
A Brief Overview of the Microstructural Engineering of Inorganic–Organic Composite Membranes Derived from Organic Chelating Ligands

Abstract
:1. Introduction
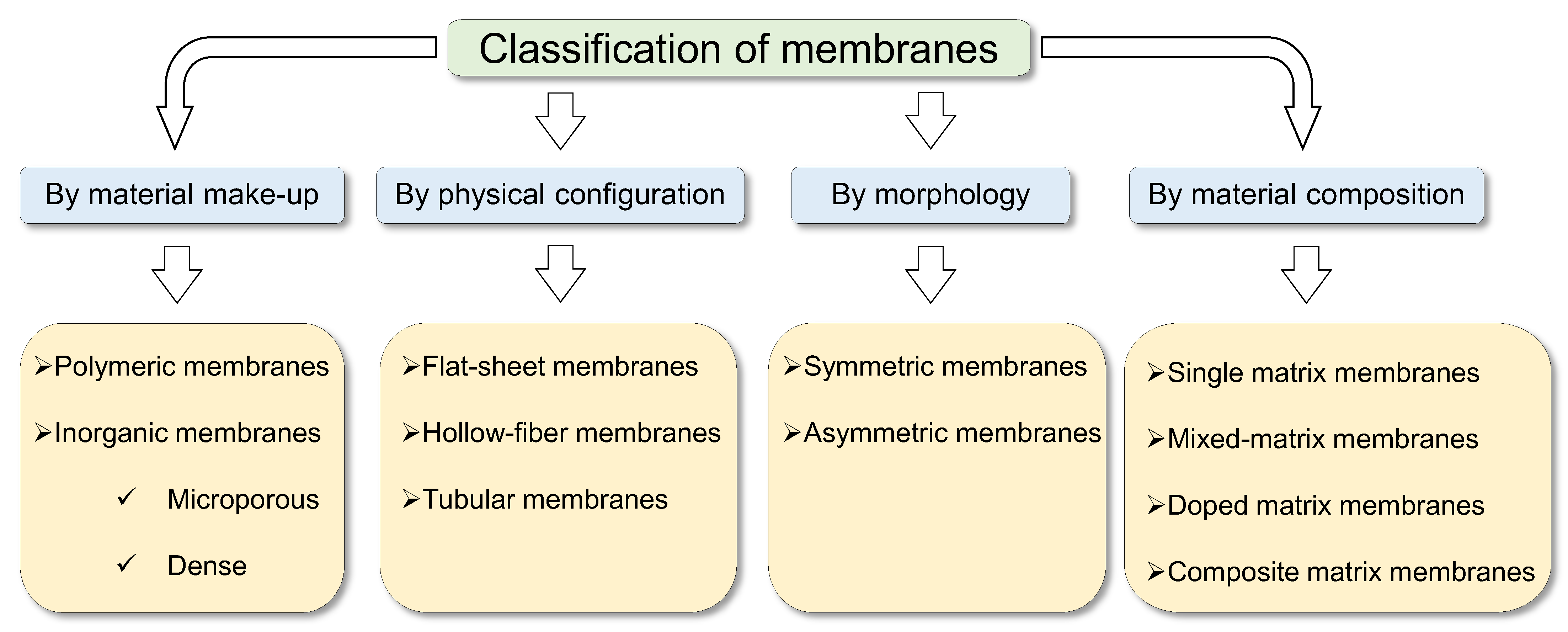
1.1. Separation Membrane Classifications by Matrix Composition
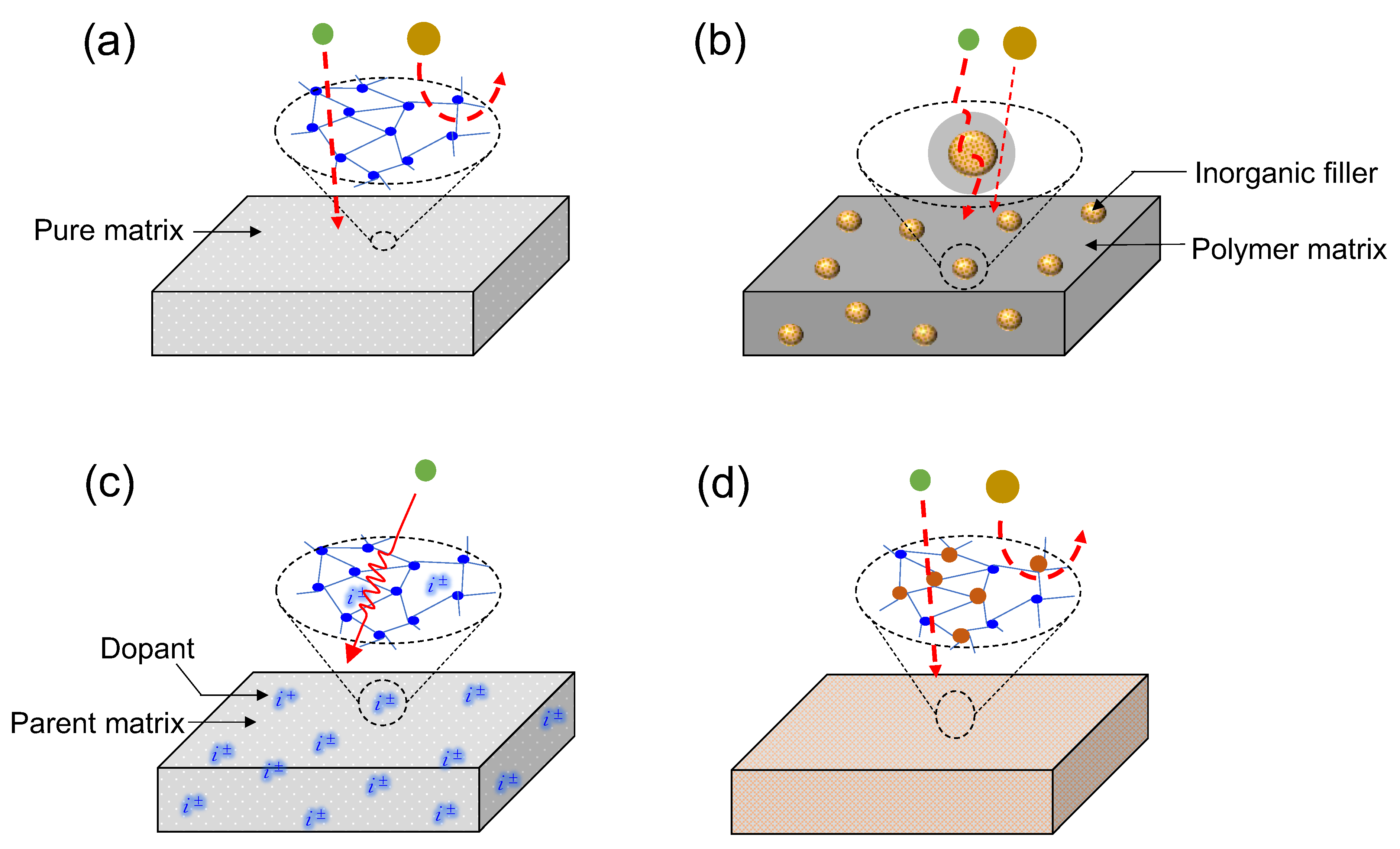
1.1.1. Single-Matrix Membranes
1.1.2. Mixed-Matrix Membranes
1.1.3. Doped Matrix Membranes
1.1.4. Composite Matrix Membranes
- Inorganic–inorganic composites
- Inorganic–organic composites
1.2. Transition-Metal Alkoxides and Organic Chelating Ligands (OCLs)
1.2.1. Transition-Metal Alkoxides
1.2.2. The Partial Charge Model
1.2.3. Gelation Time
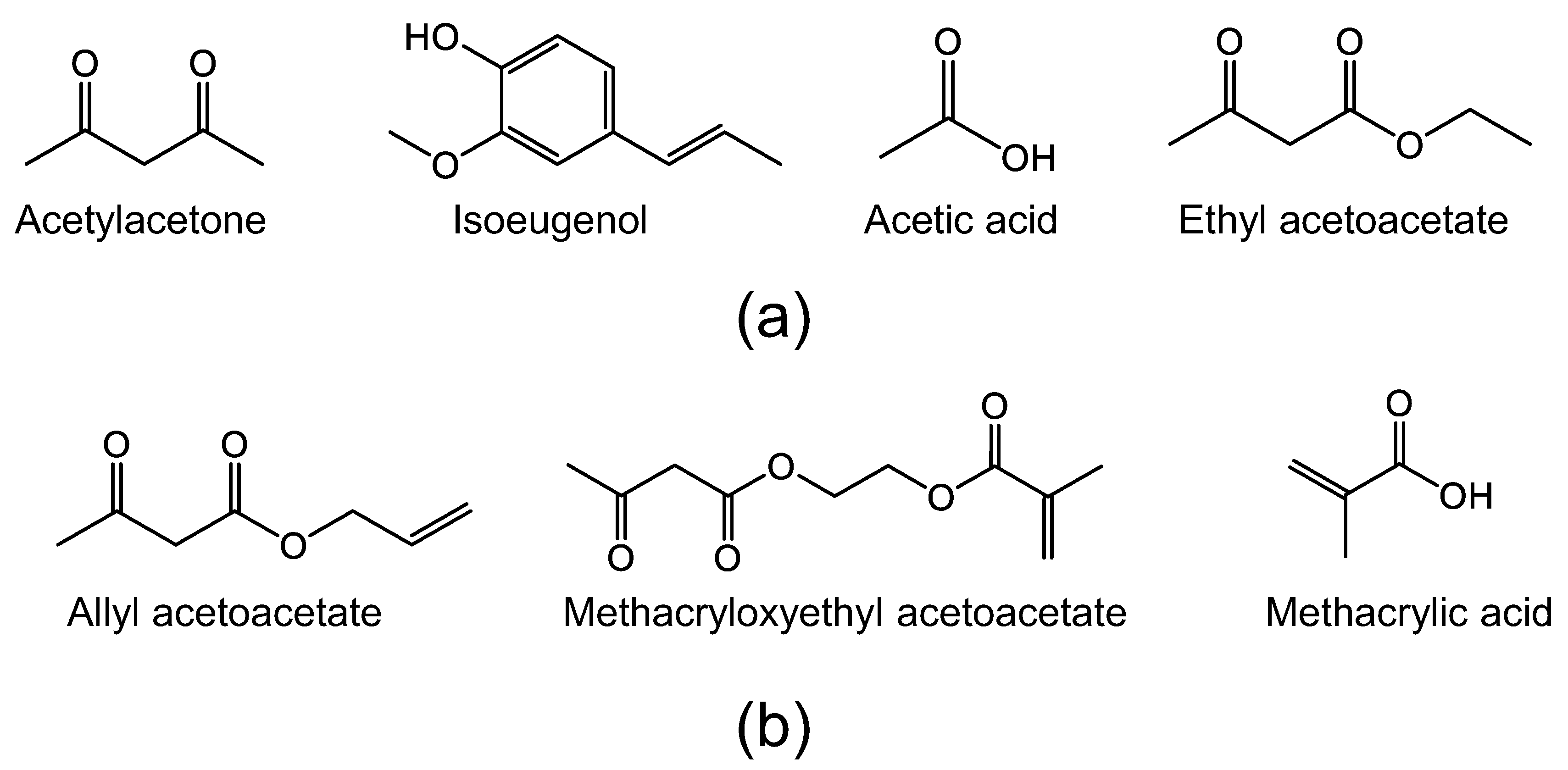
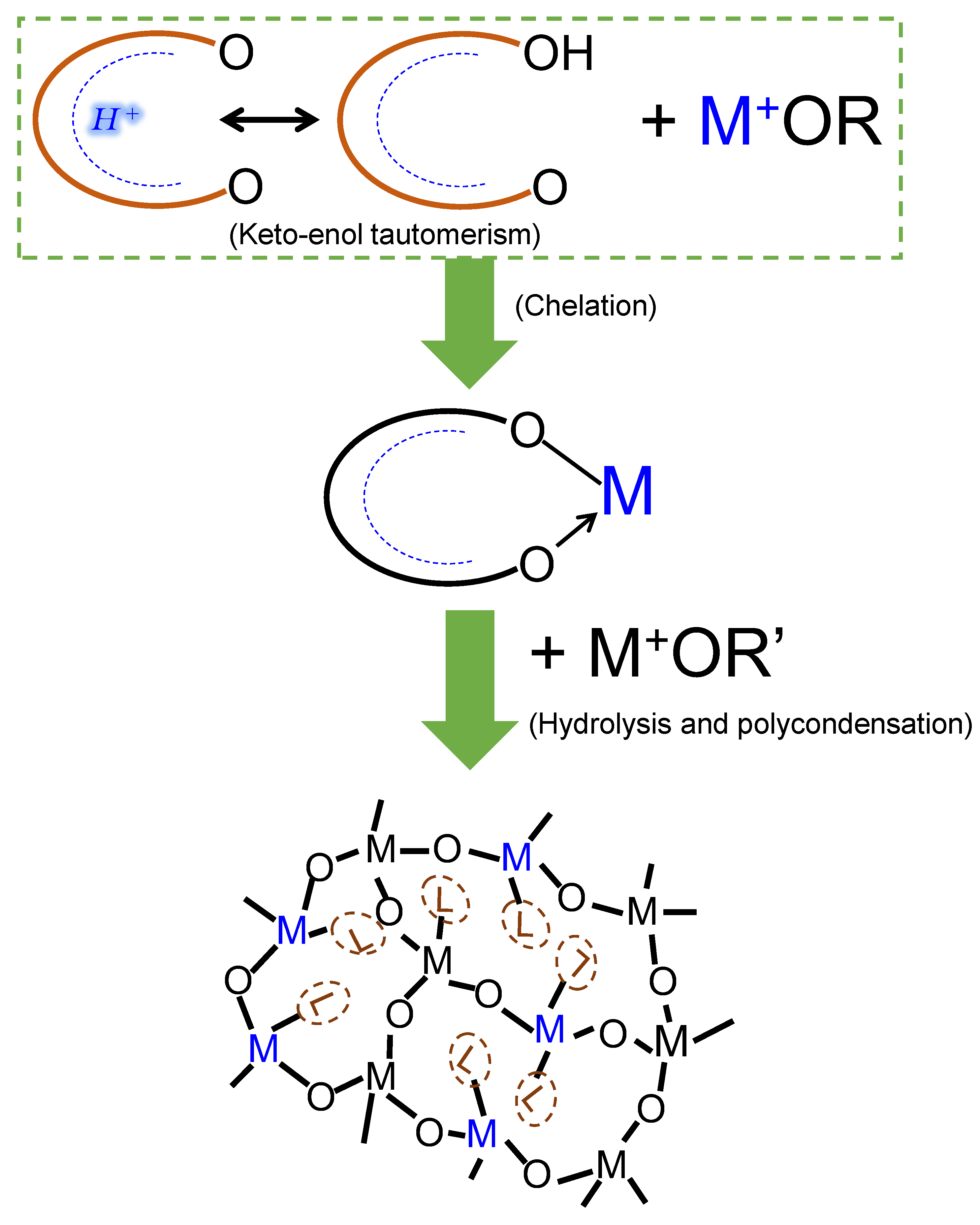
1.2.4. Organic Chelating Ligands
2. Organic Chelating Ligand-Derived Inorganic–Organic Composites
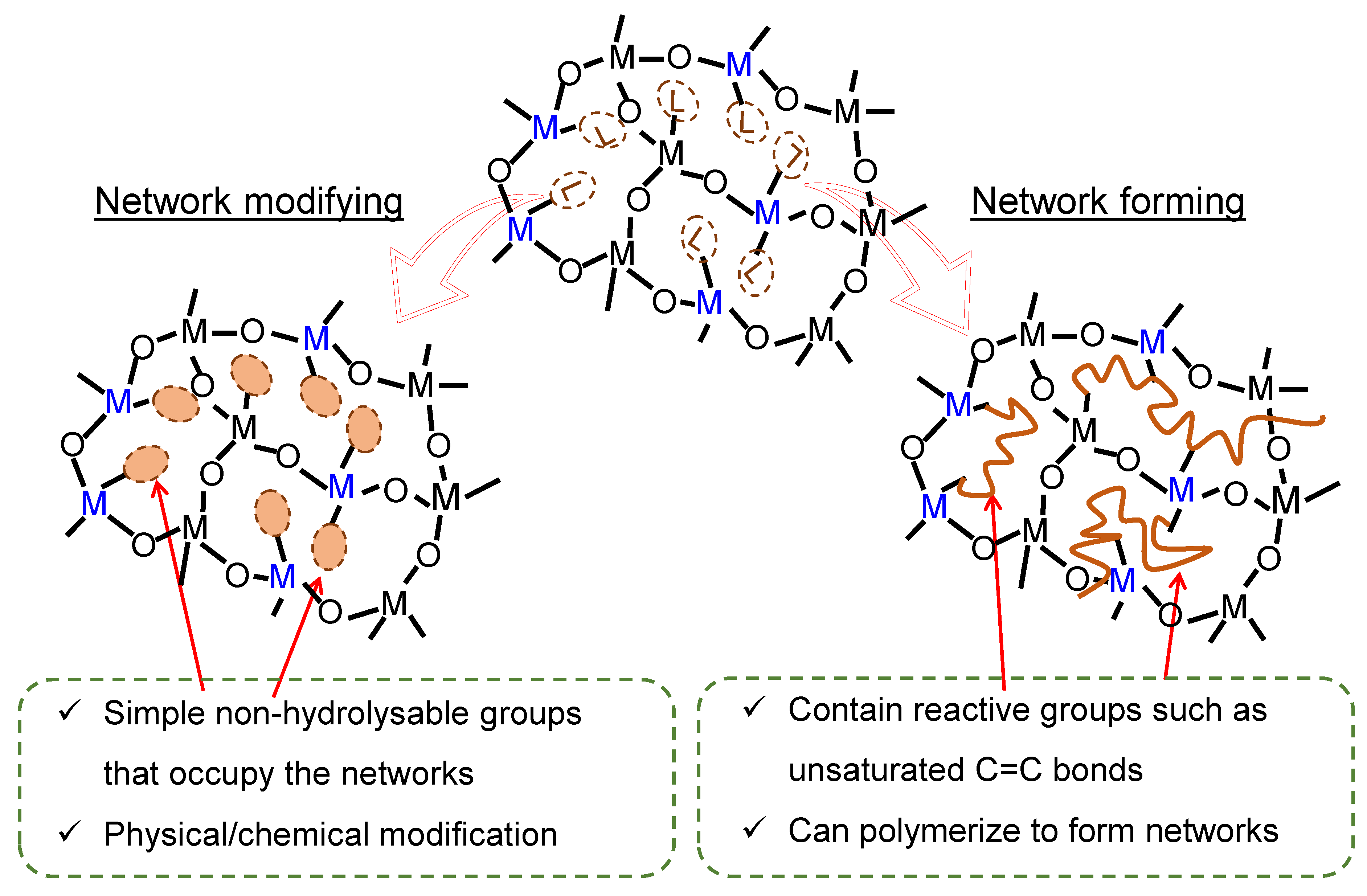
2.1. Organic Chelating Ligands as Molecular Engineering Agents
2.1.1. Network Modifiers
2.1.2. Network Formers
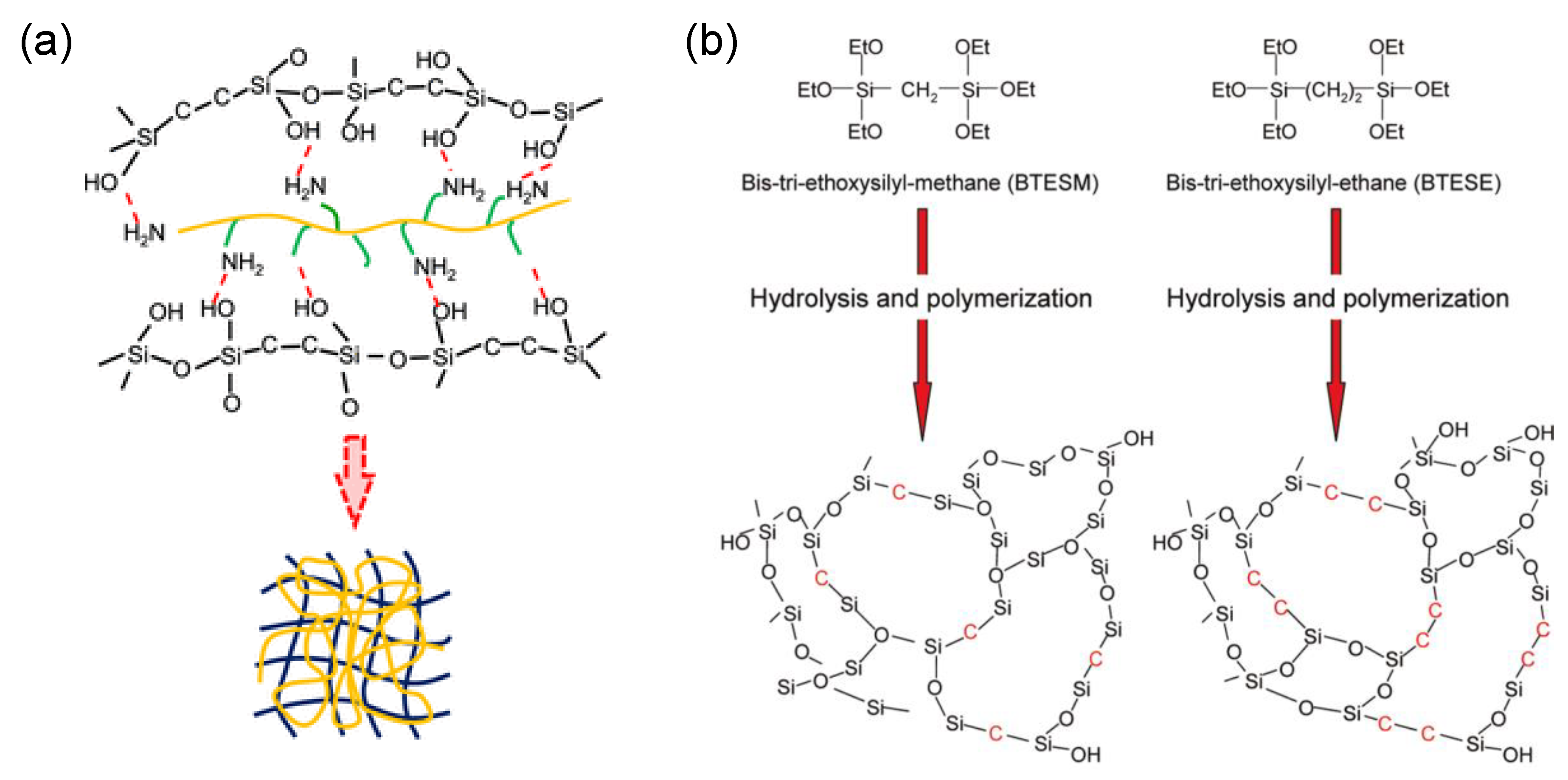
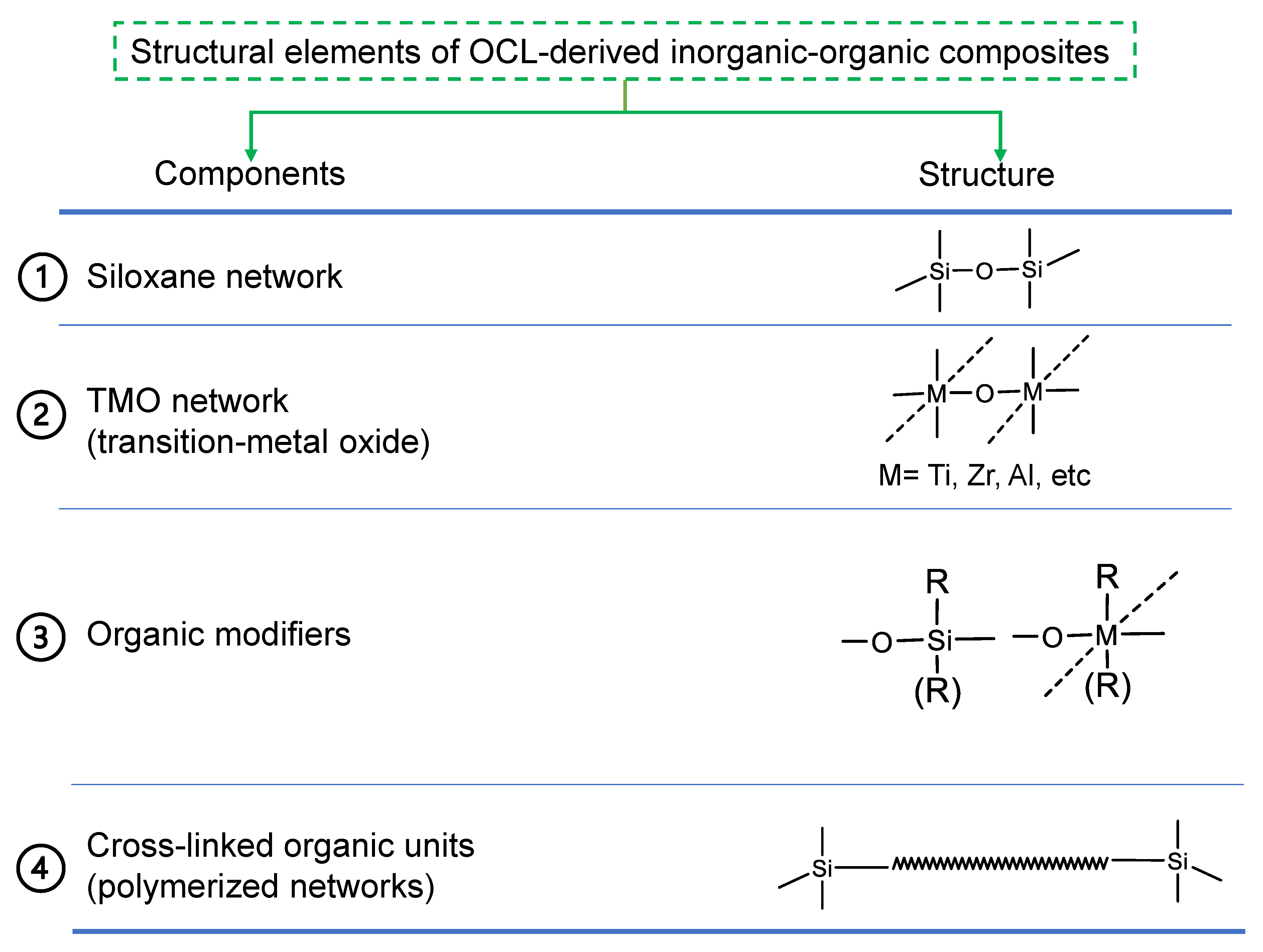
2.2. General Structural Building Blocks of Organic Chelating Ligand-Derived Inorganic–Organic Composites
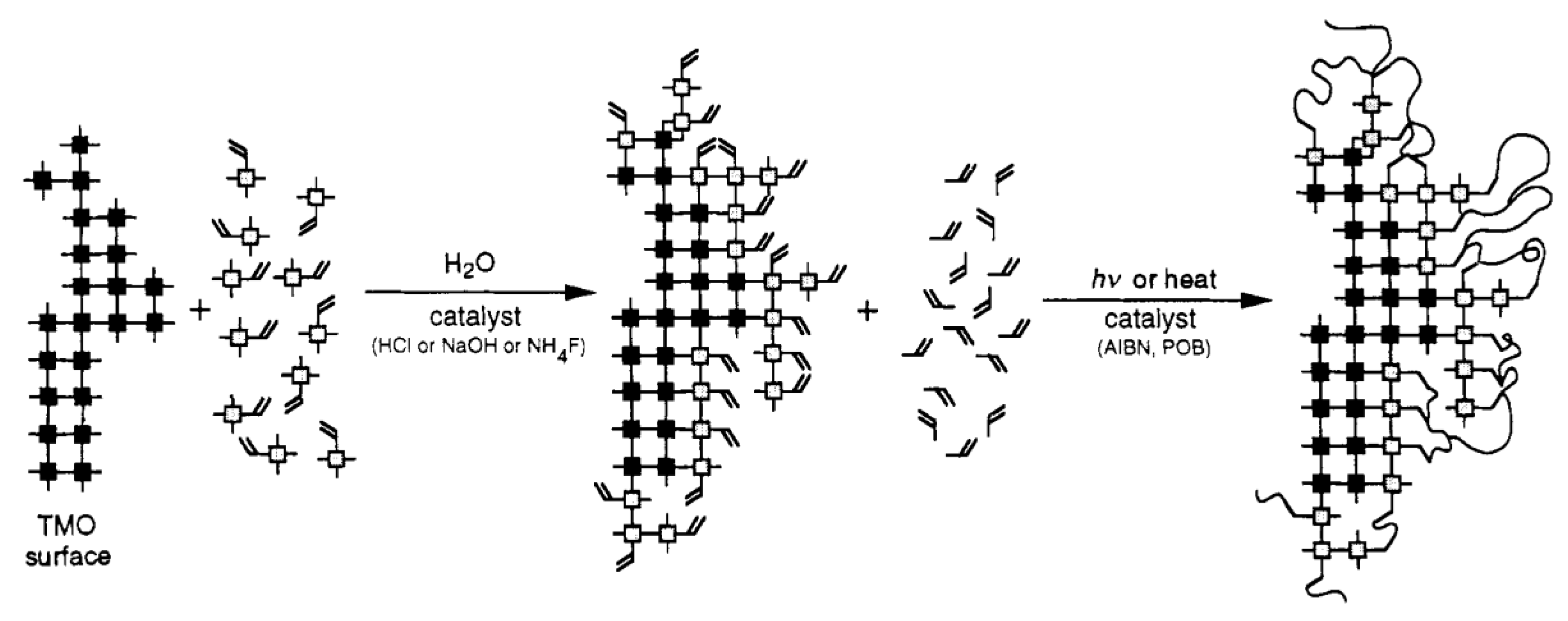
2.3. General Preparation Routes of Organic Chelating Ligand-Derived Inorganic–Organic Composites
3. Inorganic–Organic Composites and Membranes from Network-Modifying Ligands
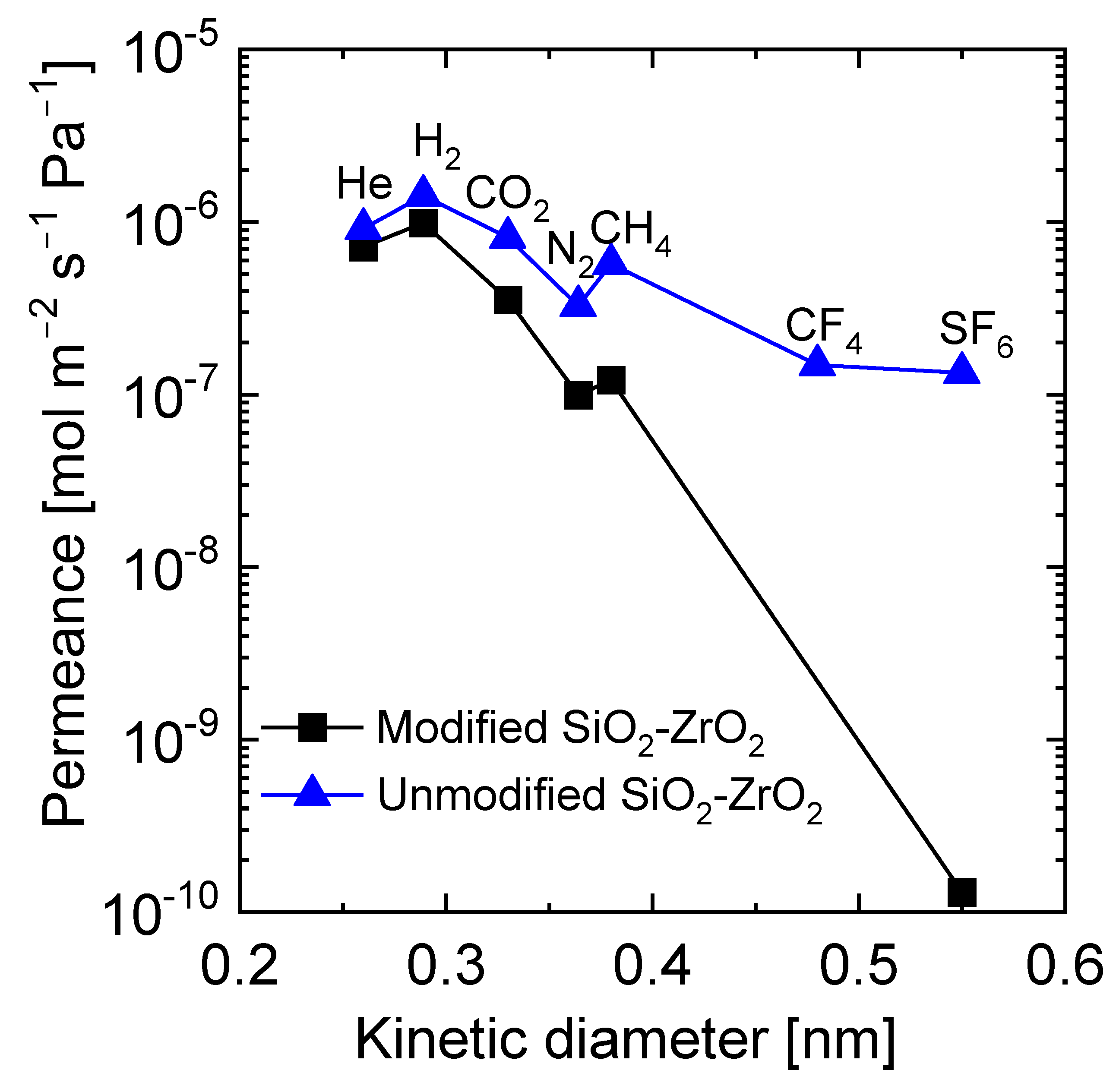
3.1. Applications for Gas Separation
3.2. Applications for Nanofiltration
4. Inorganic–Organic Composites and Membranes from Network-Forming Ligands
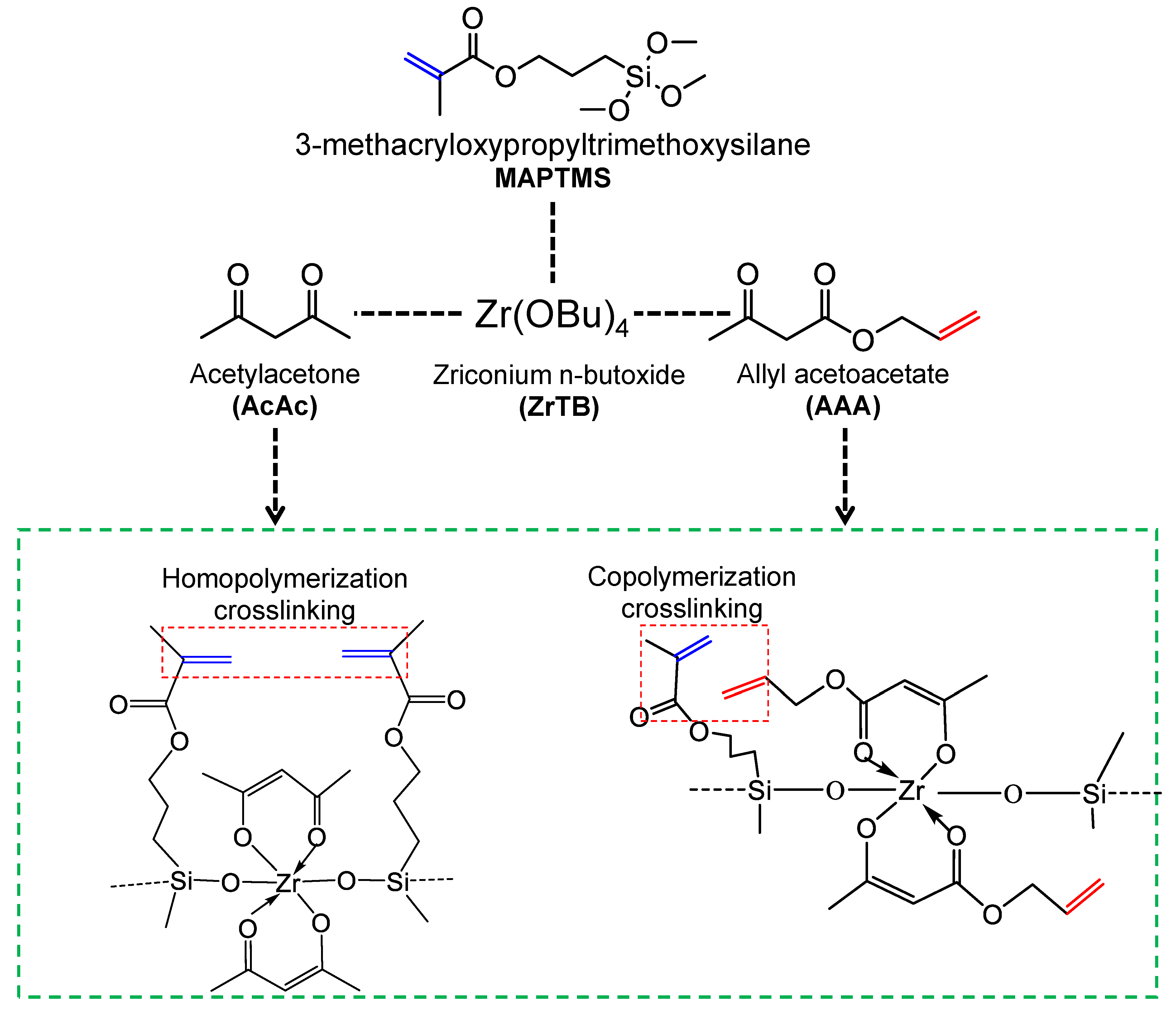
4.1. Effect of Organic Chelating Ligand Type
4.2. Effect of the Crosslinking of Reactive Organic Groups on Membrane Properties and Separation Characteristics
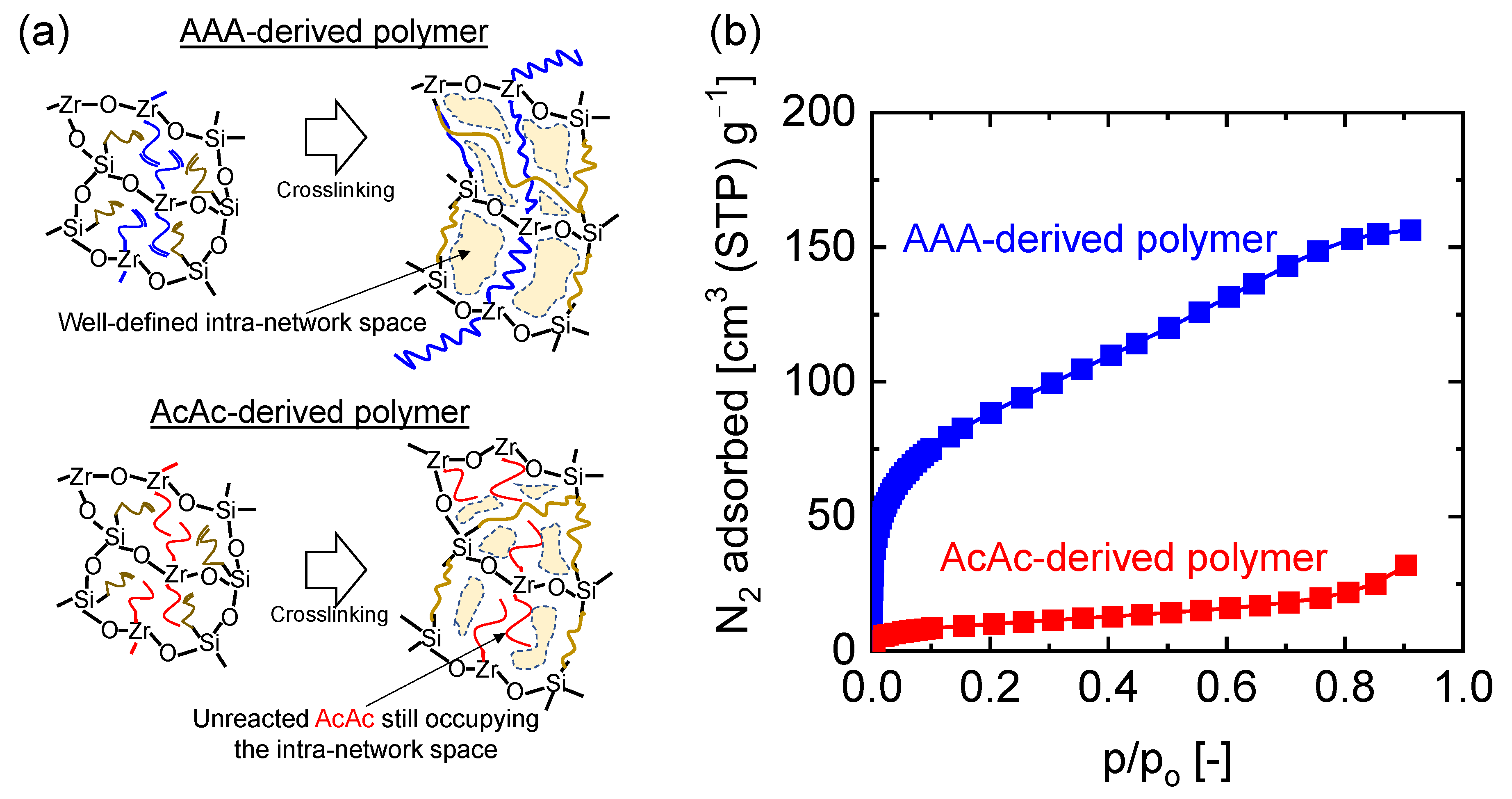
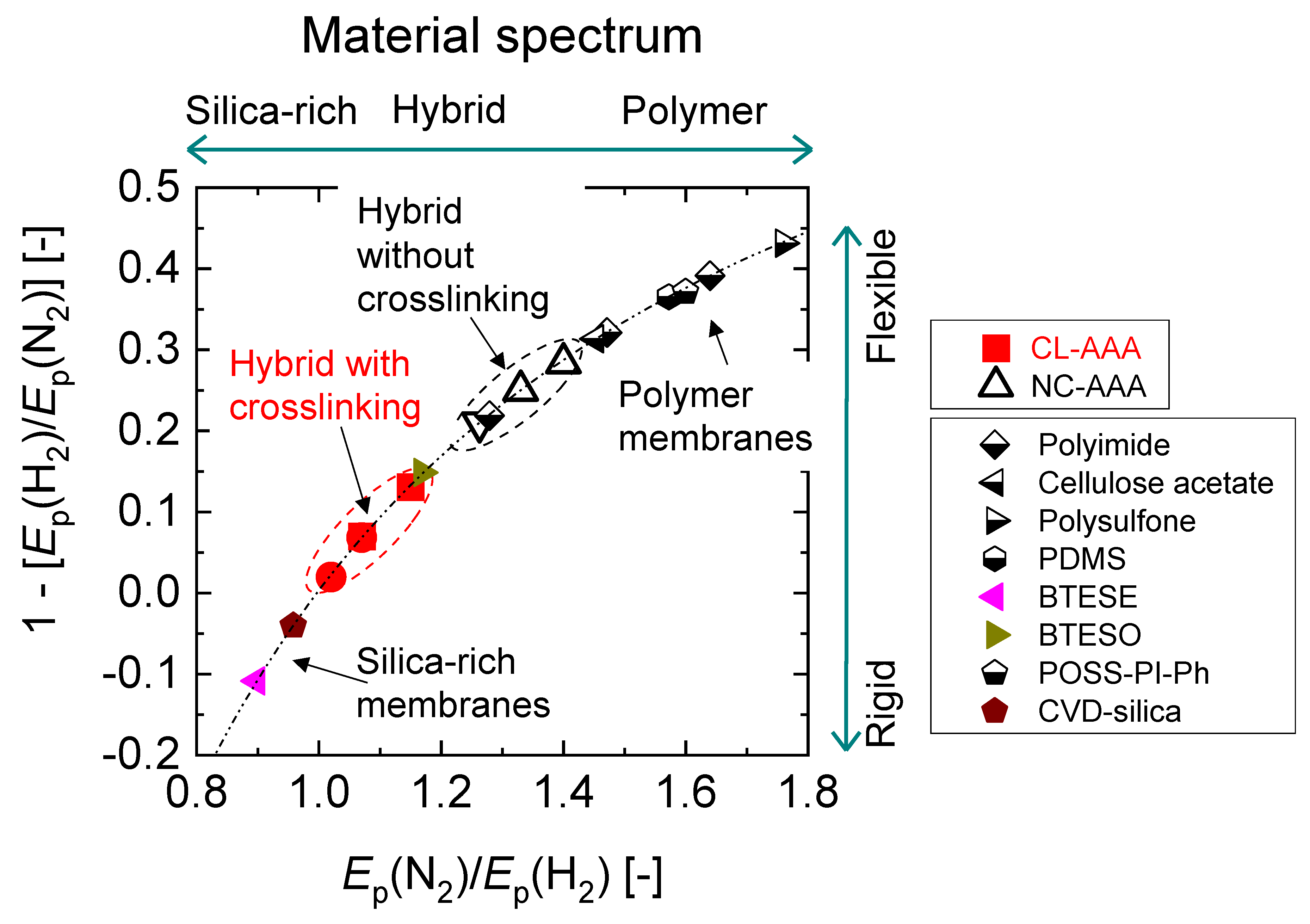
4.3. Microstructural Analysis of Organic Chelating Ligand-Derived Hybrid Polymer Membranes in Comparison to State-of-the-Art Materials
5. Carbon–Ceramic Composite Membranes from Organic Chelating Ligand-Derived Composites
5.1. Carbon–Ceramic Membranes from Network-Modifying Ligand-Derived Composites
5.2. Carbon–Ceramic Membranes from Network-Forming Ligand-Derived Composites
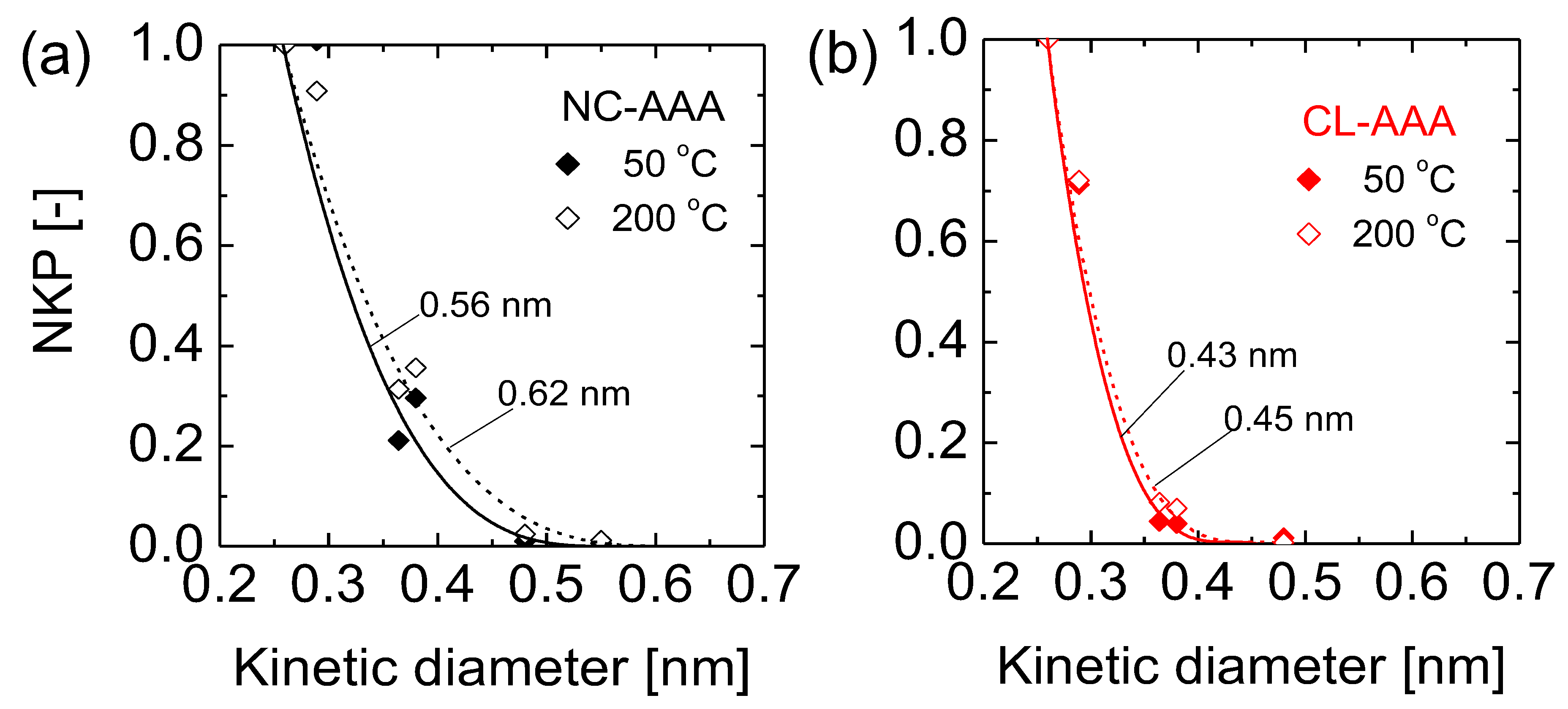
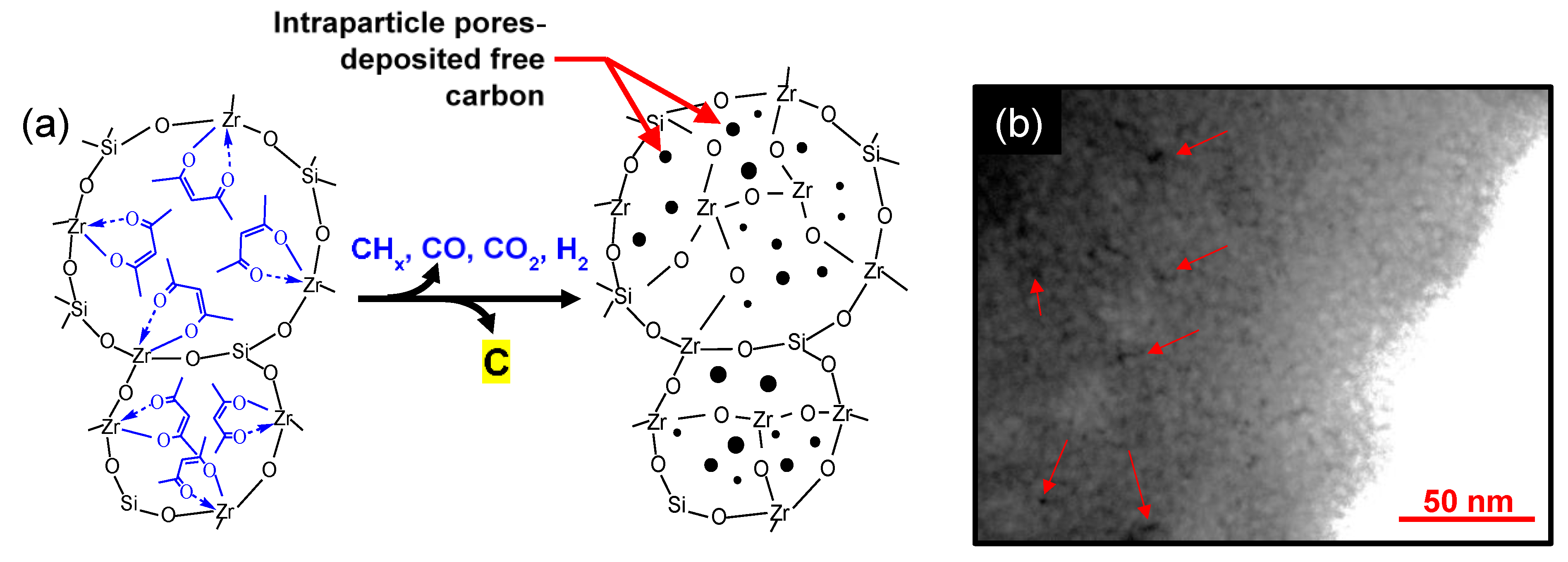
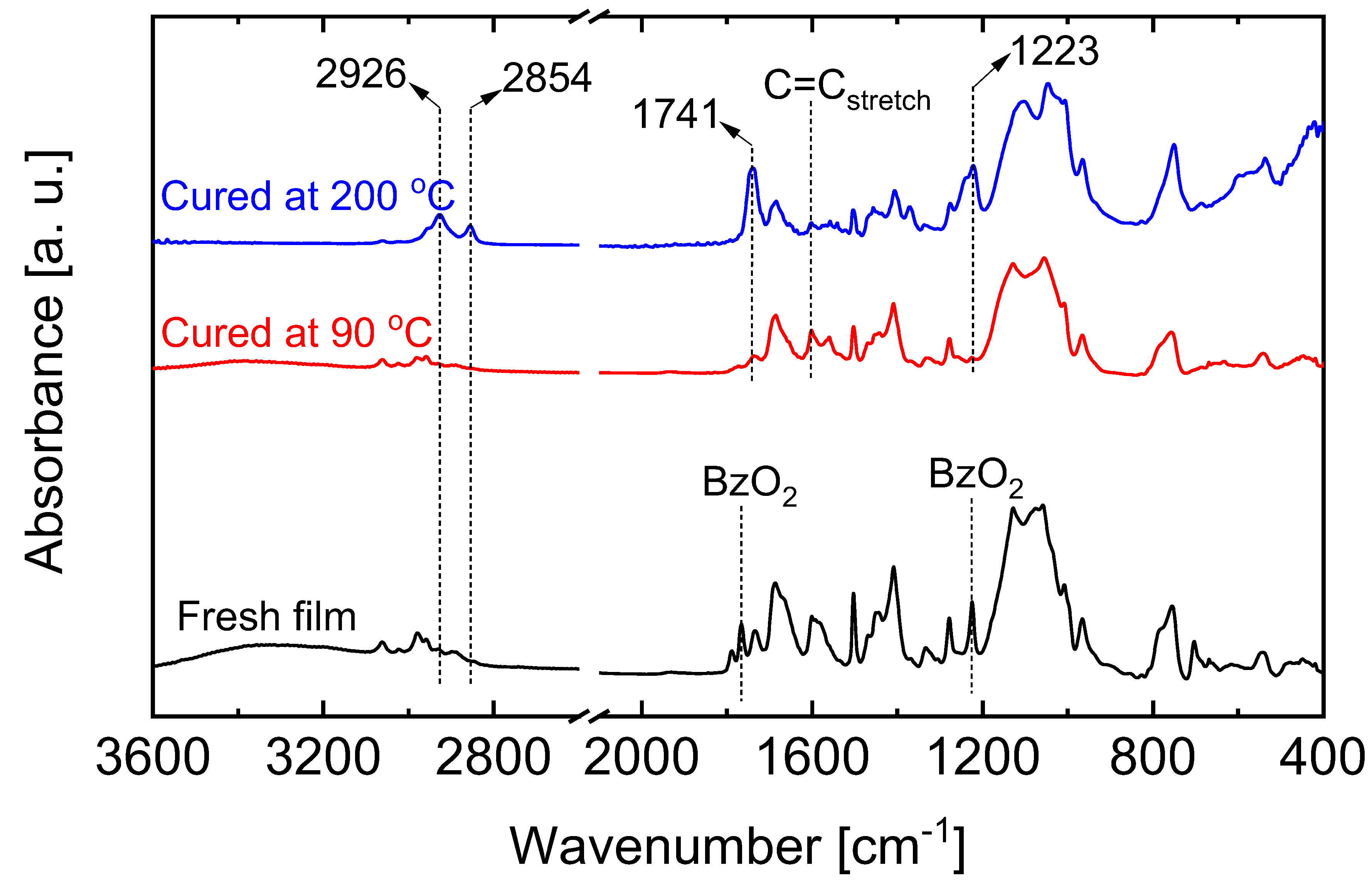
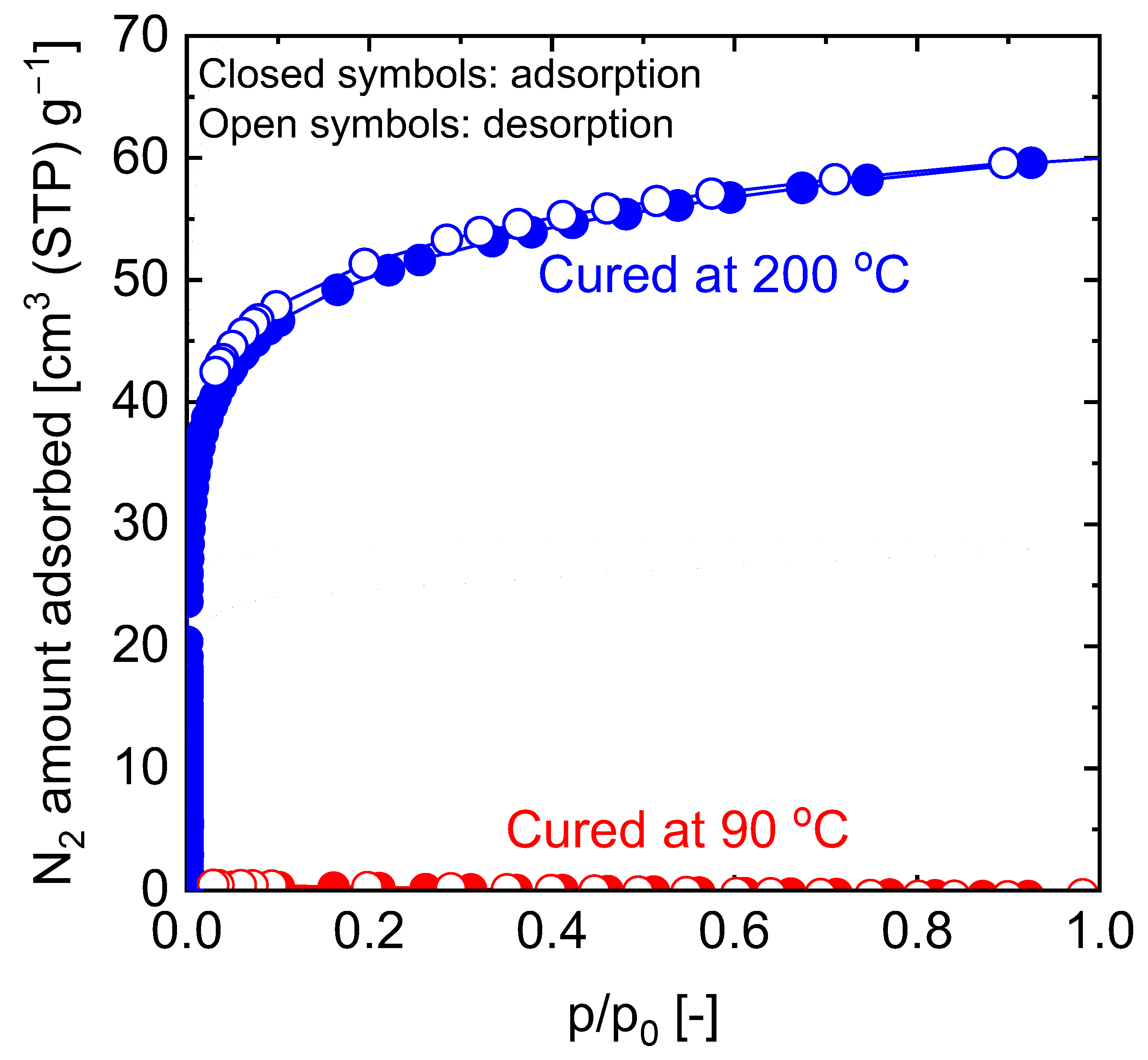
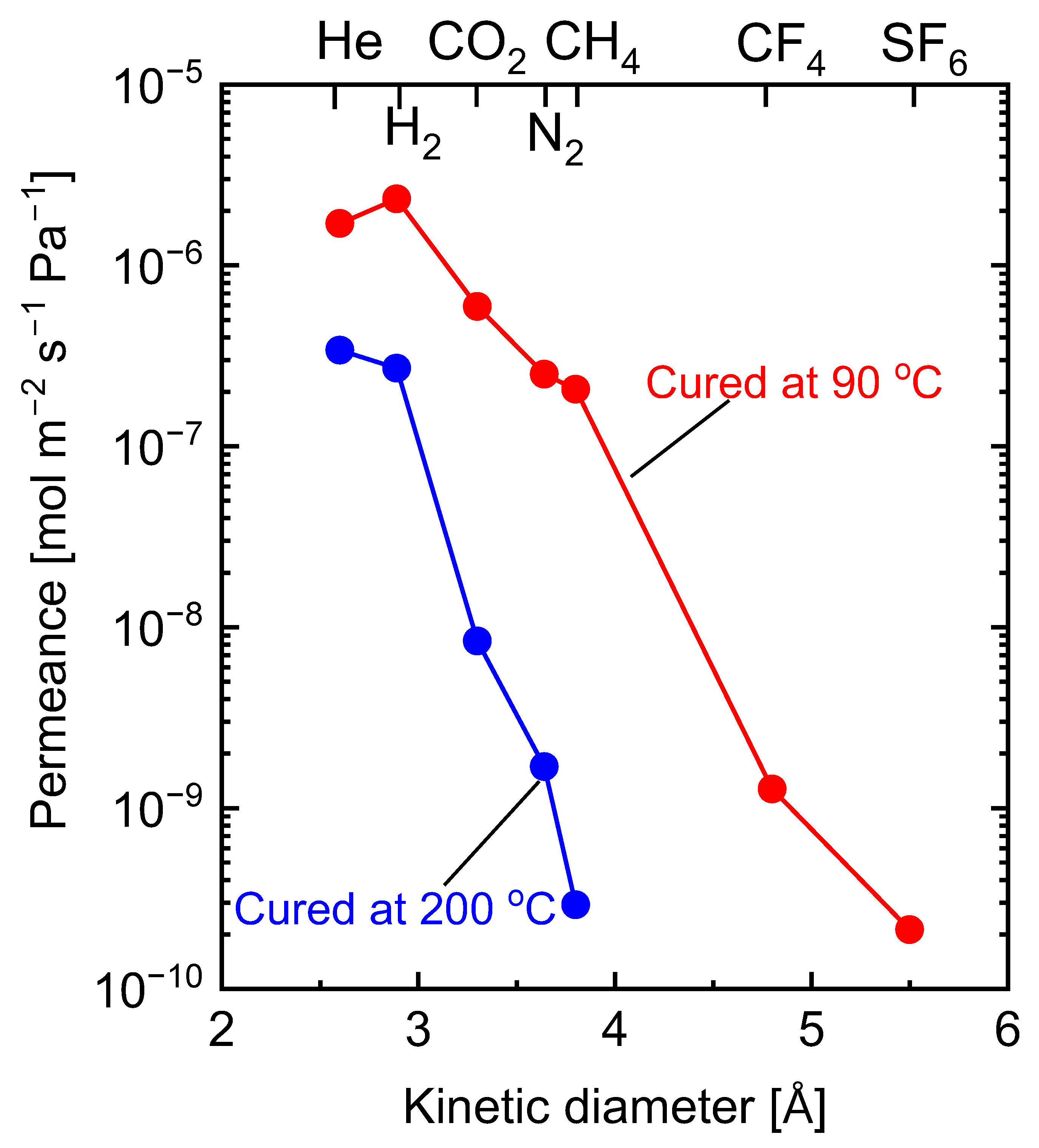
5.2.1. Effect of Curing Temperatures on Microstructural Properties
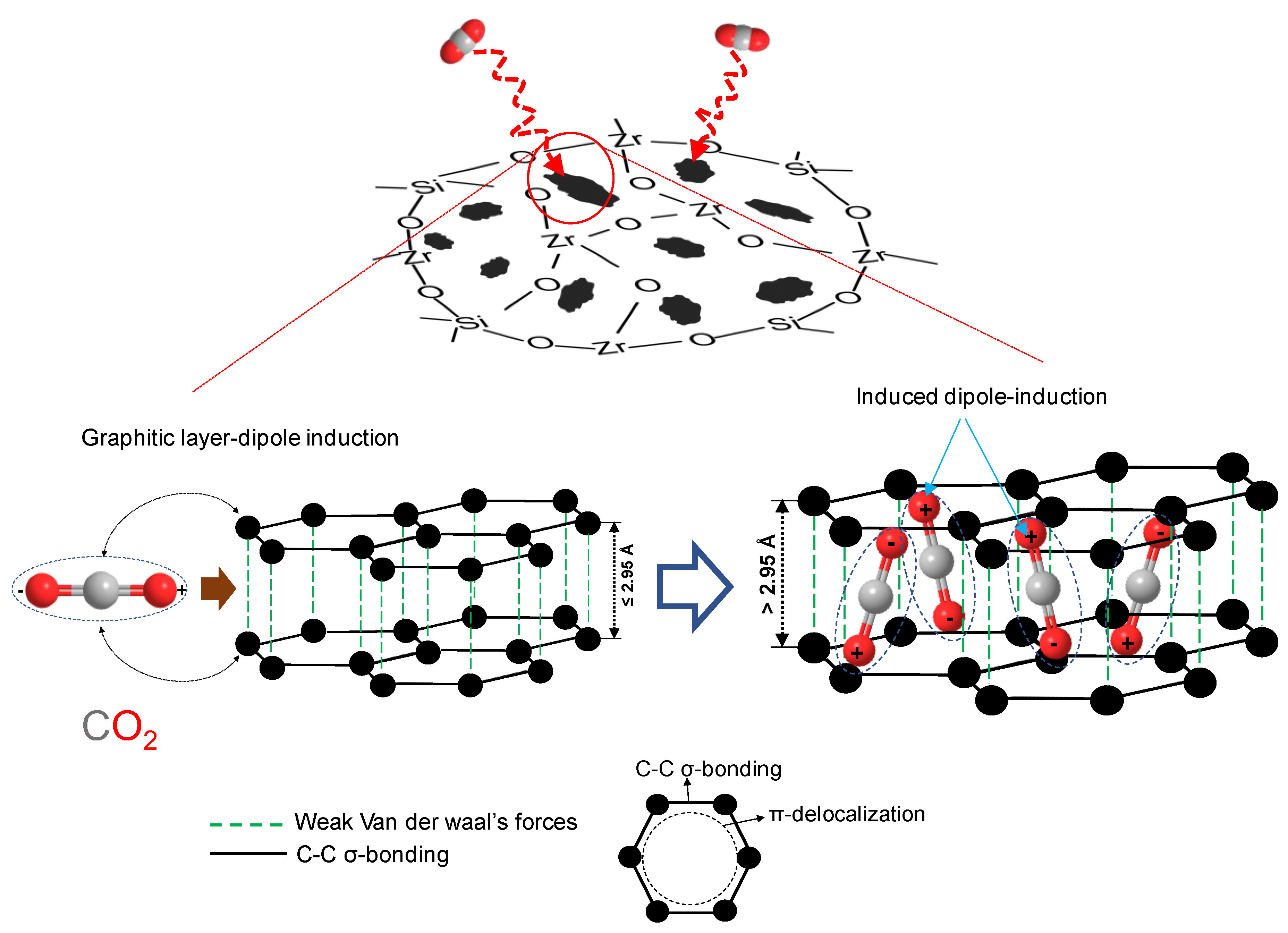
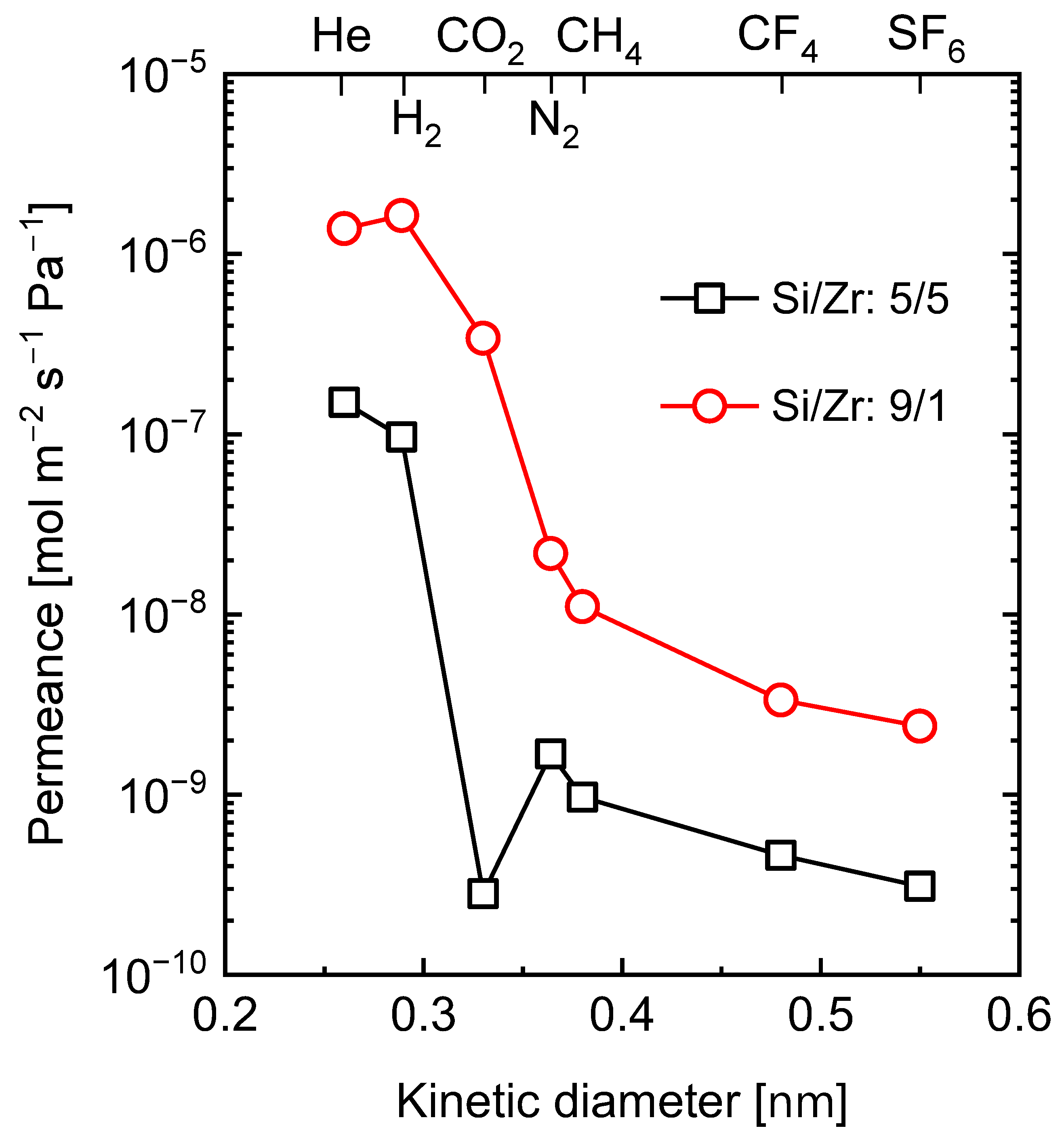
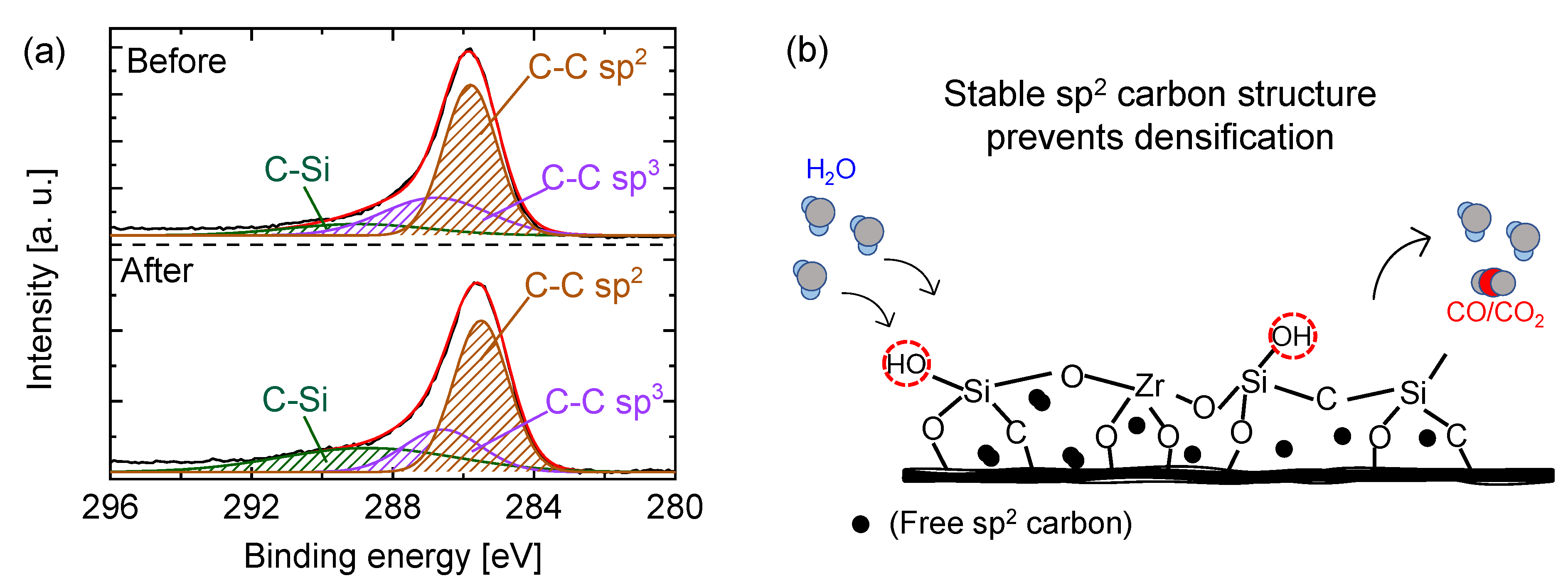
5.2.2. Carbon–Ceramic Composite Membranes’ Performance under Hydrothermal Conditions
6. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Rousseau, R.W. Handbook of Separation Process Technology; John Wiley and Sons: New York, NY, USA, 1987. [Google Scholar]
- Strathmann, H.; Giorno, L.; Drioli, E. An Introduction to Membrane Science and Technology; Consiglio Nazionale Delle Ricerche: Rome, Italy, 2006. [Google Scholar]
- Bounaceur, R.; Lape, N.; Roizard, D.; Vallieres, C.; Favre, E. Membrane processes for post-combustion carbon dioxide capture: A parametric stud. Energy 2006, 31, 2556–2570. [Google Scholar] [CrossRef]
- Charcosset, C. A review of membrane processes and renewable energies for desalination. Desalination 2009, 245, 214–231. [Google Scholar] [CrossRef]
- Strathmann, H. Membrane separation processes: Current relevance and future opportunities. AIChE J. 2001, 47, 1077–1087. [Google Scholar] [CrossRef]
- Mitchel, J.K. On the penetrativeness of fluids. J. Membr. Sci. 1995, 100, 11–16. [Google Scholar] [CrossRef]
