
Drugs That Induce or Cause Deterioration of Myasthenia Gravis: An Update


Abstract
:1. Introduction
2. Drugs That Cause de Novo MG
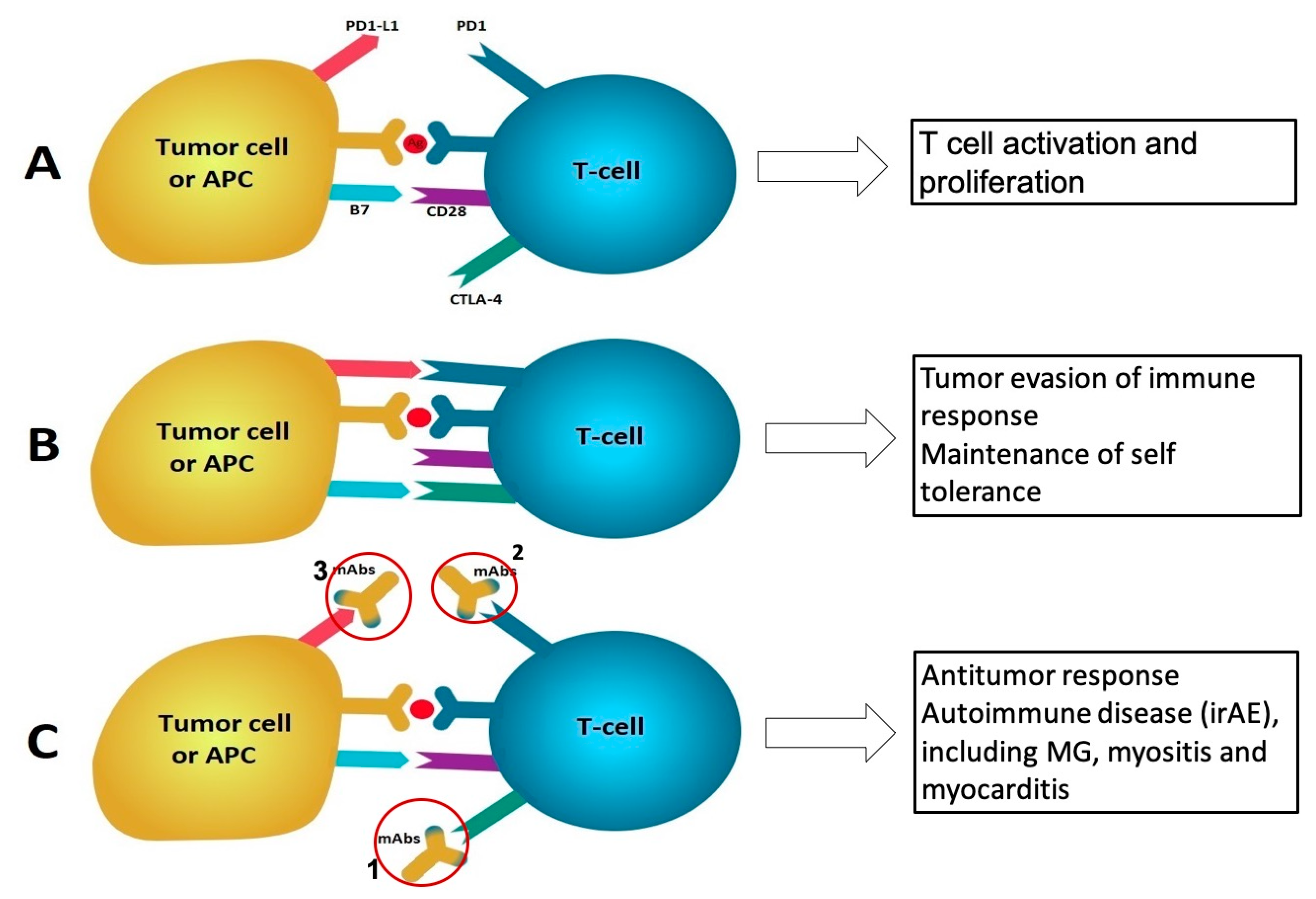
2.1. Cancer Immunotherapy
2.2. Alemtuzumab
2.3. D-Penicillamine
2.4. Tyrosine Kinase Inhibitors (TKIs)
2.5. Interferons (IFNs)
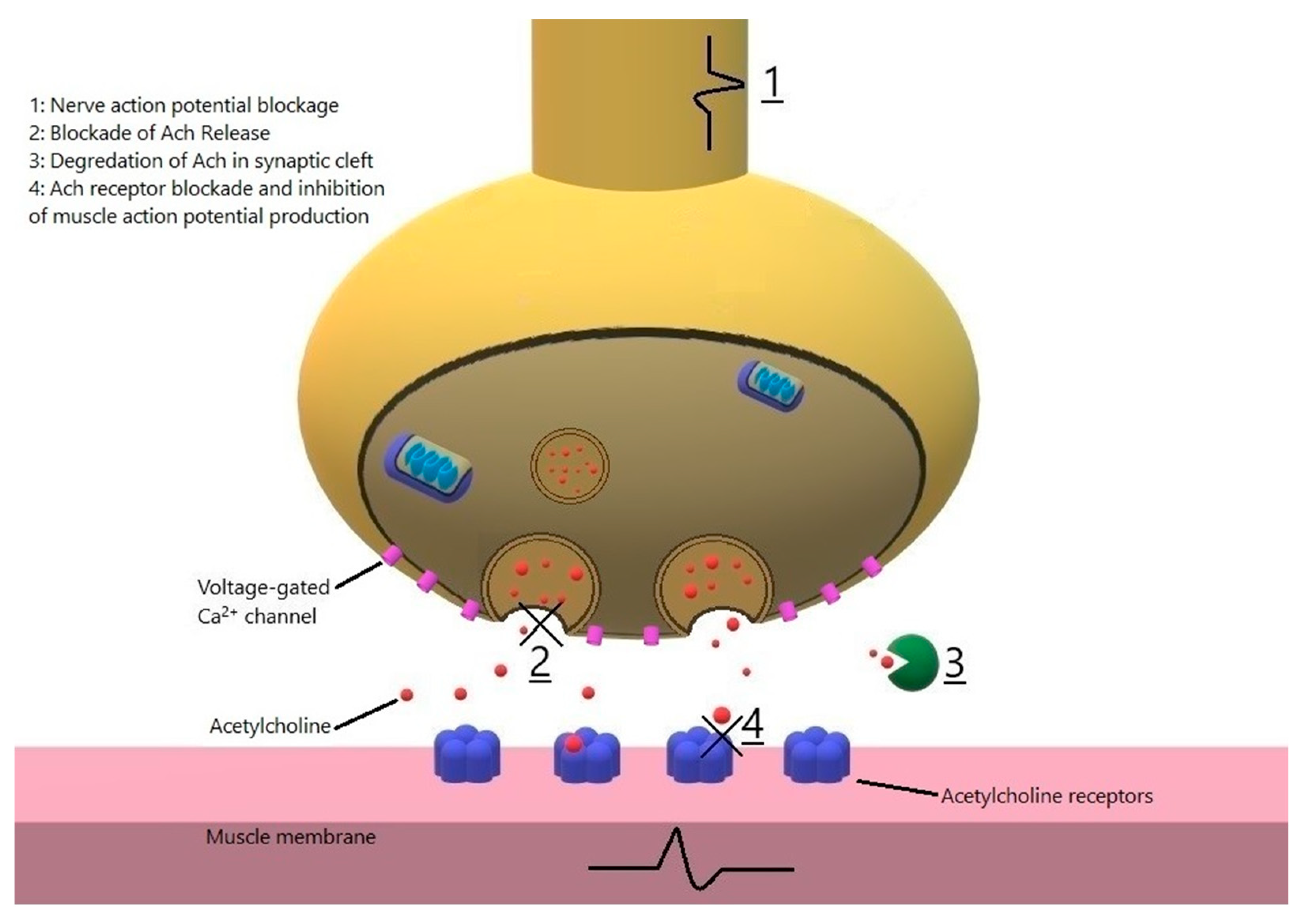
3. Drugs That Worsen MG or Cause MG-Like Symptoms through Direct Action on Neuromuscular Junction
3.1. Antibiotics
- A.
- Macrolides: In a retrospective study on 212 MG exacerbations including 141 hospitalizations, azithromycin was the most common medications associated with deterioration of MG [45]. Telithromycin, no longer available in the US market, is a ketolide antibiotic known to cause severe MG exacerbation from 1 h to days after starting its administration [47,48]. Exacerbation of MG has also been reported with erythromycin [49,50,51]. Macrolides likely affect the neuromuscular transmission directly: clarithromycin is reported to cause transient generalized MG-like symptoms, responsive to pyrodostigmine in a nonmyasthenic patient [52]. In another report, intravenous azithromycin caused severe MG exacerbation, which resolved minutes after administration of calcium gluconate, leading to the conclusion that azithromycin blocks the neuromuscular transmission at the presynaptic level [53]. Another patient developed severe MG exacerbation minutes after PO azithromycin, again implying a direct effect on neuromuscular transmission [51]. Given that severe MG exacerbation is a possible complication of macrolide antibiotics, the authors suggest that macrolides to be avoided in MG patients if there is another alternative. Patients and physicians should be aware that onset of weakness can occur shortly after exposure.
