Grade Inflammation, Insulin Resistance and Hyperinsulinemia: A Mutual Ambiguous Relationship in the Development of Metabolic Diseases" Journal of Clinical Medicine 11, no. 15: 4358. https://doi.org/10.3390/jcm11154358