- Robeson, L.M. Correlation of separation factor versus permeability for polymeric membranes. J. Membr. Sci. 1991, 62, 165–185. [Google Scholar] [CrossRef]
- Pinnau, I.; Freeman, B.D. Formation and Modification of Polymeric Membranes; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1999; pp. 1–22. [Google Scholar]
- de Vos, R.M.; Verweij, H. Improved performance of silica membranes for gas separation. J. Membr. Sci. 1998, 143, 37–51. [Google Scholar] [CrossRef]
- Hatlevik, Ø.; Gade, S.K.; Keeling, M.K.; Thoen, P.M.; Davidson, A.P.; Way, J.D. Palladium and palladium alloy membranes for hydrogen separation and production: History, fabrication strategies, and current performance. Sep. Purif. Technol. 2010, 73, 59–64. [Google Scholar] [CrossRef]
- Tang, Z.; Dong, J.; Nenoff, T.M. Internal Surface Modification of MFI-Type Zeolite Membranes for High Selectivity and High Flux for Hydrogen. Langmuir 2009, 25, 4848–4852. [Google Scholar] [CrossRef]
- Cao, G.; Lu, Y.; Delattre, L.; Brinker, C.J.; Lopez, G.P. Amorphous silica molecular sieving membranes by sol-gel processing. Adv. Mater. 1996, 8, 588–591. [Google Scholar] [CrossRef]
- Iwamoto, Y.; Sato, K.; Kato, T.; Inada, T.; Kubo, Y. A hydrogen-permselective amorphous silica membrane derived from polysilazane. J. Eur. Ceram. Soc. 2005, 25, 257–264. [Google Scholar] [CrossRef]
- Tsuru, T.; Morita, T.; Shintani, H.; Yoshioka, T.; Asaeda, M. Membrane reactor performance of steam reforming of methane using hydrogen-permselective catalytic SiO2 membranes. J. Membr. Sci. 2007, 316, 53–62. [Google Scholar] [CrossRef]
- Li, G.; Kanezashi, M.; Yoshioka, T.; Tsuru, T. Ammonia decomposition in catalytic membrane reactors: Simulation and experimental studies. AIChE J. 2013, 59, 168–179. [Google Scholar] [CrossRef]
- Tavolaro, A.; Drioli, E. Zeolite membranes. Adv. Mater. 1999, 11, 975–996. [Google Scholar] [CrossRef]
- Caro, J.; Noack, M.; Kolsch, P.; Schafer, R. Zeolite membranes—State of their development and perspective. Microporous Mesoporous Mater. 2000, 38, 3–24. [Google Scholar] [CrossRef]
- Caro, J.; Noack, M. Zeolite membranes—Recent developments and progress. Microporous Mesoporous Mater. 2008, 115, 215–233. [Google Scholar] [CrossRef]
- Rangnekar, N.; Mittal, N.; Elyassi, B.; Caro, J.; Tsapatsis, M. Zeolite membranes—A review and comparison with MOFs. Chem. Soc. Rev. 2015, 44, 7128–7154. [Google Scholar] [CrossRef] [Green Version]
- Al-Mufachi, N.A.; Rees, N.V.; Steinberger-Wilkens, R. Hydrogen selective membranes: A review of palladium-based dense metal membranes. Renew. Sustain. Energy Rev. 2015, 47, 540–551. [Google Scholar] [CrossRef]
- Peachey, N.M.; Snow, R.C.; Dye, R.C. Composite Pd/Ta metal membranes for hydrogen separation. J. Membr. Sci. 1996, 111, 123–133. [Google Scholar] [CrossRef]
- Peters, T.A.; Kaleta, T.; Stange, M.; Bredesen, R. Development of thin binary and ternary Pd-based alloy membranes for use in hydrogen production. J. Membr. Sci. 2011, 383, 124–134. [Google Scholar] [CrossRef]
- Wei, J.; Qiu, C.; Tang, C.Y.; Wang, R.; Fane, A.G. Synthesis and characterization of flat-sheet thin film composite forward osmosis membranes. J. Membr. Sci. 2011, 372, 292–302. [Google Scholar] [CrossRef]
- Pezeshk, N.; Rana, D.; Narbaitz, R.M.; Matsuura, T. Novel modified PVDF ultrafiltration flat-sheet membranes. J. Membr. Sci. 2012, 389, 280–286. [Google Scholar] [CrossRef]
- Tan, X.; Liu, S.; Li, K. Preparation and characterization of inorganic hollow fiber membranes. J. Membr. Sci. 2001, 188, 87–95. [Google Scholar] [CrossRef]
