
Modeling Adsorption, Conformation, and Orientation of the Fis1 Tail Anchor at the Mitochondrial Outer Membrane


Abstract
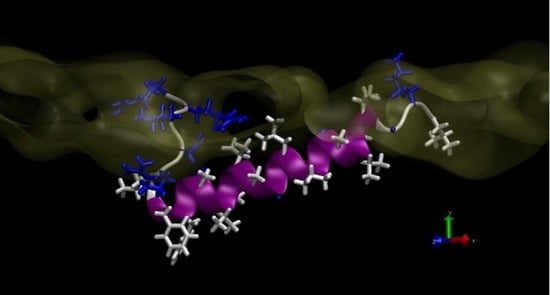
:
1. Introduction
2. Materials and Methods
2.1. Atomistic Simulations
2.1.1. Simulated Annealing
2.1.2. AA-REX
2.2. MARTINI Coarse-Grained Simulations
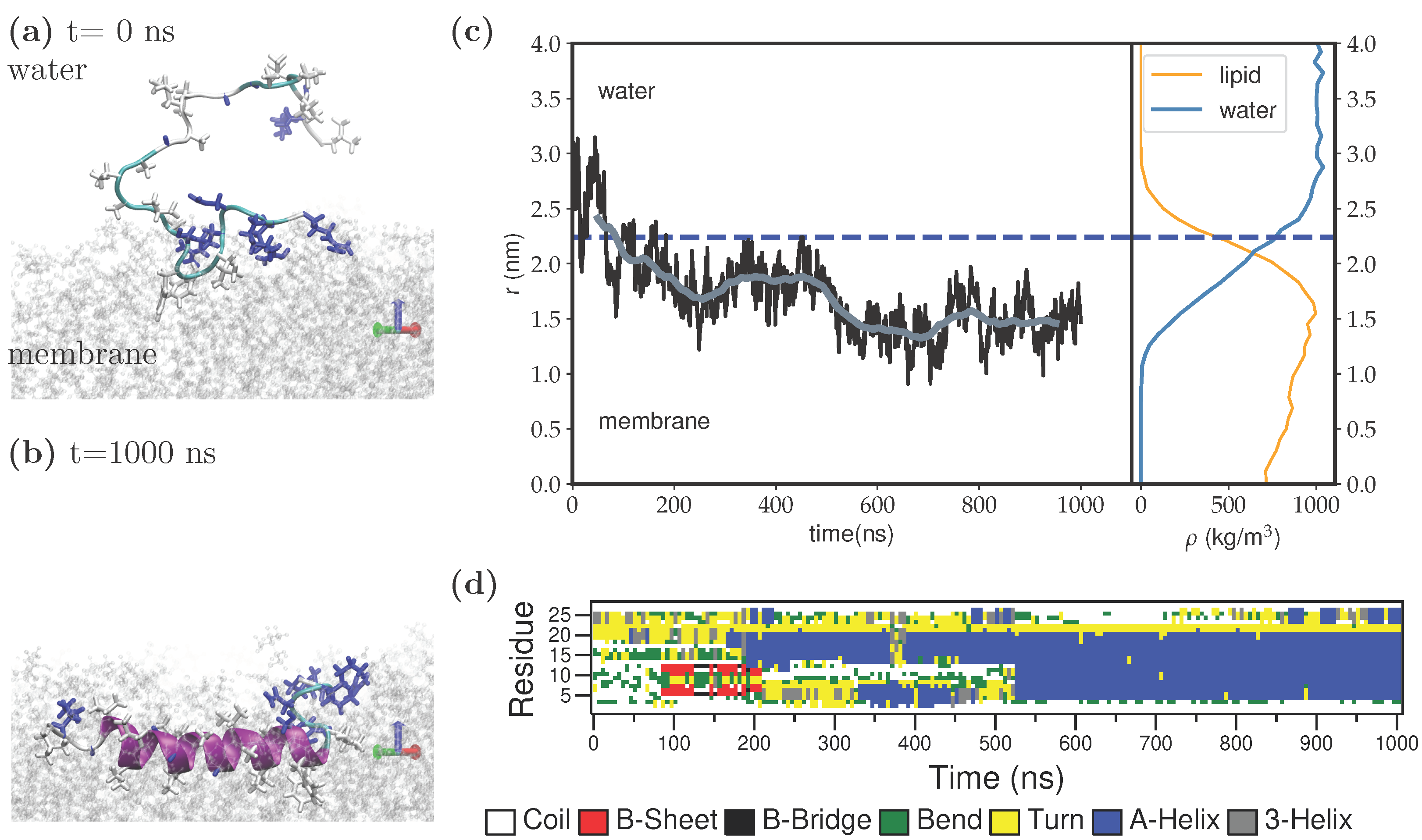
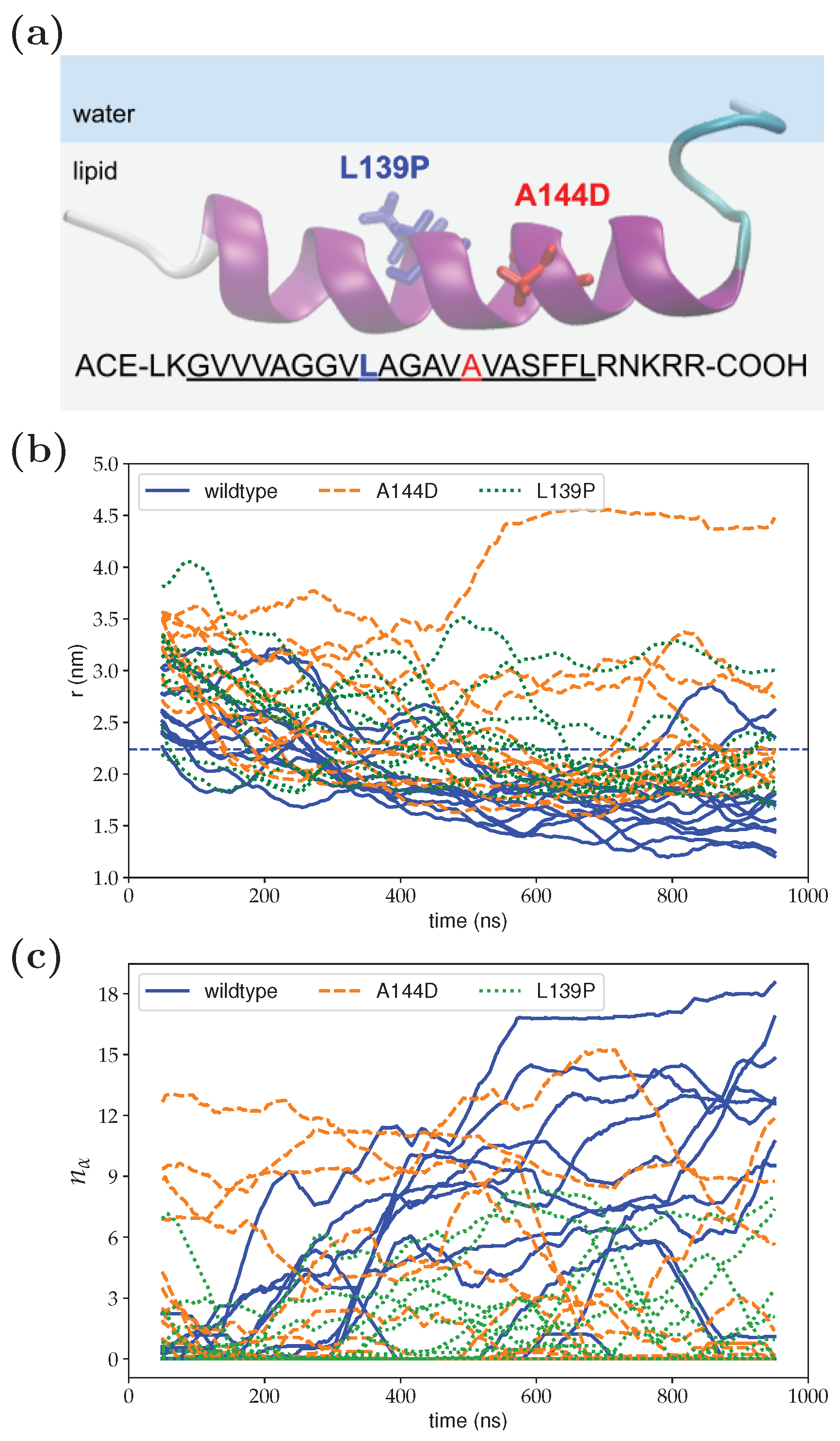
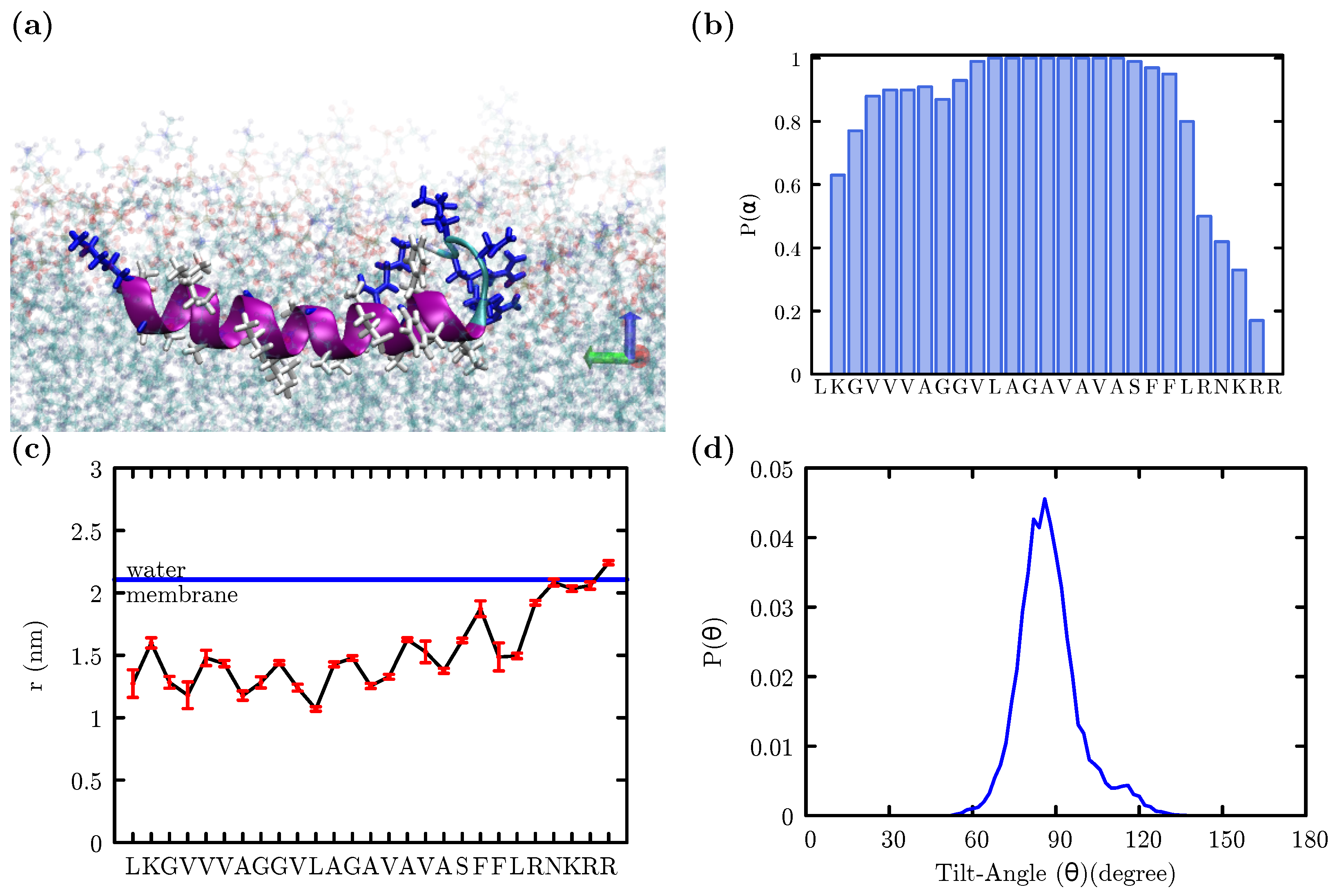
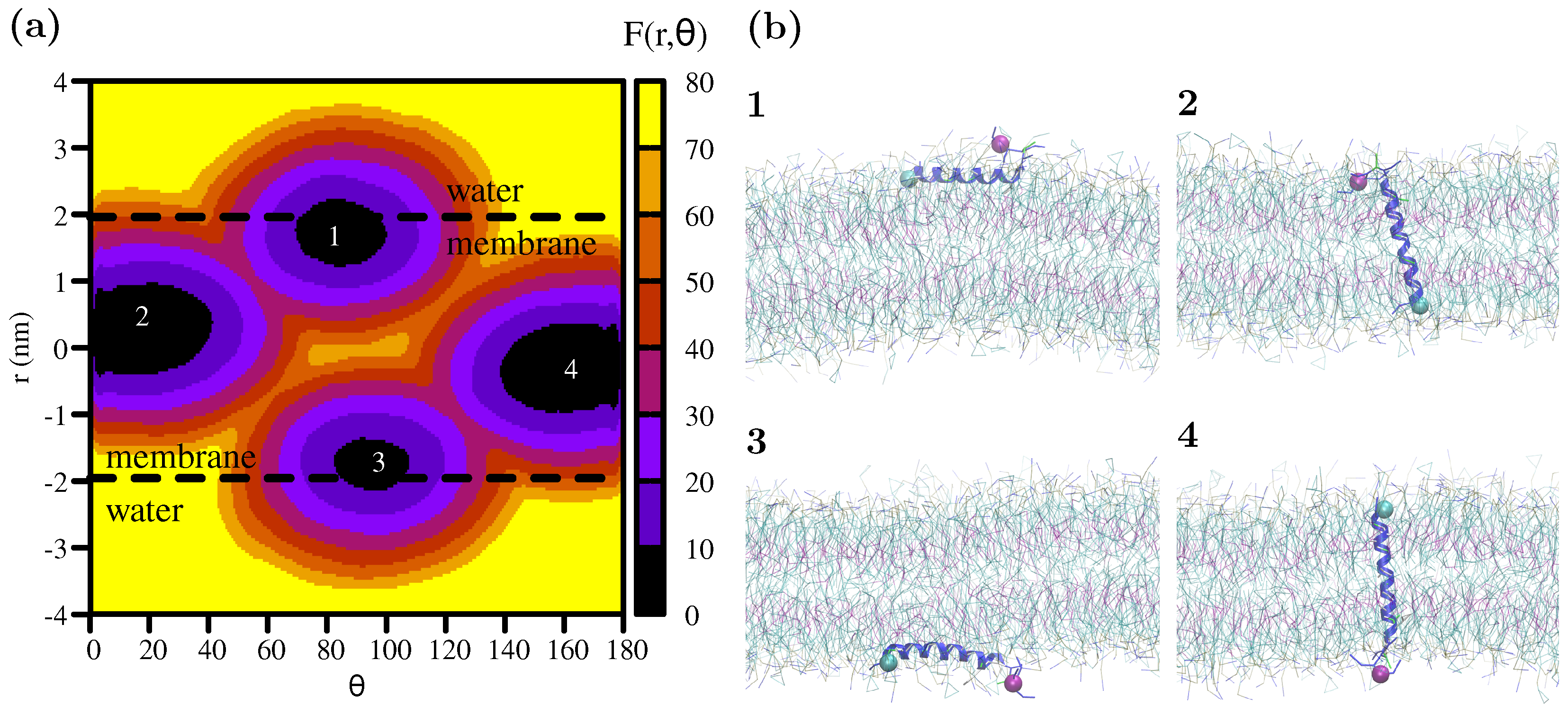
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| TA | Tail anchor |
| Fis1p | Fis1 protein |
| Fis1(TA) | Fis1 tail anchor |
| SA | Simulated annealing |
| AA-REX | Atomistic replica exchange simulation |
References
- Hegde, R.S.; Keenan, R.J. Tail-Anchored Membrane Protein Insertion into the Endoplasmic Reticulum. Nat. Rev. Mol. Cell Biol. 2011, 12, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Weill, U.; Cohen, N.; Fadel, A.; Ben-Dor, S.; Schuldiner, M. Protein Topology Prediction Algorithms Systematically Investigated in the Yeast Saccharomyces Cerevisiae. Bioessays 2019, 41, e1800252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgese, N.; Brambillasca, S.; Colombo, S. How Tails Guide Tail-Anchored Proteins to Their Destinations. Curr. Opin. Cell Biol. 2007, 19, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Rabu, C.; Schmid, V.; Schwappach, B.; High, S. Biogenesis of Tail-Anchored Proteins: The Beginning for the End? J. Cell Sci. 2009, 122, 3605–3612. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, A.; Haase, M.; Leptihn, S. Assisted and Unassisted Protein Insertion into Liposomes. Biophys. J. 2017, 113, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Borgese, N.; Coy-Vergara, J.; Colombo, S.F.; Schwappach, B. The Ways of Tails: The GET Pathway and More. Protein J. 2019, 38, 289–305. [Google Scholar] [CrossRef]
- Drwesh, L.; Rapaport, D. Biogenesis Pathways of α-Helical Mitochondrial Outer Membrane Proteins. Biol. Chem. 2020, 401, 677–686. [Google Scholar] [CrossRef]
- Mehlhorn, D.G.; Asseck, L.Y.; Grefen, C. Looking for a Safe Haven: Tail-Anchored Proteins and Their Membrane Insertion Pathways. Plant Physiol. 2021, 187, 1916–1928. [Google Scholar] [CrossRef]
- Fekkes, P.; Shepard, K.A.; Yaffe, M.P. Gag3p, an Outer Membrane Protein Required for Fission of Mitochondrial Tubules. J. Cell Biol. 2000, 151, 333–340. [Google Scholar] [CrossRef]
- Mozdy, A.D.; McCaffery, J.M.; Shaw, J.M. Dnm1p GTPase-mediated Mitochondrial Fission Is a Multi-Step Process Requiring the Novel Integral Membrane Component Fis1p. J. Cell Biol. 2000, 151, 367–380. [Google Scholar] [CrossRef]
- Tieu, Q.; Nunnari, J. Mdv1p Is a WD Repeat Protein That Interacts with the Dynamin-Related GTPase, Dnm1p, to Trigger Mitochondrial Division. J. Cell Biol. 2000, 151, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Motley, A.M.; Ward, G.P.; Hettema, E.H. Dnm1p-Dependent