- B.
- Fluoroquinolones: Worsening of MG has been reported after starting ciprofloxacin, norfloxacin and ofloxacin with a latency of 2 h to 5 days [54,55,56,57,58]. Wang et al. reported nine patients that received fluoroquinolones (4 ciprofloxacin, 4 moxifloxacin and 1 levofloxacin) and developed MG exacerbation and an average increase of 10 points in quantitative MG score with a latency of 15 min to 4 days after starting the drug [59]. Fluoroquinolones could affect the amplitude of the miniature endplate potential and current (MEPP and MEPC) by either a presynaptic or a postsynaptic mechanism. Sieb demonstrated that norfloxacin, ofloxacin, and pefloxacin decreased the amplitudes of MEPPs and MEPCs in a dose dependent fashion, which was not reversed by neostigmine, suggestive for a postsynaptic mechanism (direct blocking of AChR ion channel) [60]. Fluoroquinolones contain a quinolone moiety similar to quinine, which has also been shown to have a direct effect on the AChR ion channel [60,61] We recommend this class of drug be avoided in MG patients if a safer alternative exists.
- C.
- Aminoglycosides are well known to cause MG exacerbation and myasthenia-like symptoms in non-MG patients who are critically ill, when given in high doses, in the context of kidney failure or concomitant use of neuromuscular blockers [62,63,64]. Neomycin blocks neuromuscular transmission, demonstrated both in vivo and in vitro studies. Its effect is antagonized by neostigmine, which is effective only when the block is incomplete) and calcium, which is also effective in complete blocks [65,66]. Elmqvist and Joseffson demonstrated that neomycin blocks ACh release from the presynaptic membrane similar to magnesium, as well as decreasing the sensitivity of postsynaptic membrane to ACh [67]. Another study using isolated nerve/muscle preparations demonstrated that amikacin had magnesium-like effects on ACh release from the presynaptic membrane. [64]. The blockade was reversed completely by calcium, 4-aminopyridine and 3,4-diaminopyridine and partially by neostigmine. In yet another study, streptomycin increased MEPPs (had a stimulatory effect) at low doses, but reduced sensitivity of postsynaptic membrane to ACh resulting in reduction in twitch strength at higher doses, and inhibited ACh release from the presynaptic membrane at the highest doses [68]. Similar effects (blocking ACh release at lower doses and reducing sensitivity of postsynaptic membrane to ACh) has been reported with gentamycin [69]. Furthermore, gentamicin, when administered in high doses to a critically ill patient was reported to cause severe neuromuscular transmission disorder responsive to intravenous calcium [62]. Tobramycin, on the other hand, does not cause neuromuscular blockade when used in effective antibacterial concentrations, and may be considered as choice of an aminoglycoside antibiotic in settings that aminoglycoside-associated neuromuscular blockade may occur [70].
- D.
- Penicillins: Although penicillins are generally considered safe in MG, there are a few reports of MG exacerbation. Argov et al. reported two cases of ampicillin-induced MG exacerbation, one had recurrence of the MG symptoms after challenge with ampicillin [71]. The same authors then demonstrated severe weakness and increased decrement of the CMAP amplitudes after administering ampicillin in severely affected rabbits with experimental autoimmune MG [71]. More recently, Vacchiano et al. reported six patients with worsening MG symptoms after treatment with amoxicillin; they speculated that amoxicillin, and not the underlying infection, was the cause of exacerbation as the infections were mild [72]. All of the patients improved and were back to baseline in 1–2 months. Given the very widespread use and the limited number of the reports of adverse reaction, penicillins should be considered one of the first line antibiotics to treat infections in MG patients.
- E.
- Other antibiotics: MG exacerbation has not been reported after the use of cephalosporins, sulfa drugs (including trimethoprim/sulfamethoxazole), and clindamycin. Although an earlier in vitro study using isolated nerve and muscle preparations showed that rolitetracyline depressed muscle sensitivity to ACh, similar to d-tubocurarine [73], we did not find clinical reports of worsening MG symptomatology after use of tetracyclines. Polymyxin B has been demonstrated in in vitro studies to decrease membrane sensitivity to ACh and end-plate neuromuscular blockade [73,74]. On the other hand, the neuromuscular blocker effects of polymyxin B were reversed by calcium in the anesthetized cats, pointing to an effect on presynaptic membrane [75]. There are no clinical reports of MG exacerbation with polymyxin B. There is a report of nitrofurantoin-induced AChR negative ocular MG in a 10-year-old, which completely improved after nitrofurantoin was discontinued [76]. Given the rarity, or lack of reports of adverse reaction of cephalosporins, sulfa drugs, clindamycin, tetracyclines, polymyxin B and nitrofurantoin, these drugs can be safely administered to MG patients.
3.2. Antihypertensives and Antiarrhythmics
- A.
- Blockers of β-adrenergic and calcium channel may cause transient exacerbation of MG symptoms [77,78,79,80,81,82]. Although a case of fulminant AChR Ab + MG has been reported 2 weeks after starting acebutolol, the association appears to be incidental as the disease ran a progressive course after acebutolol was discontinued [83]. β blockers frequently cause subjective fatigue. Felodipine and nifedipine were reported to cause significant MG exacerbation which abated after stopping and returned after re-challenge with those medications [84]. On the other hand, starting verapamil or nifedipine in patients with severe generalized MG has resulted in respiratory failure [85,86,87]. In some of the aforementioned cases, MG could have been deteriorated due to postoperative state [86], or starting high dose of prednisone [87]. In a study on 10 MG patients who received 3 Hz repetitive nerve stimulation at baseline and every 5 min for 3 times after receiving IV propranolol, the degree of CMAP decrement was variable and overall not significantly different after administration of propranolol or verapamil [88]. In another crossover study testing PO and IV propranolol and verapamil vs. placebo, there was no detrimental effect of the aforementioned drugs on muscle strength or on the CMAP decrement in the repetitive nerve stimulation [89]. In the retrospective study by Gummi et al., there were more exacerbations in patients on β blockers, but not shortly after the administration of these medications, which argues for contributing factor of comorbidities that led to the use of β blockers on MG exacerbation [45]. We conclude that MG patients, especially those in remission or well controlled, who need β adrenergic or calcium channel receptor blockers can generally undergo treatment with these medications at lowest effective dose but be closely monitored, especially initially, for deterioration of MG symptomatology.
- B.
- Antiarrhythmics: In an earlier study, Sieb et al. demonstrated the effects of quinoline derivatives quinine, quinidine and chloroquine on both presynaptic and postsynaptic aspects of neuromuscular transmission [61]. Chloroquine has been associated with emergence of myasthenia through production of AChR antibodies, and more commonly exacerbation of MG through direct effect on the neuromuscular transmission [61,90,91,92]. Onset and exacerbation of MG has also been reported with hydroxychloroquine, a drug similar to chloroquine, which is widely used in the treatment of rheumatological diseases [93,94] Procainamide has caused MG-like symptoms in nonmyasthenic patients with kidney failure [95,96,97], and respiratory failure in MG patients which did not have history of respiratory symptoms prior to the use of that medication [98,99]. Yeh et al. demonstrated that procainamide alters neuromuscular transmission at both presynaptic and postsynaptic levels in rats with experimental autoimmune MG [100]. We recommend that class Ia antiarrhythmic be avoided or used with extreme caution in MG patients. Propafenone, a class Ic antiarrhythmic has been reported to cause worsening of MG symptoms within a few hours of starting the medication [101]. There have been no reports of class Ib antiarrhythmics, such as flecainide, potassium channel blockers (such as amiodarone and dofetilide), and moricizine causing worsening of symptoms in MG patients.
3.3. Cholesterol Lowering Drugs
3.4. Magnesium
3.5. Bisphosphonates
3.6. Sedatives and Analgesics
3.7. Neuromuscular Blockers and Inhalation Anesthetics
3.8. Antipsychotics and Lithium
- A.
- Antipsychotics: Chlorpromazine was the first typical antipsychotic reported to be associated with MG exacerbation [142]; it was subsequently shown to impair neuromuscular transmission both at the postsynaptic membrane and also to inhibit of ACh release from the presynaptic membrane. [143]. Similar, dose dependent effects on neuromuscular transmission were later demonstrated by Nguyen et al. for atypical antipsychotics clozapine, olanzapine, sulpiride and risperidone [144]. Pimozide, thioridazine, clozapine, olanzapine, haloperidol, quetiapine long-acting risperidone and olanzapine are also reported to cause deterioration of symptoms in MG patients [145,146,147,148]
- B.