- Khan, I.U.; Othman, M.H.D.; Ismail, A.F.; Matsuura, T.; Hashim, H.; Abdul Hadi Md Nordin, N.; Rahman, M.A.; Jaafar, J.; Jilani, A. Status and improvement of dual-layer hollow fiber membranes via coextrusion process for gas separation: A review. J. Nat. Gas Sci. Eng. 2018, 52, 215–234. [Google Scholar] [CrossRef]
- Chen, X.Y.; Kaliaguine, S.; Rodrigue, D. A Comparison between Several Commercial Polymer Hollow Fiber Membranes for Gas Separation. J. Membr. Sep. Technol. 2017, 6, 1–15. [Google Scholar] [CrossRef]
- Lei, L.; Pan, F.; Lindbrathen, A.; Zhang, X.; Hillestad, M.; Nie, Y.; Bai, L.; He, X.; Guiver, M.D. Carbon hollow fiber membranes for a molecular sieve with precise-cutoff ultramicropores for superior hydrogen separation. Nat. Comm. 2021, 12, 268. [Google Scholar] [CrossRef]
- Lu, H.; Zhu, S. Interfacial synthesis of free-standing metal–organic framework membranes. Eur. J. Inorg. Chem. 2013, 8, 1294–1300. [Google Scholar] [CrossRef]
- Caro, J. Hierarchy in inorganic membranes. Chem. Soc. Rev. 2016, 45, 3468. [Google Scholar] [CrossRef] [Green Version]
- Koros, W.J. Evolving beyond the thermal age of separation processes: Membranes can lead the way. AIChE J. 2004, 50, 2326–2334. [Google Scholar] [CrossRef]
- Koros, W.J. A more aggressive approach is needed for gas separation membrane development. Membrane 2006, 31, 157–160. [Google Scholar] [CrossRef] [Green Version]
- Abdul Hamid, M.R.; Jeong, H. -K. Recent advances on mixed-matrix membranes from gas separation: Opportunities and engineering challenges. Korean J. Chem. Eng. 2018, 35, 1577–1600. [Google Scholar] [CrossRef]
- Zimmerman, C.M.; Singh, A.; Koros, W.J. Tailoring mixed matrix composite membranes for gas separations. J. Membr. Sci. 1997, 137, 145–154. [Google Scholar] [CrossRef]
- Dong, G.; Li, H.; Chen, V. Challenges and opportunities for mixed-matrix membranes for gas separation. J. Mater. Chem. A 2013, 1, 4610–4630. [Google Scholar] [CrossRef]
- Goh, P.S.; Ismail, A.F.; Sanip, S.M.; Ng, B.C.; Aziz, M. Recent advances of inorganic fillers in mixed matrix membrane for gas separation. Sep. Purif. Technol. 2011, 81, 243–264. [Google Scholar] [CrossRef]
- Vinh-Thang, H.; Kaliaguine, S. Predictive models for mixed-matrix membrane performance: A review. Chem. Rev. 2013, 113, 4980–5028. [Google Scholar] [CrossRef] [PubMed]
- Kanezashi, M.; Asaeda, M. Hydrogen permeation characteristics and stability of Ni-doped silica membranes in steam at high temperature. J. Membr. Sci. 2006, 271, 86–93. [Google Scholar] [CrossRef]
- Uhlmann, D.; Liu, S.; Ladewig, B.P.; da Costa, J.C.D. Cobalt-doped silica membranes for gas separation. J. Membr. Sci. 2009, 326, 316–321. [Google Scholar] [CrossRef]
- Kanezashi, M.; Murata, M.; Nagasawa, H.; Tsuru, T. Fluorine doping of microporous organosilica membranes for pore size control and enhanced hydrophobic properties. ACS Omega 2018, 3, 8612–8620. [Google Scholar] [CrossRef]
- Takenaka, M.; Nagasawa, H.; Tsuru, T.; Kanezashi, M. Hydrocarbon permeation properties through microporous fluorine-doped organosilica membranes with controlled pore sizes. J. Membr. Sci. 2021, 619, 118787. [Google Scholar] [CrossRef]
- Kanezashi, M.; Matsutani, T.; Nagasawa, H.; Tsuru, T. Fluorine-induced microporous silica membranes: Dramatic improvement in hydrothermal stability and pore size controllability for highly permeable propylene/propane separation. J. Membr. Sci. 2018, 549, 111–119. [Google Scholar] [CrossRef]
- Kanezashi, M.; Hataoka, N.; Ikram, R.; Nagasawa, H.; Tsuru, T. Hydrothermal stability of fluorine-induced microporous silica membranes: Effect of steam treatment conditions. AIChE J. 2021, 67, e17292. [Google Scholar] [CrossRef]
- Yang, J.; Fan, W.; Bell, C.-M. Effect of calcination atmosphere on microstructure and H2/CO2 separation of palladium-doped silica membranes. Sep. Purif. Technol. 2019, 210, 659–669. [Google Scholar] [CrossRef]