Peroxisome Fission Requires Caf4p, Mdv1p and Fis1p. J. Cell Sci. 2008, 121, 1633–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerveny, K.L.; Jensen, R.E. The WD-repeats of Net2p Interact with Dnm1p and Fis1p to Regulate Division of Mitochondria. MBoC 2003, 14, 4126–4139. [Google Scholar] [CrossRef] [PubMed]
- Tieu, Q.; Okreglak, V.; Naylor, K.; Nunnari, J. The WD Repeat Protein, Mdv1p, Functions as a Molecular Adaptor by Interacting with Dnm1p and Fis1p during Mitochondrial Fission. J. Cell Biol. 2002, 158, 445–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karren, M.A.; Coonrod, E.M.; Anderson, T.K.; Shaw, J.M. The Role of Fis1p–Mdv1p Interactions in Mitochondrial Fission Complex Assembly. J. Cell Biol. 2005, 171, 291–301. [Google Scholar] [CrossRef]
- Habib, S.J.; Vasiljev, A.; Neupert, W.; Rapaport, D. Multiple Functions of Tail-Anchor Domains of Mitochondrial Outer Membrane Proteins. FEBS Lett. 2003, 555, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Kemper, C.; Habib, S.J.; Engl, G.; Heckmeyer, P.; Dimmer, K.S.; Rapaport, D. Integration of Tail-Anchored Proteins into the Mitochondrial Outer Membrane Does Not Require Any Known Import Components. J. Cell Sci. 2008, 121, 1990–1998. [Google Scholar] [CrossRef] [Green Version]