- Lithium: There are several reports of emergence of myasthenic symptoms shortly after starting lithium, which improved after discontinuation and later recurrence with a re-challenge [149,150,151]. On the other hand, lithium may unmask the symptoms of an AChR + MG [152,153]. Pestronk and Drachman demonstrated that lithium causes reduction in the number of AChRs in the postsynaptic membrane [154]. Others have shown lithium-induced reduction in ACh synthesis in the presynaptic membrane and its release by competing with calcium inside the presynaptic motor nerve terminal [155].
3.9. Anticonvulsants
3.10. Corticosteroids and Estrogens
3.11. Acetylcholinesterase Inhibitors
3.12. Botulinum Toxin
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Deenen, J.C.; Horlings, C.G.; Verschuuren, J.J.; Verbeek, A.L.; Van Engelen, B.G. The Epidemiology of Neuromuscular Disorders: A Comprehensive Overview of the Literature. J. Neuromuscul. Dis. 2015, 2, 73–85. [Google Scholar] [CrossRef] [Green Version]
- Carr, A.S.; Cardwell, C.; McCarron, P.O.; McConville, J. A systematic review of population based epidemiological studies in Myasthenia Gravis. BMC Neurol. 2010, 10, 46. [Google Scholar] [CrossRef] [Green Version]
- Mantegazza, R.; Baggi, F.; Antozzi, C.; Confalonieri, P.; Morandi, L.; Bernasconi, P.; Andreetta, F.; Simoncini, O.; Campanella, A.; Beghi, E.; et al. Myasthenia Gravis (MG): Epidemiological Data and Prognostic Factors. Ann. N. Y. Acad. Sci. 2003, 998, 413–423. [Google Scholar] [CrossRef]
- Gilhus, N.E.; Verschuuren, J.J. Myasthenia gravis: Subgroup classification and therapeutic strategies. Lancet Neurol. 2015, 14, 1023–1036. [Google Scholar] [CrossRef]
- Wolfe, G.I.; Oh, S.J. Clinical Phenotype of Muscle-specific Tyrosine Kinase-antibody-positive Myasthenia Gravis. Ann. N. Y. Acad. Sci. 2008, 1132, 71–75. [Google Scholar] [CrossRef]
- Oger, J.; Frykman, H. An update on laboratory diagnosis in myasthenia gravis. Clin. Chim. Acta 2015, 449, 43–48. [Google Scholar] [CrossRef]
- Tzartos, J.S.; Zhang, B.; Belimezi, M.; Ragheb, S.; Bealmear, B.; Lewis, R.A.; Xiong, W.-C.; Lisak, R.P.; Tzartos, S.J.; Mei, L. Autoantibodies to Lipoprotein-Related Protein 4 in Patients With Double-Seronegative Myasthenia Gravis. Arch. Neurol. 2012, 69, 445–451. [Google Scholar] [CrossRef] [Green Version]
- Gallardo, E.; Martínez-Hernández, E.; Titulaer, M.J.; Huijbers, M.G.; Martínez, M.A.; Ramos, A.; Querol, L.; Díaz-Manera, J.; Rojas-García, R.; Hayworth, C.R.; et al. Cortactin autoantibodies in myasthenia gravis. Autoimmun. Rev. 2014, 13, 1003–1007. [Google Scholar] [CrossRef]
- Gasperi, C.; Melms, A.; Schoser, B.; Zhang, Y.; Meltoranta, J.; Risson, V.; Schaeffer, L.; Schalke, B.; Kröger, S.; Schwartz, S.R.; et al. Anti-agrin autoantibodies in myasthenia gravis. Neurology 2014, 82, 1976–1983. [Google Scholar] [CrossRef]
- Argov, Z.; Mastaglia, F.L. Drug therapy: Disorders of neuromuscular transmission caused by drugs. N. Engl. J. Med. 1979, 301, 409–413. [Google Scholar]
- A Naranjo, C.; Busto, U.; Sellers, E.M.; Sandor, P.; Ruiz, I.; Roberts, E.A.; Janecek, E.; Domecq, C.; Greenblatt, D.J. A method for estimating the probability of adverse drug reactions. Clin. Pharmacol. Ther. 1981, 30, 239–245. [Google Scholar] [CrossRef]
- Safa, H.; Johnson, D.H.; Trinh, V.A.; Rodgers, T.E.; Lin, H.; Suarez-Almazor, M.E.; Faak, F.; Saberian, C.; Yee, C.; Davies, M.A.; et al. Immune checkpoint inhibitor related myasthenia gravis: Single center experience and systematic review of the literature. J. Immunother. Cancer 2019, 7, 319. [Google Scholar] [CrossRef]
- Puwanant, A.; Isfort, M.; Lacomis, D.; Živković, S.A. Clinical spectrum of neuromuscular complications after immune checkpoint inhibition. Neuromuscul. Disord. 2019, 29, 127–133. [Google Scholar] [CrossRef]
- Postow, M.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Yuen, C.; Fleming, G.; Meyers, M.; Soliven, B.; Rezania, K. Myasthenia gravis induced by avelumab. Immunotherapy 2019, 11, 1181–1185. [Google Scholar] [CrossRef]
- Thakolwiboon, S.; Karukote, A.; Wilms, H. De Novo Myasthenia Gravis Induced by Atezolizumab in a Patient with Urothelial Carcinoma. Cureus 2019, 11, e5002. [Google Scholar] [CrossRef]
- Berrih-Aknin, S.; Le Panse, R. Myasthenia gravis: A comprehensive review of immune dysregulation and etiological mechanisms. J. Autoimmun. 2014, 52, 90–100. [Google Scholar] [CrossRef]
- Suzuki, S.; Ishikawa, N.; Konoeda, F.; Seki, N.; Fukushima, S.; Takahashi, K.; Uhara, H.; Hasegawa, Y.; Inomata, S.; Otani, Y.; et al. Nivolumab-related myasthenia gravis with myositis and myocarditis in Japan. Neurology 2017, 89, 1127–1134. [Google Scholar] [CrossRef]
- Lipe, D.N.; Galvis-Carvajal, E.; Rajha, E.; Wechsler, A.H.; Gaeta, S. Immune checkpoint inhibitor-associated myasthenia gravis, myositis, and myocarditis overlap syndrome. Am. J. Emerg. Med. 2021, 46, 51–55. [Google Scholar] [CrossRef]
- Arora, P.; Talamo, L.; Dillon, P.; Gentzler, R.D.; Millard, T.; Salerno, M.; Gaughan, E.M. Severe combined cardiac and neuromuscular toxicity from immune checkpoint blockade: An institutional case series. Cardio-Oncol. 2020, 6, 1–11. [Google Scholar] [CrossRef]
- Maeda, O.; Yokota, K.; Atsuta, N.; Katsuno, M.; Akiyama, M.; Ando, Y. Nivolumab for the treatment of malignant melanoma in a patient with pre-existing myasthenia gravis. Nagoya J. Med Sci. 2016, 78, 119–122. [Google Scholar] [PubMed]
- Mitsune, A.; Yanagisawa, S.; Fukuhara, T.; Miyauchi, E.; Morita, M.; Ono, M.; Tojo, Y.; Ichinose, M. Relapsed Myasthenia Gravis after Nivolumab Treatment. Intern. Med. 2018, 57, 1893–1897. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.G.; Milner-Brown, H.S.; Mirka, A. Prednisone-induced worsening of neuromuscular function in myasthenia gravis. Neurol. 1986, 36, 729. [Google Scholar] [CrossRef] [PubMed]
- Lünemann, J.D.; Nimmerjahn, F.; Dalakas, M.C. Intravenous immunoglobulin in neurology—Mode of action and clinical efficacy. Nat. Rev. Neurol. 2015, 11, 80–89. [Google Scholar] [CrossRef]
- Pariani, N.; Willis, M.; Muller, I.; Healy, S.; Nasser, T.; McGowan, A.; Lyons, G.; Jones, J.; Chatterjee, K.; Dayan, C.; et al. Alemtuzumab-Induced Thyroid Dysfunction Exhibits Distinctive Clinical and Immunological Features. J. Clin. Endocrinol. Metab. 2018, 103, 3010–3018. [Google Scholar] [CrossRef]
- Killestein, J.; van Oosten, B. Emerging safety issues in alemtuzumab-treated MS patients. Mult. Scler. 2019, 25, 1206–1208. [Google Scholar] [CrossRef] [Green Version]
- Midaglia, L.; Gratacòs, M.; Caronna, E.; Raguer, N.; Sastre-Garriga, J.; Montalban, X.; Tintoré, M. Myasthenia gravis following alemtuzumab therapy for multiple sclerosis. Neurology 2018, 91, 622–624. [Google Scholar] [CrossRef]
- Barrons, R.W. Drug-induced neuromuscular blockade and myasthenia gravis. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1997, 17, 1220–1232. [Google Scholar]
- Vincent, A.; Palace, J.; Hilton-Jones, D. Myasthenia gravis. Lancet 2001, 357, 2122–2128. [Google Scholar] [CrossRef]
- Katz, L.J.; Lesser, R.L.; Merikangas, J.R.; Silverman, J.P. Ocular myasthenia gravis after D-penicillamine administration. Br. J. Ophthalmol. 1989, 73, 1015–1018. [Google Scholar] [CrossRef] [PubMed]