- Anggarini, U.; Yu, L.; Nagasawa, H.; Kanezashi, M.; Tsuru, T. Metal-induced microporous aminosilica creates a highly permeable gas-separation membrane. Mater. Chem. Front. 2021, 5, 3029–3042. [Google Scholar] [CrossRef]
- Anggarini, U.; Yu, L.; Nagasawa, H.; Kanezashi, M.; Tsuru, T. Microporous nickel-coordinated aminosilica membranes for improved pervaporation performance of methanol/toluene separation. ACS Appl. Mater. Interfaces 2021, 13, 23247–23259. [Google Scholar] [CrossRef] [PubMed]
- Kanezashi, M.; Sano, M.; Yoshioka, T.; Tsuru, T. Extremely thin Pd–silica mixed-matrix membranes with nano-dispersion for improved hydrogen permeability. Chem. Commun. 2010, 46, 6171–6173. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Hirano, Y.; Fujii, H.; Tsuru, T.; Asaeda, M. Hydrothermal stability and performance of silica-zirconia membranes for hydrogen separation in hydrothermal conditions. J. Chem. Eng. Jpn. 2001, 34, 523–530. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.-S.; Ryu, C.-H.; Hwang, G.-J. Obtention of ZrO2–SiO2 hydrogen permselective membrane by chemical vapor deposition method. Chem. Eng. J. 2013, 232, 302–309. [Google Scholar] [CrossRef]
- Ahn, S.-J.; Takagaki, A.; Sugawara, T.; Kikuchi, R.; Oyama, S.T. Permeation properties of silica-zirconia composite membranes supported on porous alumina substrates. J. Membr. Sci. 2017, 526, 409–416. [Google Scholar] [CrossRef]
- Anisah, S.; Puthai, W.; Kanezashi, M.; Nagasawa, H.; Tsuru, T. Preparation, characterization, and evaluation of TiO2-ZrO2 nanofiltration membranes fired at different temperatures. J. Membr. Sci. 2018, 564, 691–699. [Google Scholar] [CrossRef]
- Anisah, S.; Kanezashi, M.; Nagasawa, H.; Tsuru, T. Hydrothermal stability and permeation properties of TiO2-ZrO2 (5/5) nanofiltration membranes at high temperatures. Sep. Purif. Technol. 2019, 212, 1001–1012. [Google Scholar] [CrossRef]
- Guo, H.; Zhao, S.; Wu, X.; Qi, H. Fabrication and characterization of TiO2/ZrO2 ceramic membranes for nanofiltration. Microporous Mesoporous Mater. 2018, 260, 125–131. [Google Scholar] [CrossRef]
- Guizard, C.; Ayral, A.; Julbe, A. Potentiality of organic solvents filtration with ceramic membranes. A comparison with polymer membranes. Desalination 2002, 147, 275–280. [Google Scholar] [CrossRef]
- Asaeda, M.; Sakou, Y.; Yang, J.; Shimasaki, K. Stability and performance of porous silica–zirconia composite membranes for pervaporation of aqueous organic solutions. J. Membr. Sci. 2002, 209, 163–175. [Google Scholar] [CrossRef]
- Puthai, W.; Kanezashi, M.; Nagasawa, H.; Wakamura, K.; Ohnishi, H.; Tsuru, T. Effect of firing temperature on the water permeability of SiO2–ZrO2 membranes for nanofiltration. J. Membr. Sci. 2016, 497, 348–356. [Google Scholar] [CrossRef] [Green Version]
- Puthai, W.; Kanezashi, M.; Nagasawa, H.; Tsuru, T. SiO2-ZrO2 nanofiltration membranes of different Si/Zr molar ratios: Stability in hot water and acid/alkaline solutions. J. Membr. Sci. 2017, 524, 700–711. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, C.; Ribot, F.; Lebeau, B. Molecular design of hybrid organic-inorganic nanocomposites synthesized via sol-gel chemistry. J. Chem. Mater. 1999, 9, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Nakahiro, K.; Yu, L.; Nagasawa, H.; Tsuru, T.; Kanezashi, M. Pore structure controllability and CO2 permeation properties of silica-derived membranes with a dual-network structure. Ind. Eng. Chem. Res. 2021, 60, 8527–8537. [Google Scholar] [CrossRef]