- Krumpe, K.; Frumkin, I.; Herzig, Y.; Rimon, N.; Özbalci, C.; Brügger, B.; Rapaport, D.; Schuldiner, M. Ergosterol Content Specifies Targeting of Tail-Anchored Proteins to Mitochondrial Outer Membranes. Mol. Biol. Cell 2012, 23, 3927–3935. [Google Scholar] [CrossRef]
- Keskin, A.; Akdoğan, E.; Dunn, C.D. Evidence for Amino Acid Snorkeling from a High-Resolution, In Vivo Analysis of Fis1 Tail-Anchor Insertion at the Mitochondrial Outer Membrane. Genetics 2017, 205, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Vitali, D.G.; Drwesh, L.; Cichocki, B.A.; Kolb, A.; Rapaport, D. The Biogenesis of Mitochondrial Outer Membrane Proteins Show Variable Dependence on Import Factors. iScience 2020, 23, 100779. [Google Scholar] [CrossRef] [Green Version]
- Horie, C.; Suzuki, H.; Sakaguchi, M.; Mihara, K. Targeting and Assembly of Mitochondrial Tail-Anchored Protein Tom5 to the TOM Complex Depend on a Signal Distinct from That of Tail-Anchored Proteins Dispersed in the Membrane. J. Biol. Chem. 2003, 278, 41462–41471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cichocki, B.A.; Krumpe, K.; Vitali, D.G.; Rapaport, D. Pex19 Is Involved in Importing Dually Targeted Tail-Anchored Proteins to Both Mitochondria and Peroxisomes. Traffic 2018, 19, 770–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setoguchi, K.; Otera, H.; Mihara, K. Cytosolic Factor- and TOM-independent Import of C-tail-anchored Mitochondrial Outer Membrane Proteins. EMBO J. 2006, 25, 5635–5647. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, N.; Garipler, G.; Akdoğan, E.; Dunn, C.D. Activation of the Pleiotropic Drug Resistance Pathway Can Promote Mitochondrial DNA Retention by Fusion-Defective Mitochondria in Saccharomyces Cerevisiae. G3 2014, 4, 1247–1258. [Google Scholar] [CrossRef] [Green Version]
- Doan, K.N.; Grevel, A.; Mårtensson, C.U.; Ellenrieder, L.; Thornton, N.; Wenz, L.S.; Opaliński, Ł; Guiard, B.; Pfanner, N.; Becker, T. The Mitochondrial Import Complex MIM Functions as Main Translocase for α-Helical Outer Membrane Proteins. Cell Rep. 2020, 31, 107567. [Google Scholar] [CrossRef]
- Blobel, G. Intracellular Protein Topogenesis. Proc. Natl. Acad. Sci. USA 1980, 77, 1496–1500. [Google Scholar] [CrossRef] [Green Version]
- Beilharz, T.; Egan, B.; Silver, P.A.; Hofmann, K.; Lithgow, T. Bipartite Signals Mediate Subcellular Targeting of Tail-Anchored Membrane Proteins in Saccharomyces Cerevisiae. J. Biol. Chem. 2003, 278, 8219–8223. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, J.; Jo, S.; Brooks, C.L.; Lee, H.S.; Im, W. CHARMM-GUI Ligand Reader and Modeler for CHARMM Force Field Generation of Small Molecules. J. Comput. Chem. 2017, 38, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Hockney, R.W.; Goel, S.P.; Eastwood, J.W. Quiet High-Resolution Computer Models of a Plasma. J. Comput. Phys. 1974, 14, 148–158. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Kučerka, N.; Nagle, J.F.; Sachs, J.N.; Feller, S.E.; Pencer, J.; Jackson, A.; Katsaras, J. Lipid Bilayer Structure Determined by the Simultaneous Analysis of Neutron and X-Ray Scattering Data. Biophys. J. 2008, 95, 2356–2367. [Google Scholar] [CrossRef] [Green Version]