- Poulas, K.; Koutsouraki, E.; Kordas, G.; Kokla, A.; Tzartos, S.J. Anti-MuSK- and anti-AChR-positive myasthenia gravis induced by d-penicillamine. J. Neuroimmunol. 2012, 250, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Albers, J.W.; Hodach, R.J.; Kimmel, D.W.; Treacy, W.L. Penicillamine-associated myasthenia gravis. Neurol. 1980, 30, 1246. [Google Scholar] [CrossRef]
- Liu, G.T.; Bienfang, D.C. Penicillamine-induced ocular myasthenia gravis in rheumatoid arthritis. J. Clin. Neuroophthalmol. 1990, 10, 201–205. [Google Scholar] [PubMed]
- Hill, M.; Moss, P.; Wordsworth, P.; Newsom-Davis, J.; Willcox, N. T cell responses to D-penicillamine in drug-induced myasthenia gravis: Recognition of modified DR1:peptide complexes. J. Neuroimmunol. 1999, 97, 146–153. [Google Scholar] [CrossRef]
- Desai, A.; Sriwastava, S.; Gadgeel, S.M.; Lisak, R.P. New onset myasthenia gravis in a patient with non small cell lung cancer treated with lorlatinib a novel anti-cancer agent. J. Neurol. Sci. 2018, 392, 100–101. [Google Scholar] [CrossRef]
- Sanford, D.; Macdonald, M.; Nicolle, M.; Xenocostas, A. Development of myasthenia gravis in a patient with chronic myeloid leukemia during treatment with nilotinib. Hematol. Rep. 2014, 6, 5288. [Google Scholar] [CrossRef]
- Kopp, C.R.; Jandial, A.; Mishra, K.; Sandal, R.; Malhotra, P. Myasthenia gravis unmasked by imatinib. Br. J. Haematol. 2019, 184, 321. [Google Scholar] [CrossRef] [Green Version]
- Zaloum, A.; Falet, J.R.; Elkrief, A.; Chalk, C. Myasthenia gravis following dabrafenib and trametinib for metastatic melanoma. Neurology 2020, 94, 322–323. [Google Scholar] [CrossRef] [PubMed]
- Lehky, T.J.; Iwamoto, F.M.; Kreisl, T.N.; Floeter, M.K.; Fine, H.A. Neuromuscular junction toxicity with tandutinib induces a myasthenic-like syndrome. Neurology 2011, 76, 236–241. [Google Scholar] [CrossRef] [Green Version]
- Rönnblom, L.; Pascual, V. The innate immune system in SLE: Type I interferons and dendritic cells. Lupus 2008, 17, 394–399. [Google Scholar] [CrossRef] [Green Version]
- Di Domizio, J.; Cao, W. Fueling autoimmunity: Type I interferon in autoimmune diseases. Expert Rev. Clin. Immunol. 2013, 9, 201–210. [Google Scholar] [CrossRef]
- Mehrizi, M.F.R.; Gearhart, T.R.; Pascuzzi, R.M. Medications and Myasthenia Gravis (A Reference for Health Care Professionals). 2012. Available online: https://www.myasthenia.org/portals/0/draft_medications_and_myasthenia_gravis_for_MGFA_website_8%2010%2012.pdf (accessed on 4 February 2021).
- Baik, S.J.; Kim, T.H.; Kim, H.I.; Rhie, J.Y. Myasthenia Crisis Induced by Pegylated-Interferon in Patient with Chronic Hepatitis C: A Case Report. Medicine (Baltimore) 2016, 95, e3782. [Google Scholar] [CrossRef] [PubMed]
- Jani-Acsadi, A.; Lisak, R.P. Myasthenic crisis: Guidelines for prevention and treatment. J. Neurol. Sci. 2007, 261, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Gummi, R.R.; Kukulka, N.A.; Deroche, C.B.; Govindarajan, R. Factors associated with acute exacerbations of myasthenia gravis. Muscle Nerve 2019, 60, 693–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilhus, N.E.; Romi, F.; Hong, Y.; Skeie, G.O. Myasthenia gravis and infectious disease. J. Neurol. 2018, 265, 1251–1258. [Google Scholar] [CrossRef]
- Perrot, X.; Bernard, N.; Vial, C.; Antoine, J.C.; Laurent, H.; Vial, T.; Confavreux, C.; Vukusic, S. Myasthenia gravis exacerbation or unmasking associated with telithromycin treatment. Neurology 2006, 67, 2256–2258. [Google Scholar] [CrossRef]
- Jennett, A.M.; Bali, D.; Jasti, P.; Shah, B.; Browning, L.A. Telithromycin and Myasthenic Crisis. Clin. Infect. Dis. 2006, 43, 1621–1622. [Google Scholar] [CrossRef] [Green Version]
- May, E.F.; Calvert, P.C. Aggravation of myasthenia gravis by erythromycin. Ann. Neurol. 1990, 28, 577–579. [Google Scholar] [CrossRef]
- Absher, J.R.; Bale, J.F., Jr. Aggravation of myasthenia gravis by erythromycin. J. Pediatr. 1991, 119 Pt 1, 155–156. [Google Scholar] [CrossRef]
- Cadisch, R.; Streit, E.; Hartmann, K. Exacerbation of pseudoparalytic myasthenia gravis following azithromycin (Zithromax). Schweiz. Med. Wochenschr. 1996, 126, 308–310. [Google Scholar]
- Pijpers, E.; Van Rijswijk, R.E.N.; Kohlen, B.T.; Schrey, G. A Clarithromycin-Induced Myasthenic Syndrome. Clin. Infect. Dis. 1996, 22, 175–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradhan, S.; Pardasani, V.; Ramteke, K. Azithromycin-induced myasthenic crisis: Reversibility with calcium gluconate. Neurol. India 2009, 57, 352–353. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.; Safani, M.; Keesey, J. Possible Exacerbation of MYASTHENIA gravis by ciprofloxacin. Lancet 1988, 331, 882. [Google Scholar] [CrossRef]
- Mumford, C.J.; Ginsberg, L. Ciprofloxacin and myasthenia gravis. BMJ 1990, 301, 818. [Google Scholar] [CrossRef] [Green Version]
- Rauser, E.H.; Ariano, R.E.; Anderson, B.A. Exacerbation of Myasthenia Gravis by Norfloxacin. DICP 1990, 24, 207–208. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, E.; Ribeiro, J.A.; Polónia, J.; Pontes, C. Probable exacerbation of myasthenia gravis by ofloxacin. J. Neurol. 1993, 240, 508. [Google Scholar] [CrossRef] [PubMed]
- Roquer, J.; Cano, A.; Seoane, J.L.; Serradell, A. Myasthenia gravis and ciprofloxacin. Acta Neurol. Scand. 1996, 94, 419–420. [Google Scholar] [CrossRef]
- Wang, S.-H.; Xie, Y.-C.; Jiang, B.; Zhang, J.-Y.; Qu, Y.; Zhao, Y.; Li, Y.; Qiao, S.-S.; Xu, C.-L. Fluoroquinolone associated myasthenia gravis exacerbation: Clinical analysis of 9 cases. Zhonghua Yi Xue Za Zhi 2013, 93, 1283–1286. [Google Scholar]
- Sieb, J.P. Fluoroquinolone antibiotics block neuromuscular transmission. Neurology 1998, 50, 804–807. [Google Scholar] [CrossRef]
- Sieb, J.P.; Milone, M.; Engel, A.G. Effects of the quinoline derivatives quinine, quinidine, and chloroquine on neuromuscular transmission. Brain Res. 1996, 712, 179–189. [Google Scholar] [CrossRef]
- Warner, W.A.; Sanders, E. Neuromuscular blockade associated with gentamicin therapy. JAMA 1971, 215, 1153–1154. [Google Scholar] [CrossRef]
- Hall, D.R.; McGibbon, D.H.; Evans, C.C.; Meadows, G.A. Gentamicin, tubocurarine, lignocaine and neuromuscular blockade. A case report. Br. J. Anaesth. 1972, 44, 1329–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, Y.N.; Marshall, I.G.; Harvey, A.L. Some effects of the aminoglycoside antibiotic amikacin on neuromuscular and autonomic transmission. Br. J. Anaesth. 1978, 50, 109–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pittinger, C.B.; Long, J.P. Neuromuscular blocking action of neomycin sulfate. Antibiot. Chemother. 1958, 8, 198–203. [Google Scholar]
- Corrado, A.P.; Ramos, A.O.; De Escobar, C.T. Neuro-muscular blockade by neomycin, potentiation by ether anesthesia and d-tubucurarine and antagonism by calcium and prostigmine. Arch. Int. Pharmacodyn. Ther. 1959, 121, 380–394. [Google Scholar] [PubMed]
- Elmqvist, D.; Josefsson, J.-O. The Nature of the Neuromuscular Block Produced by Neomycine. Acta Physiol. Scand. 1962, 54, 105–110. [Google Scholar] [CrossRef]
- Dretchen, K.; Sokoll, M.; Gergis, S.; Long, J. Relative effects of streptomycin on motor nerve terminal and endplate. Eur. J. Pharmacol. 1973, 22, 10–16. [Google Scholar] [CrossRef]