- Castricum, H.L.; Sah, A.; Kreiter, R.; Blank, D.H.; Vente, J.F.; Johan, E. Hydrothermally stable molecular separation membranes from organically linked silica. J. Mater. Chem. 2008, 18, 2150–2158. [Google Scholar] [CrossRef]
- Kanezashi, M.; Kawano, M.; Yoshioka, T.; Tsuru, T. Organic–inorganic hybrid silica membranes with controlled silica network size for propylene/propane separation. Ind. Eng. Chem. Res. 2012, 51, 944–953. [Google Scholar] [CrossRef]
- Yu, L.; Kanezashi, M.; Nagasawa, H.; Oshita, J.; Naka, A.; Tsuru, T. Pyrimidine-bridged organoalkoxysilane membrane for high-efficiency CO2 transport via mild affinity. Sep. Purif. Technol. 2017, 178, 232–241. [Google Scholar] [CrossRef]
- Lin, B.; Heijman, S.G.; Shang, R.; Rietveld, L.C. Integration of oxalic acid chelation and Fenton process for synergistic relaxation-oxidation of persistent gel-like fouling of ceramic nanofiltration membranes. J. Membr. Sci. 2021, 636, 119553. [Google Scholar] [CrossRef]
- Safaei, M.; Foroughi, M.M.; Ebrahimpoor, N.; Jahani, S.; Omidi, A.; Khatami, M. A review on metal-organic frameworks: Synthesis and applications. TrAC Trends Anal. Chem. 2019, 118, 401–425. [Google Scholar] [CrossRef]
- Ghasempour, H.; Wang, K.-Y.; Powell, J.A.; ZareKarizi, F.; Lv, X.-L.; Morsali, A.; Zhou, H.-C. Metal–organic frameworks based on multicarboxylate linkers. Coord. Chem. Rev. 2021, 426, 213542. [Google Scholar] [CrossRef]
- Milea, C.; Bogatu, C.; Duta, A. The influence of parameters in silica sol-gel process. Bull. Transylvania Univ. Bras. 2011, 4, 53. [Google Scholar]
- Ward, D.A.; Ko, E.I. Preparing catalytic materials by the sol-gel method. Ind. Eng. Chem. Res. 1995, 34, 421–433. [Google Scholar] [CrossRef]
- Livage, J.; Sanchez, C. Sol-gel chemistry. J. Non-Cryst. Solids 1992, 145, 11–19. [Google Scholar] [CrossRef]
- Sanchez, C.; Livage, J.; Henry, M.; Babonneau, F. Chemical modification of alkoxide precursors. J. Non-Cryst. Solids 1988, 100, 65–76. [Google Scholar] [CrossRef]
- Klopman, G. A semiempirical treatment of molecular structures. I. Electronegativity and atomic terms. J. Am. Chem. Soc. 1964, 86, 1463–1469. [Google Scholar] [CrossRef]
- Stockmayer, W.H. Theory of molecular size distribution and gel formation in branched-chain polymers. J. Chem. Phys. 1943, 11, 45–55. [Google Scholar] [CrossRef]
- Errington, R.J.; Ridland, J.; Clegg, W.; Coxall, R.A.; Sherwood, J.M. β-Diketonate derivatives of titanium alkoxides: X-ray crystal structures and solution dynamics of the binuclear complexes [{Ti (OR)3 (dik)}2]. Polyhedron 1998, 17, 659–674. [Google Scholar] [CrossRef]
- Chen, H.-J.; Wang, L.; Chiu, W.-Y. Chelation and solvent effect on the preparation of titania colloids. Mater. Chem. Phys. 2007, 101, 12–19. [Google Scholar] [CrossRef]
- Kurajica, S.; Škorić, I.; Lozić, I.; Mandić, V. Ethyl acetoacetate ligand distribution in the course of titanium n-butoxide chelation. Mater. Chem. Phys. 2014, 147, 1058–1067. [Google Scholar] [CrossRef]
- Hoebbel, D.; Reinert, T.; Schmidt, H.; Arpac, E. On the hydrolytic stability of organic ligands in Al-, Ti-and Zr-alkoxide complexes. J. Sol-Gel Sci. Technol. 1997, 10, 115–126. [Google Scholar] [CrossRef]
- Schmidt, H. Preparation, application and potential of ORMOCERs. In Sol-Gel: Science and Technology—Proceedings of the Winter School on Glass and Ceramics From Gels; Aegerter, M.A., Ed.; World Scientific: Singapore, 1989; pp. 432–469. [Google Scholar]
- Sanchez, C.; Soler-Illia, G.D.A.; Ribot, F.; Lalot, T.; Mayer, C.R.; Cabuil, V. Designed hybrid organic− inorganic nanocomposites from functional nanobuilding blocks. Chem. Mater. 2001, 13, 3061–3083. [Google Scholar] [CrossRef]
- Stockmayer, W.H. Theory of molecular size distribution and gel formation in branched polymers II. General cross linking. J. Chem. Phys. 1944, 12, 125–131. [Google Scholar] [CrossRef]