- Simbeni, R.; Pon, L.; Zinser, E.; Paltauf, F.; Daum, G. Mitochondrial Membrane Contact Sites of Yeast. Characterization of Lipid Components and Possible Involvement in Intramitochondrial Translocation of Phospholipids. J. Biol. Chem. 1991, 266, 10047–10049. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Williams, T.; Kelley, C.; Bersch, C.; Bröker, H.B.; Campbell, J.; Cunningham, R.; Denholm, D.; Elber, G.; Fearick, R.; Grammes, C.; et al. Gnuplot 5.2; An Interactive Plotting Program; Available online: http://www.gnuplot.info/docs_5 (accessed on 28 June 2022).
- Rosta, E.; Hummer, G. Error and Efficiency of Replica Exchange Molecular Dynamics Simulations. J. Chem. Phys. 2009, 131, 165102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stelzl, L.S.; Hummer, G. Kinetics from Replica Exchange Molecular Dynamics Simulations. J. Chem. Theory Comput. 2017, 13, 3927–3935. [Google Scholar] [CrossRef]
- Patriksson, A.; van der Spoel, D. A Temperature Predictor for Parallel Tempering Simulations. Phys. Chem. Chem. Phys. PCCP 2008, 10, 2073–2077. [Google Scholar] [CrossRef]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MARTINI Website. Available online: http://cgmartini.nl/ (accessed on 30 September 2018).