- Torda, T. The nature of gentamicin-induced neuromuscular block. Br. J. Anaesth. 1980, 52, 325–329. [Google Scholar] [CrossRef] [Green Version]
- de Rosayro, M.; Healy, T.E. Tobramycin and neuromuscular transmission in the rat isolated phrenic nerve-diaphragm preparation. Br. J. Anaesth. 1978, 50, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Argov, Z.; Brenner, T.; Abramsky, O. Ampicillin May Aggravate Clinical and Experimental Myasthenia Gravis. Arch. Neurol. 1986, 43, 255–256. [Google Scholar] [CrossRef]
- Vacchiano, V.; Solli, P.; Bartolomei, I.; Lai, G.; Liguori, R.; Salvi, F. Exacerbation of myasthenia gravis after amoxicillin therapy: A case series. Neurol. Sci. 2020, 41, 2255–2257. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.M.; Collier, B. The site of the neuromuscular block produced by polymyxin B and rolitetracycline. Can. J. Physiol. Pharmacol. 1976, 54, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Fiekers, J.F. Neuromuscular block produced by polymyxin B: Interaction with end-plate channels. Eur. J. Pharmacol. 1981, 70, 77–81. [Google Scholar] [CrossRef]
- Lee, C.; Chen, D.; Nagel, E.L. Neuromuscular block by antibiotics: Polymyxin B. Anesth. Analg. 1977, 56, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, B.N.; Chronister, T.E.; Stark, B.I.; Saran, B.R. Ocular myasthenia and nitrofurantoin. Am. J. Ophthalmol. 2000, 130, 531–533. [Google Scholar] [CrossRef]
- Herishanu, Y.; Rosenberg, P. Letter: Beta-Blockers and myasthenia gravis. Ann. Intern. Med. 1975, 83, 834–835. [Google Scholar] [CrossRef]
- Weber, J.C. Beta-adrenoreceptor antagonists and diplopia. Lancet 1982, 2, 826–827. [Google Scholar] [CrossRef]
- Rozen, M.S.; Whan, F.M. Prolonged curarization associated with propranolol. Med J. Aust. 1972, 1, 467–468. [Google Scholar] [CrossRef] [PubMed]
- Coppeto, J.R. Timolol-associated myasthenia gravis. Am. J. Ophthalmol. 1984, 98, 244–245. [Google Scholar] [CrossRef]
- Shaivitz, S.A. Timolol and Myasthenia Gravis. JAMA 1979, 242, 1611. [Google Scholar] [CrossRef] [PubMed]
- Verkijk, A. Worsening of myasthenia gravis with timolol maleate eyedrops. Ann. Neurol. 1985, 17, 211–212. [Google Scholar] [CrossRef] [PubMed]
- Confavreux, C.; Charles, N.; Aimard, G. Fulminant Myasthenia Gravis Soon after Initiation of Acebutolol Therapy. Eur. Neurol. 1990, 30, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Pina Latorre, M.A.; Cobeta, J.C.; Rodilla, F.; Navarro, N.; Zabala, S. Influence of calcium antagonist drugs in myasthenia gravis in the elderly. J. Clin. Pharm. Ther. 1998, 23, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Howard, J., Jr. Adverse Drug Effects on Neuromuscular Transmission. Semin. Neurol. 1990, 10, 89–102. [Google Scholar] [CrossRef]
- Swash, M.; Ingram, D.A. Adverse effect of verapamil in myasthenia gravis. Muscle Nerve 1992, 15, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Nogues, M.A.; Rivero, A. Arterial hypertension, nifedipine, and myasthenia gravis. Muscle Nerve 1993, 16, 797. [Google Scholar] [PubMed]
- Jonkers, I.; Swerup, C.; Pirskanen, R.; Bjelak, S.; Matell, G. Acute effects of intravenous injection of beta-adrenoreceptor- and calcium channel at antagonists and agonists in myasthenia gravis. Muscle Nerve 1996, 19, 959–965. [Google Scholar] [CrossRef]
- Matell, G.; Bjelak, S.; Jonkers, I.; Pirskanen, R.; Van Vliet, J.; Swerup, C. Calcium channel and beta-receptor antagonists and agonists in MG. Ann. N. Y. Acad. Sci. 1998, 841, 785–788. [Google Scholar] [CrossRef]
- Hearn, J.; Tiliakos, N.A. Myasthenia Gravis Caused by Penicillamine and Chloroquine Therapy for Rheumatoid Arthritis. South. Med. J. 1986, 79, 1185–1186. [Google Scholar] [CrossRef]
- Schumm, F.; Wietholter, H.; Fateh-Moghadam, A. Myasthenia syndrome during chloroquine treatment (author’s transl). Dtsch. Med. Wochenschr. 1981, 106, 1745–1747. [Google Scholar] [CrossRef]
- Sghirlanzoni, A.; Mantegazza, R.; Mora, M.; Pareyson, D.; Cornelio, F. Chloroquine myopathy and myasthenia-like syndrome. Muscle Nerve 1988, 11, 114–119. [Google Scholar] [CrossRef]
- Varan, O.; Kucuk, H.; Tufan, A. Myasthenia gravis due to hydroxychloroquine. Reumatismo 2015, 67, 125. [Google Scholar] [CrossRef] [Green Version]
- Elavarasi, A.; Goyal, V. Hydroxychloroquine and Myasthenia Gravis-Can One Take This Risk? Ann. Indian Acad. Neurol. 2020, 23, 360–361. [Google Scholar]
- Miller, C.D.; Oleshansky, M.A.; Gibson, K.F.; Cantilena, L.R. Procainamide-Induced Myasthenia-Like Weakness and Dysphagia. Ther. Drug Monit. 1993, 15, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.J.; Elmore, R.S.; Sarala, P.K.; Kuba, T. Procainamide-induced myasthenia-like syndrome. Muscle Nerve 1986, 9, 670–672. [Google Scholar]
- Niakan, E.; Bertorini, T.E.; Acchiardo, S.R.; Werner, M.F. Procainamide-Induced Myasthenia-like Weakness in a Patient With Peripheral Neuropathy. Arch. Neurol. 1981, 38, 378–379. [Google Scholar] [CrossRef] [PubMed]
- Godley, P.J.; Morton, T.A.; Karboski, J.A.; Tami, J.A. Procainamide-Induced Myasthenic Crisis. Ther. Drug Monit. 1990, 12, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Drachman, D.A.; Skom, J.H. Procainamide--a Hazard in Myasthenia Gravis. Arch. Neurol. 1965, 13, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Yeh, T.-M.; Tami, J.A.; Krolick, K.A. Exacerbated muscle dysfunction by procainamide in rats with experimental myasthenia gravis. Drug Chem. Toxicol. 1992, 15, 53–65. [Google Scholar] [CrossRef]
- Lecky, B.R.; Weir, D.; Chong, E. Exacerbation of myasthenia by propafenone. J. Neurol. Neurosurg. Psychiatry 1991, 54, 377. [Google Scholar] [CrossRef] [Green Version]
- Parmar, B.; Francis, P.J.; Ragge, N.K. Statins, fibrates, and ocular myasthenia. Lancet 2002, 360, 717. [Google Scholar] [CrossRef]
- Cartwright, M.S.; Jeffery, D.R.; Nuss, G.R.; Donofrio, P.D. Statin-associated exacerbation of myasthenia gravis. Neurology 2004, 63, 2188. [Google Scholar] [CrossRef] [PubMed]
- Purvin, V.; Kawasaki, A.; Smith, K.H.; Kesler, A. Statin-associated myasthenia gravis: Report of 4 cases and review of the literature. Medicine (Baltimore) 2006, 85, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Gale, J.; Danesh-Meyer, H.V. Statins can induce myasthenia gravis. J. Clin. Neurosci. 2014, 21, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.J.; Dhall, R.; Young, A.; Morgan, M.B.; Lu, L.; Claussen, G.C. Statins may aggravate myasthenia gravis. Muscle Nerve 2008, 38, 1101–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gras-Champel, V.; Batteux, B.; Masmoudi, K.; Liabeuf, S. Statin-induced myasthenia: A disproportionality analysis of the WHO’s VigiBase pharmacovigilance database. Muscle Nerve 2019, 60, 382–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youssef, S.; Stüve, O.; Patarroyo, J.C.; Ruiz, P.J.; Radosevich, J.L.; Hur, E.M.; Bravo, M.; Mitchell, D.J.; Sobel, R.A.; Steinman, L.; et al. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nat. Cell Biol. 2002, 420, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, W.-M.; Gao, C.; Sun, N.-L. Effects of atorvastatin on the Th1/Th2 polarization of ongoing experimental autoimmune myocarditis in Lewis rats. J. Autoimmun. 2005, 25, 258–263. [Google Scholar] [CrossRef]