- Benfer, S.; Popp, U.; Richter, H.; Siewert, C.; Tomandl, G. Development and characterization of ceramic nanofiltration membranes. Sep. Purif. Technol. 2001, 22, 231–237. [Google Scholar] [CrossRef]
- Haas, K.H. Hybrid inorganic–organic polymers based on organically modified Si-Alkoxides. Adv. Eng. Mater. 2000, 2, 571–582. [Google Scholar] [CrossRef]
- Sanchez, C.; Julián, B.; Belleville, P.; Popall, M. Applications of hybrid organic–inorganic nanocomposites. J. Mater. Chem. 2005, 15, 3559–3592. [Google Scholar] [CrossRef]
- Judeinstein, P.; Sanchez, C. Hybrid organic–inorganic materials: A land of multidisciplinarity. J. Mater. Chem. 1996, 6, 511–525. [Google Scholar] [CrossRef]
- Fukumoto, T.; Yoshioka, T.; Nagasawa, H.; Kanezashi, M.; Tsuru, T. Development and gas permeation properties of microporous amorphous TiO2–ZrO2–organic composite membranes using chelating ligands. J. Membr. Sci. 2014, 461, 96–105. [Google Scholar] [CrossRef]
- Spijksma, G.I.; Huiskes, C.; Benes, N.E.; Kruidhof, H.; Blank, D.H.; Kessler, V.G.; Bouwmeester, H.J. Microporous Zirconia–Titania Composite Membranes Derived from Diethanolamine-Modified Precursors. Adv. Mater. 2006, 18, 2165–2168. [Google Scholar] [CrossRef]
- Tachibana, T.; Yoshioka, T.; Nakagawa, K.; Shintani, T.; Kamio, E.; Matsuyama, H. Gas Permeation Characteristics of TiO2-ZrO2-Aromatic Organic Chelating Ligand (aOCL) Composite Membranes. Membranes 2020, 10, 388. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Vivar, J.; Mendoza-Serna, R. SiO2-TiO2 membranes by the sol-gel process. Silicon Chem. 2006, 3, 59–64. [Google Scholar] [CrossRef]
- Lawal, S.; Kanezashi, M.; Nagasawa, H.; Tsuru, T. Development of an acetylacetonate-modified silica-zirconia composite membrane applicable to gas separation. J. Membr. Sci. 2020, 599, 117844. [Google Scholar] [CrossRef]
- Sada, Y.; Yoshioka, T.; Nakagawa, K.; Shintani, T.; Iesako, R.; Kamio, E.; Matsuyama, H. Preparation and characterization of organic chelate ligand (OCL)-templated TiO2–ZrO2 nanofiltration membranes. J. Membr. Sci. 2019, 591, 117304. [Google Scholar] [CrossRef]
- Iesako, R.; Yoshioka, T.; Nakagawa, K.; Shintani, T.; Matsuoka, A.; Kamio, E.; Matsuyama, H. Organic solvent permeation characteristics of TiO2-ZrO2 composite nanofiltration membranes prepared using organic chelating ligand to control pore size and surface property. Sep. Purif. Technol. 2022, 297, 121458. [Google Scholar] [CrossRef]
- Amberg-Schwab, S.; Katschorek, H.; Weber, U.; Burger, A.; Hänsel, R.; Steinbrecher, B.; Harzer, D. Inorganic-organic polymers as migration barriers against liquid and volatile compounds. J. Sol-Gel Sci. Technol. 2003, 26, 699–703. [Google Scholar] [CrossRef]
- Le Guével, X.; Palazzesi, C.; Prosposito, P.; Della Giustina, G.; Brusatin, G. Influence of chelating agents on the photopolymerization of hybrid Ti-based waveguides. J. Mater. Chem. 2008, 18, 3556–3562. [Google Scholar] [CrossRef]
- Rodič, P.; Zanna, S.; Milošev, I.; Marcus, P. Degradation of Sol-Gel Acrylic Coatings Based on Si and Zr Investigated Using Electrochemical Impedance, Infrared and X-Ray Photoelectron Spectroscopies. Front. Mater. Sci. 2021, 8, 756447. [Google Scholar] [CrossRef]
- Lawal, S.O.; Nagasawa, H.; Tsuru, T.; Kanezashi, M. Facile development of microstructure-engineered, ligand-chelated SiO2–ZrO2 composite membranes for molecular separations. Mol. Syst. Des. Eng. 2021, 6, 429–444. [Google Scholar] [CrossRef]