- Barducci, A.; Bussi, G.; Parrinello, M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil-Ley, A.; Bussi, G. Enhanced Conformational Sampling Using Replica Exchange with Collective-Variable Tempering. J. Chem. Theory Comput. 2015, 11, 1077–1085. [Google Scholar] [CrossRef]
- Tribello, G.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New Feathers for an Old Bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Sugita, Y.; Okamoto, Y. Replica Exchange Molecular Dynamics Method for Protein Folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- García, A.E.; Sanbonmatsu, K.Y. Alpha-Helical Stabilization by Side Chain Shielding of Backbone Hydrogen Bonds. Proc. Natl. Acad. Sci. USA 2002, 99, 2782–2787. [Google Scholar] [CrossRef] [Green Version]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.J. The MARTINI Coarse-Grained Force Field: Extension to Proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef]
- Chetwynd, A.; Wee, C.L.; Hall, B.A.; Sansom, M.S.P. The Energetics of Transmembrane Helix Insertion into a Lipid Bilayer. Biophys. J. 2010, 99, 2534–2540. [Google Scholar] [CrossRef] [Green Version]
- Hall, B.A.; Chetwynd, A.P.; Sansom, M.S.P. Exploring Peptide-Membrane Interactions with Coarse-Grained MD Simulations. Biophys. J. 2011, 100, 1940–1948. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, G.; Nandekar, P.P.; Yu, X.; Wade, R.C. On the Application of the MARTINI Coarse-Grained Model to Immersion of a Protein in a Phospholipid Bilayer. J. Chem. Phys. 2015, 143, 243139. [Google Scholar] [CrossRef]
- Nørholm, M.H.H.; Shulga, Y.V.; Aoki, S.; Epand, R.M.; von Heijne, G. Flanking Residues Help Determine Whether a Hydrophobic Segment Adopts a Monotopic or Bitopic Topology in the Endoplasmic Reticulum Membrane. J. Biol. Chem. 2011, 286, 25284–25290. [Google Scholar] [CrossRef] [Green Version]
- Entova, S.; Billod, J.M.; Swiecicki, J.M.; Martín-Santamaría, S.; Imperiali, B. Insights into the Key Determinants of Membrane Protein Topology Enable the Identification of New Monotopic Folds. eLife 2018, 7, e40889. [Google Scholar] [CrossRef]
- Romero-García, J.; Biarnés, X.; Planas, A. Essential Mycoplasma Glycolipid Synthase Adheres to the Cell Membrane by Means of an Amphipathic Helix. Sci. Rep. 2019, 9, 7085. [Google Scholar] [CrossRef] [Green Version]
- Herce, H.D.; Garcia, A.E. Cell Penetrating Peptides: How Do They Do It? J. Biol. Phys. 2008, 33, 345. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipid | Percentage | Charge |
|---|---|---|
| DOPC (PC) | 46% | 0 |
| DOPE (PE) | 33% | 0 |
| POPI (PI) | 10% | −1 |
| Cardiolipin | 6% | −2 |
| DOPA (PA) | 4% | −1 |
| DOPS (PS) | 1% | −1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ozgur, B.; Dunn, C.D.; Sayar, M. Modeling Adsorption, Conformation, and Orientation of the Fis1 Tail Anchor at the Mitochondrial Outer Membrane. Membranes 2022, 12, 752. https://doi.org/10.3390/membranes12080752
Ozgur B, Dunn CD, Sayar M. Modeling Adsorption, Conformation, and Orientation of the Fis1 Tail Anchor at the Mitochondrial Outer Membrane. Membranes. 2022; 12(8):752. https://doi.org/10.3390/membranes12080752
Chicago/Turabian StyleOzgur, Beytullah, Cory D. Dunn, and Mehmet Sayar. 2022. "Modeling Adsorption, Conformation, and Orientation of the Fis1 Tail Anchor at the Mitochondrial Outer Membrane" Membranes 12, no. 8: 752. https://doi.org/10.3390/membranes12080752