- Balasa, B.; Sarvetnick, N. Is pathogenic humoral autoimmunity a Th1 response? Lessons from (for) myasthenia gravis. Immunol. Today 2000, 21, 19–23. [Google Scholar] [CrossRef]
- Cao, Y.; Amezquita, R.A.; Kleinstein, S.H.; Stathopoulos, P.; Nowak, R.J.; O’Connor, K.C. Autoreactive T Cells from Patients with Myasthenia Gravis Are Characterized by Elevated IL-17, IFN-γ, and GM-CSF and Diminished IL-10 Production. J. Immunol. 2016, 196, 2075–2084. [Google Scholar] [CrossRef] [Green Version]
- Ragbourne, S.C.; Crook, M.A. Use of lipid-lowering medications in myasthenia gravis: A case report and literature review. J. Clin. Lipidol. 2015, 9, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Del Castillo, J.; Engbaek, L. The nature of the neuromuscular block produced by magnesium. J. Physiol. 1954, 124, 370–384. [Google Scholar] [CrossRef] [Green Version]
- Cohen, B.A.; London, R.S.; Goldstein, P.J. Myasthenia gravis and preeclampsia. Obstet. Gynecol. 1976, 48 (Suppl. 1), 35S–37S. [Google Scholar] [PubMed]
- Bashuk, R.G.; Krendel, D.A. Myasthenia gravis presenting as weakness after magnesium administration. Muscle Nerve 1990, 13, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Klair, J.S.; Rochlani, Y.M.; Meena, N.K. Myasthenia gravis masquerading as dysphagia: Unveiled by magnesium infusion. BMJ Case Rep. 2014, 2014. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Idowu, O.; Malik, I.; Nates, J.L. Acute Respiratory Failure Induced by Magnesium Replacement in a 62-Year-Old Woman with Myasthenia Gravis. Tex. Hear. Inst. J. 2015, 42, 495–497. [Google Scholar] [CrossRef] [Green Version]
- Kesikburun, S.; Güzelküçük, Ü.; Alay, S.; Yavuz, F.; Tan, A.K. Exacerbation of myasthenia gravis by alendronate. Osteoporos. Int. 2014, 25, 2319–2320. [Google Scholar] [CrossRef]
- Raja, V.; Sandanshiv, P.; Neugebauer, M. Risedronate induced transient ocular myasthenia. J. Postgrad. Med. 2007, 53, 274–275. [Google Scholar] [CrossRef]
- Van den Bersselaar, L.R.; Snoeck, M.M.J.; Gubbels, M.; Riazi, S.; Kamsteeg, E.J.; Jungbluth, H.; Voermans, N.C. Anaesthesia and neuromuscular disorders: What a neurologist needs to know. Pract. Neurol. 2020, 21. [Google Scholar] [CrossRef]
- Srivastava, V.K.; Agrawal, S.C.; Ahmed, M.R.; Sharma, S.K. Anesthetic Management of a Patient with Myasthenia Gravis for Meningioma Surgery-A Case Report. Kathmandu Univ. Med J. 2015, 13, 80–82. [Google Scholar] [CrossRef] [Green Version]
- Stiru, O.; Stefan, M.; Geana, R.C.; Sorostinean, D.; Radu, R.; Filipescu, D.; Chioncel, O.; Iliescu, V.A. Mitral Valve Replacement with Thymectomy in a Patient with Ocular Myasthenia Gravis: Case Report. Hear. Surg. Forum 2019, 22, E340–E342. [Google Scholar] [CrossRef] [PubMed]
- Baftiu, N.; Hadri, B.; Morina, M.; Mustafa, A. Anesthesia for Trans Sternal Thymectomy: Modified Non-muscle Relaxant Technique. Med. Arch. 2011, 65, 317–318. [Google Scholar] [CrossRef] [PubMed]
- Gorback, M.S.; Moon, R.E.; Massey, J.M. Extubation after transsternal thymectomy for myasthenia gravis: A prospective analysis. South Med J. 1991, 84, 701–706. [Google Scholar] [CrossRef]
- Anderson, H.J.; Churchill-Davidson, H.C.; Richardson, A.T. Bronchial neoplasm with myasthenia; prolonged apnoea after administration of succinylcholine. Lancet 1953, 265, 1291–1293. [Google Scholar] [CrossRef]
- Eisenkraft, J.B.; Book, W.J.; Mann, S.M.; Papatestas, A.E.; Hubbard, M. Resistance to succinylcholine in myasthenia gravis: A dose-response study. Anesthesiology 1988, 69, 760–763. [Google Scholar] [CrossRef]
- Levitan, R. Safety of succinylcholine in myasthenia gravis. Ann. Emerg. Med. 2005, 45, 225–226. [Google Scholar] [CrossRef]
- Buzello, W.; Noeldge, G.; Krieg, N.; Brobmann, G.F. Vecuronium for Muscle Relaxation in Patients with Myasthenia Gravis. Anesthesiology 1986, 64, 507–509. [Google Scholar] [CrossRef]
- Itoh, H.; Shibata, K.; Nitta, S. Difference in sensitivity to vecuronium between patients with ocular and generalized myasthenia gravis. Br. J. Anaesth. 2001, 87, 885–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, H.; Shibata, K.; Nitta, S. Sensitivity to Vecuronium in Seropositive and Seronegative Patients with Myasthenia Gravis. Anesthesia Analg. 2002, 95, 109–113. [Google Scholar] [CrossRef]
- Abel, M.; Eisenkraft, J.B. Anesthetic implications of myasthenia gravis. Mt. Sinai J. Med. A J. Transl. Pers. Med. 2002, 69, 31–37. [Google Scholar]
- Gissen, A.J.; Karis, J.H.; Nastuk, W.L. Effect of halothane on neuromuscular transmission. JAMA 1966, 197, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Rowbottom, S.J. Isoflurane for Thymectomy in Myasthenia Gravis. Anaesth. Intensiv. Care 1989, 17, 444–447. [Google Scholar] [CrossRef]
- Nilsson, E.; Müller, K. Neuromuscular effects of isoflurane in patients with myasthenia gravis. Acta Anaesthesiol. Scand. 1990, 34, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Tsukagoshi, H.; Kurosaki, D.; Sugaya, T.; Yoshikawa, D.; Shimada, H. Neuromuscular effects of sevoflurane in patients with myasthenia gravis. J. Anesth. 1996, 10, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Sungur, Z.; Sentürk, M. Anaesthesia for thymectomy in adult and juvenile myasthenic patients. Curr. Opin. Anaesthesiol. 2016, 29, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Kas, J.; Kiss, D.; Simon, V.; Svastics, E.; Major, L.; Szobor, A. Decade-long experience with surgical therapy of myasthenia gravis: Early complications of 324 transsternal thymectomies. Ann. Thorac. Surg. 2001, 72, 1691–1697. [Google Scholar] [CrossRef]
- Gritti, P.; Sgarzi, M.; Carrara, B.; Lanterna, L.A.; Novellino, L.; Spinelli, L.; Khotcholava, M.; Poli, G.; Lorini, F.L.; Sonzogni, V. A standardized protocol for the perioperative management of myasthenia gravis patients. Experience with 110 patients. Acta Anaesthesiol. Scand. 2011, 56, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Moriyama, S.; Aoki, S.; Yoshizawa, S.; Tomita, M.; Kojima, T.; Mori, Y.; Takeuchi, N.; So, M.-H.; Yano, M.; et al. Estimation of the success rate of anesthetic management for thymectomy in patients with myasthenia gravis treated without muscle relaxants: A retrospective observational cohort study. J. Anesthesia 2015, 29, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Carron, M.; De Cassai, A.; Linassi, F. Sugammadex in the management of myasthenic patients undergoing surgery: Beyond expectations. Ann. Transl. Med. 2019, 7, S307. [Google Scholar] [CrossRef]
- Mouri, H.; Jo, T.; Matsui, H.; Fushimi, K.; Yasunaga, H. Effect of Sugammadex on Postoperative Myasthenic Crisis in Myasthenia Gravis Patients: Propensity Score Analysis of a Japanese Nationwide Database. Anesth. Analg. 2020, 130, 367–373. [Google Scholar] [CrossRef]
- McQuillen, M.P.; Gross, M.; Sykesville; Johns, R.J. Chlorpromazine-Induced Weakness in Myasthenia Gravis. Arch. Neurol. 1963, 8, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Argov, Z.; Yaari, Y. The action of chlorpromazine at an isolated cholinergic synapse. Brain Res. 1979, 164, 227–236. [Google Scholar] [CrossRef]
- Nguyen, Q.-T.; Yang, J.; Miledi, R. Effects of atypical antipsychotics on vertebrate neuromuscular transmission. Neuropharmacology 2002, 42, 670–676. [Google Scholar] [CrossRef]