- Grzybowska, K.; Wojnarowska, Z.; Grzybowski, A.; Paluch, M.; Giussi, J.M.; Cortizo, M.S.; Blaszczyk-Lezak, I.; Mijangos, C. Effect of polymer structure on the molecular dynamics and thermal behavior of poly (allyl acetoacetate) and copolymers. Polymer 2014, 55, 1040–1047. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, A.; Kumagai, T.; Aota, H.; Kawasaki, H.; Arakawa, R. Reassessment of free-radical polymerization mechanism of allyl acetate based on end-group determination of resulting oligomers by MALDI-TOF-MS spectrometry. Polym. J. 2009, 41, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Giussi, J.M.; Blaszczyk-Lezak, I.; Sanz, B.; Allegretti, P.E.; Mijangos, C.; Cortizo, M.S. Tautomeric acetoacetate monomers as building units of functional copolymers. Eur. Polym. J. 2014, 59, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Kumbar, S.M.; Selvam, T.; Gellermann, C.; Storch, W.; Ballweg, T.; Breu, J.; Sextl, G. ORMOCERs (organic–inorganic hybrid copolymers)-zeolite Beta (BEA) nanocomposite membranes for gas separation applications. J. Membr. Sci. 2010, 347, 132–140. [Google Scholar] [CrossRef]
- Lee, H.R.; Kanezashi, M.; Shimomura, Y.; Yoshioka, T.; Tsuru, T. Evaluation and fabrication of pore-size-tuned silica membranes with tetraethoxydimethyl disiloxane for gas separation. AIChE J. 2011, 57, 2755–2765. [Google Scholar] [CrossRef]
- Castricum, H.L.; Paradis, G.G.; Mittelmeijer-Hazeleger, M.C.; Kreiter, R.; Vente, J.F.; Ten Elshof, J.E. Tailoring the separation behavior of hybrid organosilica membranes by adjusting the structure of the organic bridging group. Adv. Funct. Mater. 2011, 21, 2319–2329. [Google Scholar] [CrossRef] [Green Version]
- Kanezashi, M.; Yoneda, Y.; Nagasawa, H.; Tsuru, T.; Yamamoto, K.; Ohshita, J. Gas permeation properties for organosilica membranes with different Si/C ratios and evaluation of microporous structures. AIChE J. 2017, 63, 4491–4498. [Google Scholar] [CrossRef]
- Park, H.B.; Lee, Y.M. Pyrolytic carbon–silica membrane: A promising membrane material for improved gas separation. J. Membr. Sci. 2003, 213, 263–272. [Google Scholar] [CrossRef]
- Li, L.; Wang, C.; Wang, N.; Cao, Y.; Wang, T. The preparation and gas separation properties of zeolite/carbon hybrid membranes. J. Membr. Sci. 2015, 50, 2561–2570. [Google Scholar] [CrossRef]
- Wang, Q.; Yokoji, M.; Nagasawa, H.; Yu, L.; Kanezashi, M.; Tsuru, T. Microstructure evolution and enhanced permeation of SiC membranes derived from allylhydridopolycarbosilane. J. Membr. Sci. 2020, 612, 118392. [Google Scholar] [CrossRef]
- Wang, Q.; Kawano, Y.; Yu, L.; Nagasawa, H.; Kanezashi, M.; Tsuru, T. Development of high-performance sub-nanoporous SiC-based membranes derived from polytitanocarbosilane. J. Membr. Sci. 2020, 598, 117688. [Google Scholar] [CrossRef]
- Lawal, S.O.; Yu, L.; Nagasawa, H.; Tsuru, T.; Kanezashi, M. A carbon–silica–zirconia ceramic membrane with CO2 flow-switching behaviour promising versatile high-temperature H2/CO2 separation. J. Mater. Chem. A 2020, 8, 23563–23573. [Google Scholar] [CrossRef]
- Lawal, S.O.; Nagasawa, H.; Tsuru, T.; Kanezashi, M. Enhancement of the H2-permselectivity of a silica-zirconia composite membrane enabled by ligand-ceramic to carbon-ceramic transformation. J. Membr. Sci. 2022, 642, 119948. [Google Scholar] [CrossRef]
- Duke, M.C.; Da Costa, J.D.; Do, D.D.; Gray, P.G.; Lu, G.Q. Hydrothermally robust molecular sieve silica for wet gas separation. Adv. Funct. Mater. 2006, 16, 1215–1220. [Google Scholar] [CrossRef]
- Chung, D. Review graphite. J. Mater. Sci. 2002, 37, 1475–1489. [Google Scholar] [CrossRef]
- Fam, S.B.; Uyar, T.; Ishida, H.; Hacaloglu, J. Investigation of polymerization of benzoxazines and thermal degradation characteristics of polybenzoxazines via direct pyrolysis mass spectrometry. Polym. Int. 2012, 61, 1532–1541. [Google Scholar] [CrossRef] [Green Version]