- Al-Hashel, J.Y.; Ismail, I.I.; John, J.K.; Ibrahim, M.; Ali, M. Worsening of myasthenia gravis after administration of injectable long-acting risperidone for treatment of schizophrenia; first case report and a call for caution. Neuromuscul. Disord. 2016, 26, 309–311. [Google Scholar] [CrossRef]
- She, S.; Yingjun, Z.; Zhang, B.; Zheng, Y. Worsening of Myasthenia Gravis After Administration of Antipsychotics for Treatment of Schizophrenia. J. Clin. Psychopharmacol. 2017, 37, 620–622. [Google Scholar] [CrossRef]
- Wittbrodt, E.T. Drugs and myasthenia gravis. An update. Arch. Intern. Med. 1997, 157, 399–408. [Google Scholar] [CrossRef]
- Chiu, Y.H.; Yang, A.C.; Chern, C.H.; How, C.K. Myasthenic crisis may mimic antipsychotic-induced extrapyramidal syndromes. J Neuropsychiatry Clin. Neurosci. 2011, 23, E36–E37. [Google Scholar] [CrossRef]
- Neil, J.F.; Himmelhoch, J.M.; Licata, S.M. Emergence of Myasthenia Gravis during Treatment with Lithium Carbonate. Arch. Gen. Psychiatry 1976, 33, 1090–1092. [Google Scholar] [CrossRef]
- Lipton, I.D. Myasthenia Gravis Unmasked by Lithium Carbonate. J. Clin. Psychopharmacol. 1987, 7, 57. [Google Scholar] [CrossRef]
- Granacher, R.P. Neuromuscular problems associated with lithium. Am. J. Psychiatry 1977, 134, 702. [Google Scholar] [CrossRef]
- Alevizos, B.; Gatzonis, S.; Anagnostara, C. Myasthenia gravis disclosed by lithium carbonate. J. Neuropsychiatry Clin. Neurosci. 2006, 18, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Ronzière, T.; Auzou, P.; Ozsancak, C.; Magnier, P.; Sénant, J.; Hannequin, D. Myasthenic syndrome induced by lithium. Presse Médicale 2000, 29, 1043–1044. [Google Scholar]
- Pestronk, A.; Drachman, D. Lithium reduces the number of acetylcholine receptors in skeletal muscle. Science 1980, 210, 342–343. [Google Scholar] [CrossRef] [PubMed]
- Vizi, E.S.; Illés, P.; Rónai, A.; Knoll, J. Effect of lithium on acetylcholine release and synthesis. Neuropharmacology 1972, 11, 521–530. [Google Scholar] [CrossRef]
- Ozawa, T.; Nakajima, T.; Furui, E.; Fukuhara, N. A case of myasthenia gravis associated with long-term phenytoin therapy. Rinsho Shinkeigaku 1996, 36, 1262–1264. [Google Scholar] [PubMed]
- Kurian, M.A.; King, M.D. Antibody positive myasthenia gravis following treatment with carbamazepine--a chance association? Neuropediatrics 2003, 34, 276–277. [Google Scholar]
- Zaidat, O.O.; Kaminski, H.J.; Berenson, F.; Katirji, B. Neuromuscular transmission defect caused by carbamazepine1. Muscle Nerve 1999, 22, 1293–1296. [Google Scholar] [CrossRef]
- Boneva, N.; Brenner, T.; Argov, Z. Gabapentin may be hazardous in myasthenia gravis. Muscle Nerve 2000, 23, 1204–1208. [Google Scholar] [CrossRef]
- Scheschonka, A.; Beuche, W. Treatment of post-herpetic pain in myasthenia gravis: Exacerbation of weakness due to gabapentin. Pain 2003, 104, 423–424. [Google Scholar] [CrossRef]
- Sheen, V.L.; Ohaegbulam, C.; Rencus, T.; Tandon, D. Gabapentin-induced exacerbation of myasthenia gravis. Muscle Nerve 2010, 42, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haroutiunian, S.; Lecht, S.; Zur, A.A.; Hoffman, A.; Davidson, E.; Haroutounian, S. The Challenge of Pain Management in Patients With Myasthenia Gravis. J. Pain Palliat. Care Pharmacother. 2009, 23, 242–260. [Google Scholar] [CrossRef]
- Grob, D.; Harvey, A.M. Effect of adrenocorticotropic hormone (ACTH) and cortisone administration in patients with myasthenia gravis and report of onset of myasthenia gravis during prolonged cortisone administration. Bull. Johns Hopkins Hosp. 1952, 91, 124–136. [Google Scholar]
- Shy, G.M.; Brendler, S.; Rabinovitch, R.; McEachern, D. Effects of cortisone in certain neuromuscular disorders. J. Am. Med Assoc. 1950, 144, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Namba, T.; Brunner, N.G.; Shapiro, M.S.; Grob, D. Corticotropin therapy in myasthenia gravis: Effects, indications, and limitations. Neurology 1971, 21, 1008–1018. [Google Scholar] [CrossRef] [PubMed]
- Brunner, N.G.; Namba, T.; Grob, D. Corticosteroids in management of severe, generalized myasthenia gravis: Effectiveness and comparison with corticotropin therapy. Neurology 1972, 22, 603. [Google Scholar] [CrossRef]
- Jenkins, R. Treatment of myasthenia gravis with prednisone. Lancet 1972, 299, 765–767. [Google Scholar] [CrossRef]
- Seybold, M.E.; Drachman, D.B. Gradually Increasing Doses of Prednisone in Myasthenia Gravis. N. Engl. J. Med. 1974, 290, 81–84. [Google Scholar] [CrossRef]
- Nagane, Y.; Suzuki, S.; Suzuki, N.; Utsugisawa, K. Early Aggressive Treatment Strategy against Myasthenia Gravis. Eur. Neurol. 2011, 65, 16–22. [Google Scholar] [CrossRef]
- Díez-Porras, L.; Homedes, C.; Alberti, M.A.; Vélez-Santamaría, V.; Casasnovas, C. Intravenous immunoglobulins may prevent prednisone-exacerbation in myasthenia gravis. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Bae, J.S.; Go, S.M.; Kim, B.J. Clinical predictors of steroid-induced exacerbation in myasthenia gravis. J. Clin. Neurosci. 2006, 13, 1006–1010. [Google Scholar] [CrossRef]
- Lotan, I.; Hellmann, M.A.; Wilf-Yarkoni, A.; Steiner, I. Exacerbation of myasthenia gravis following corticosteroid treatment: What is the evidence? A systematic review. J. Neurol. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.I.; Goldner, M.M.; Sanders, N.B. Short-term effects of prednisolone on neuromuscular transmission in normal rats and those with experimental autoimmune myasthenia gravis. J. Neurol. Sci. 1979, 41, 223–234. [Google Scholar] [CrossRef]
- Delpy, L.; Douin-Echinard, V.; Garidou, L.; Bruand, C.; Saoudi, A.; Guéry, J.-C. Estrogen Enhances Susceptibility to Experimental Autoimmune Myasthenia Gravis by Promoting Type 1-Polarized Immune Responses. J. Immunol. 2005, 175, 5050–5057. [Google Scholar] [CrossRef] [Green Version]
- Vacca, J.B.; Knight, W.A. Estrogen therapy in myasthenia gravis; report of two cases. MO Med. 1957, 54, 337–340. [Google Scholar]
- Li, Y.; Xiao, B.; Xiao, L.; Zhang, N.; Yang, H. Myasthenia Gravis Accompanied by Premature Ovarian Failure and Aggravation by Estrogen. Intern. Med. 2010, 49, 611–613. [Google Scholar] [CrossRef] [Green Version]
- Yoo, Y.J.; Han, S.B.; Yang, H.K.; Hwang, J.-M. Case Report: Ocular Myasthenia Gravis Associated with In Vitro Fertilization Procedures. Optom. Vis. Sci. 2018, 95, 475–478. [Google Scholar] [CrossRef]
- Skeie, G.O.; Apostolski, S.; Evoli, A.; Gilhus, N.E.; Illa, I.; Harms, L.; Hilton-Jones, D.; Melms, A.; Verschuuren, J.; Horge, H.W. Guidelines for treatment of autoimmune neuromuscular transmission disorders. Eur. J. Neurol. 2010, 17, 893–902. [Google Scholar] [CrossRef]
- Chroni, E.; Punga, A.R. Neurophysiological characteristics of MuSK antibody positive Myasthenia Gravis mice: Focal denervation and hypersensitivity to acetylcholinesterase inhibitors. J. Neurol. Sci. 2012, 316, 150–157. [Google Scholar] [CrossRef]
- Morsch, M.; Reddel, S.W.; Ghazanfari, N.; Toyka, K.V.; Phillips, W.D. Pyridostigmine but not 3,4-diaminopyridine exacerbates ACh receptor loss and myasthenia induced in mice by muscle-specific kinase autoantibody. J. Physiol. 2013, 591, 2747–2762. [Google Scholar] [CrossRef] [Green Version]
- Maggi, L.; Mantegazza, R. Treatment of myasthenia gravis: Focus on pyridostigmine. Clin. Drug Investig. 2011, 31, 691–701. [Google Scholar] [CrossRef]
- Engel, A.G.; Lambert, E.H.; Santa, T. Study of long-term anticholinesterase therapy. Effects on neuromuscular transmission and on motor end-plate fine structure. Neurology 1973, 23, 1273–1281. [Google Scholar] [CrossRef]
- Punga, A.R.; Liik, M. Botulinum toxin injections associated with suspected myasthenia gravis: An underappreciated cause of MG-like clinical presentation. Clin. Neurophysiol. Pract. 2020, 5, 46–49. [Google Scholar] [CrossRef]
- Parikh, R.; Lavin, P.J.M. Cosmetic Botulinum Toxin Type A Induced Ptosis Presenting as Myasthenia. Ophthalmic Plast. Reconstr. Surg. 2011, 27, 470. [Google Scholar] [CrossRef]
- Alaraj, A.M.; Oystreck, D.T.; Bosley, T.M. Variable Ptosis after Botulinum Toxin Type A Injection with Positive Ice Test Mimicking Ocular Myasthenia Gravis. J. Neuro-Ophthalmol. 2013, 33, 169–171. [Google Scholar] [CrossRef]
- Iwase, T.; Iwase, C. Systemic effect of local and small-dose botulinum toxin injection to unmask subclinical myasthenia gravis. Graefe’s Arch. Clin. Exp. Ophthalmol. 2006, 244, 415–416. [Google Scholar] [CrossRef]
- Dressler, D. Subclinical Myasthenia Gravis causing increased sensitivity to botulinum toxin therapy. J. Neural Transm. 2010, 117, 1293–1294. [Google Scholar] [CrossRef]
- Timmermans, G.; Depierreux, F.; Wang, F.; Hansen, I.; Maquet, P. Cosmetic Injection of Botulinum Toxin Unmasking Subclinical Myasthenia Gravis: A Case Report and Literature Review. Case Rep. Neurol. 2019, 11, 244–251. [Google Scholar] [CrossRef]
- Tarsy, D.; Bhattacharyya, N.; Borodic, G. Myasthenia gravis after botulinum toxin a for Meige syndrome. Mov. Disord. 2000, 15, 736–738. [Google Scholar] [CrossRef]
- Watts, J.; Brew, B.; Tisch, S. Myasthenia gravis exacerbation with low dose ocular botulinum toxin for epiphoria. J. Clin. Neurosci. 2015, 22, 1979–1981. [Google Scholar] [CrossRef]
- Emmerson, J. Botulinum toxin for spasmodic torticollis in a patient with myasthenia gravis. Mov. Disord. 1994, 9, 367. [Google Scholar] [CrossRef]
- Gonçalves, M.R.R.; Barbosa, E.R.; Zambon, A.A.; Marchiori, P.E. Treatment of cervical dystonia with botulinum toxin in a patient with myasthenia gravis. Arq. Neuro-Psiquiatr. 1999, 57, 683–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasano, A.; Bentivoglio, A.R.; Ialongo, T.; Soleti, F.; Evoli, A. Treatment with botulinum toxin in a patient with myasthenia gravis and cervical dystonia. Neurology 2005, 64, 2155–2156. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Matsuda, A.; Kitsukawa, Y.; Tanaka, K.; Nishizawa, M.; Tagawa, A. Botulinum toxin treatment for blepharospasm associated with myasthenia gravis. Mov. Disord. 2007, 22, 1363–1364. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Drug | Mechanism | ADR Probability | Comments |
|---|---|---|---|
| Immune Checkpoint inhibitors | T cell activation Increased ratio of T effector to T regulatory cells, B cell activation, autoantibody production, cytokines such as IL-17 | Definite | Avoid after emergence of life-threatening MG If to be used in MG patients, pre-treat with steroids, IVIG, or plasmapheresis |
| D-Penicillamine | Modification of MHC or other molecules on the surface of antigen-presenting cells | Definite | Discontinue and avoid if MG occurs |
| Tyrosine kinase inhibitors * | ? Unspecified immune dysregulation Inhibition of neuromuscular transmission (with tandutinib) | Doubtful (probable with tandutinib) | Not contraindicated, association rarely reported |
| Interferons | Immune dysregulation through changes in cytokines, natural killer cells, alteration of lymphocyte profiles | Possible | Not contraindicated, association rarely reported |
| Statins * | Shift in T cell polarization Superimposed myopathy Mitochondrial toxicity | Probable | Discontinue and avoid in rare cases of emergence or exacerbation of MG |
| Drug | Mechanism | ADR Probability | Comments |
|---|---|---|---|
| Macrolides | Impair neuromuscular transmission, possibly at presynaptic level | Definite | Avoid in MG patients if there is another alternative, otherwise closely monitor |
| Fluoroquinolones | Impair neuromuscular transmission, pre and postsynaptic levels | Probable | Avoid in MG patients if there is another alternative, otherwise closely monitor |
| Aminoglycosides | Impair neuromuscular transmission, pre and postsynaptic levels | Definite | Avoid in MG patients if there is another alternative, otherwise closely monitor |
| Penicillins | Unclear, impaired neuromuscular transmission in an animal model | Probable | Can be used in MG patients as MG exacerbation is rare |
| β-adrenergic blockers | Unclear effect on neuromuscular transmission | Possible | Can be used in stable MG patients, monitor closely, especially early after starting. |
| L type Calcium channel blockers | Unclear effect on neuromuscular transmission | Possible | Can be used in stable MG patients, monitor closely, especially early after starting. |
| Class Ia antiarrhythmics | Impair neuromuscular transmission, pre and postsynaptic levels | Definite | Avoid in MG patients if there is another alternative, otherwise closely monitor |
| Magnesium | Presynaptic (blocks release of ACh) and postsynaptic | Definite | Caution and close monitoring are advised in magnesium replacement (specially parenteral) in MG patients |
| Neuromuscular blockers and inhalation anesthetics | Postsynaptic neuromuscular block | Definite | Nondepolarizing NMBs and inhalation anesthetics better be avoided; if used, observe close postop monitoring, consider using acetylcholinesterase inhibitor and sugammadex |
| Antipsychotics | Impair neuromuscular transmission at presynaptic and postsynaptic levels | Possible | Can be used in MG patients as MG exacerbation is rarely reported |
| Lithium * | Presynaptic: reduction in ACh synthesis and release; postsynaptic: reduction in number of AChRs | Probable | Can be used in MG patients as MG exacerbation is rarely reported |
| Corticosteroids | Unknown; possible direct effect on neuromuscular transmission at high doses | Definite | Avoid starting high doses, if high doses are to be started, consider pretreatment with IVIG or plasmapheresis |
| Botulinum toxin | Presynaptic reduction in ACh release | Definite | Avoid if possible, may be offered with caution and slow dose titration to patients with mild/stable MG who also have blepharospasm or cervical dystonia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheikh, S.; Alvi, U.; Soliven, B.; Rezania, K. Drugs That Induce or Cause Deterioration of Myasthenia Gravis: An Update. J. Clin. Med. 2021, 10, 1537. https://doi.org/10.3390/jcm10071537
Sheikh S, Alvi U, Soliven B, Rezania K. Drugs That Induce or Cause Deterioration of Myasthenia Gravis: An Update. Journal of Clinical Medicine. 2021; 10(7):1537. https://doi.org/10.3390/jcm10071537
Chicago/Turabian StyleSheikh, Shuja, Usman Alvi, Betty Soliven, and Kourosh Rezania. 2021. "Drugs That Induce or Cause Deterioration of Myasthenia Gravis: An Update" Journal of Clinical Medicine 10, no. 7: 1537. https://doi.org/10.3390/jcm10071537