- Lawal, S.O.; Nagasawa, H.; Tsuru, T.; Kanezashi, M. Design of carbon–ceramic composite membranes with tunable molecular cut-offs from a carboxylic benzoxazine ligand chelated to silica–zirconia. Mol. Syst. Des. Eng. 2022, 7, 1030–1038. [Google Scholar] [CrossRef]
- Lawal, S.O.; Nagasawa, H.; Tsuru, T.; Kanezashi, M. Hydrothermal stability of hydrogen-selective carbon–ceramic membranes derived from polybenzoxazine-modified silica–zirconia. Membranes 2022, 13, 30. [Google Scholar] [CrossRef]
- Shaer, C.; Oppenheimer, L.; Lin, A.; Ishida, H. Advanced carbon materials derived from polybenzoxazines: A review. Polymers 2021, 13, 3775. [Google Scholar] [CrossRef]
- Fotou, G.; Lin, Y.; Pratsinis, S.E. Hydrothermal stability of pure and modified microporous silica membranes. J. Membr. Sci. 1995, 30, 2803–2808. [Google Scholar] [CrossRef]
- Imai, H.; Morimoto, H.; Tominaga, A.; Hirashima, H. Structural changes in sol-gel derived SiO2 and TiO2 films by exposure to water vapor. J. Sol-Gel Sci. Technol. 1997, 10, 45–54. [Google Scholar] [CrossRef]


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Membrane † | Modifying Ligand | Appl. | Permeance [10−7 mol m−2 s−1 Pa−1] | Gas pair Selectivity [-] | Water Perm. [LMH bar−1] | MWCO [g/mol] | Ref. |
|---|---|---|---|---|---|---|---|
| TiO2-ZrO2 | Isoeugenol | GS | 2.0 (He) 1.4 (CO2) | 65 (He/N2) 46 (CO2/N2) | - | - | [83] |
| TiO2-ZrO2 | Diethanolamine (DEA) | GS | 1.8 (He) 0.16 (CO2) | 12 (He/N2) 1.1 (CO2/N2) | - | - | [83] |
| TiO2-ZrO2 | Diethanolamine (DEA) | GS | 3.0 (H2) | 54 (H2/butane) | - | - | [84] |
| TiO2-ZrO2 | Methyl gallate | GS | 8.6 (He) | 62 (He/N2) 111 (He/CH4) | - | - | [85] |
| TiO2-ZrO2 | Ethyl ferrulate | GS | 26.9 (He) | 10.6 (He/N2) 13.6 (He/CH4) | - | - | [85] |
| SiO2-TiO2 | Acetylacetone | GS | - - | 2.43 (He/N2) 2.16 (N2/CO2) | - | - | [86] |
| SiO2-TiO2 | Isoeugenol | GS | - - | 2.39 (He/N2) 2.22 (N2/CO2) | - | - | [86] |
| SiO2-ZrO2 | Acetylacetone | GS | 10 (H2) | 7600 (H2/SF6) | - | - | [87] |
| TiO2-ZrO2 | Ethyl acetoacetate | NF | - | - | 5.6 | 760 | [88] |
| TiO2-ZrO2 | 2,3-dihydroxynaphthalene | NF | - | - | 9.2 | 670 | [88] |
| TiO2-ZrO2 | 3,5-di-tert-butylcatechol | NF | - | - | - | 500 | [89] |
| Author(s) | Inorganic unit | Reactive Organic Species Precursor/Organic Modifier | Hydrolysis/Condensation Conditions | Crosslinking/Curing Conditions | Application(s) |
|---|---|---|---|---|---|
| Amberg-Schwab et al. [90] | Si-O-Zr Si-O-Al |
|
|
| Transparent barrier coatings |
| Le Guevel et al. [91] | Si-O-Ti |
|
|
| Optical waveguides |
| Rodic et al. [92] | Si-O-Zr |
|
|
| Anti-corrosion coating |
| Pyrolysis Temperature [°C] | C 1s Binding Energy [eV] | Sp3 [%] | Sp2 [%] | Sp3/Sp2 [-] |
|---|---|---|---|---|
| 90 | 284.4 | 62 | - | - |
| 550 | 284.3 | 53 | 6.1 | 8.7 |
| 750 | 283.3 | 29 | 57 | 0.51 |
| 850 | 283.2 | 27 | 58 | 0.47 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lawal, S.O.; Kanezashi, M. A Brief Overview of the Microstructural Engineering of Inorganic–Organic Composite Membranes Derived from Organic Chelating Ligands. Membranes 2023, 13, 390. https://doi.org/10.3390/membranes13040390
Lawal SO, Kanezashi M. A Brief Overview of the Microstructural Engineering of Inorganic–Organic Composite Membranes Derived from Organic Chelating Ligands. Membranes. 2023; 13(4):390. https://doi.org/10.3390/membranes13040390
Chicago/Turabian StyleLawal, Sulaiman Oladipo, and Masakoto Kanezashi. 2023. "A Brief Overview of the Microstructural Engineering of Inorganic–Organic Composite Membranes Derived from Organic Chelating Ligands" Membranes 13, no. 4: 390. https://doi.org/10.3390/membranes13040390