
Interaction of Human Serum Albumin with Uremic Toxins: The Need of New Strategies Aiming at Uremic Toxins Removal

 ,
,  ,
,  and
and 
Abstract
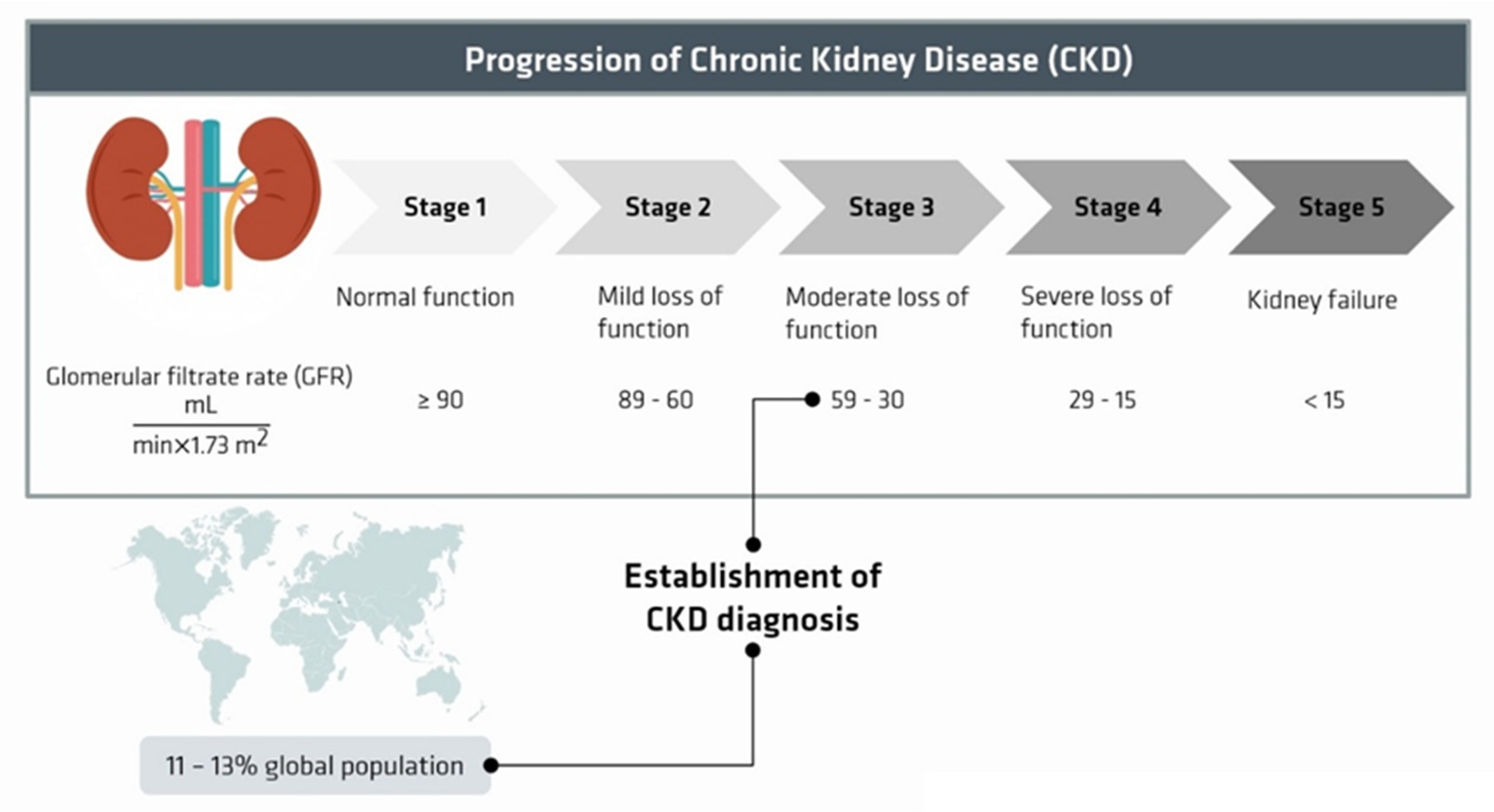
:1. Introduction
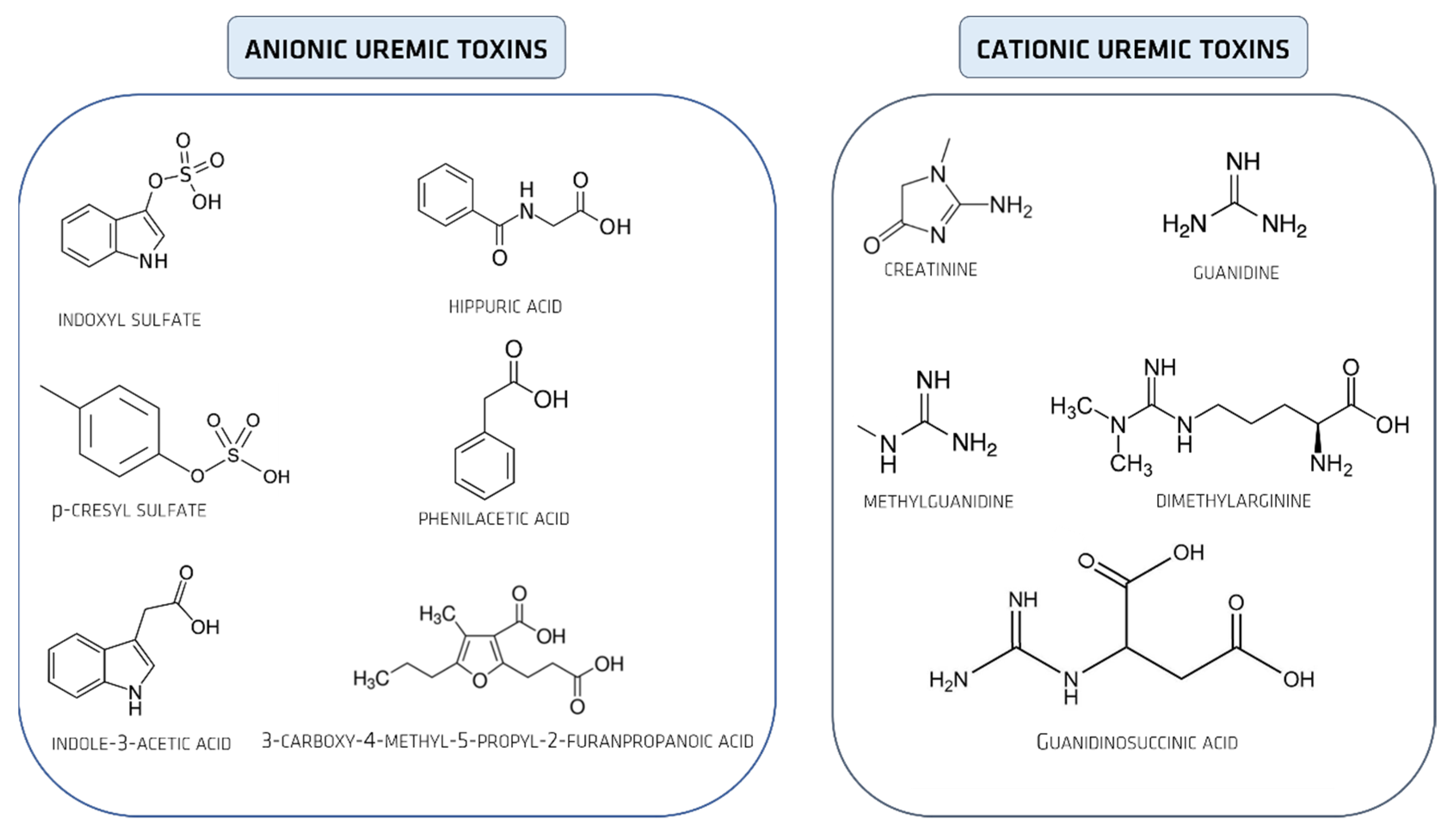
2. Uremic Toxins
Uremic Toxins Classification
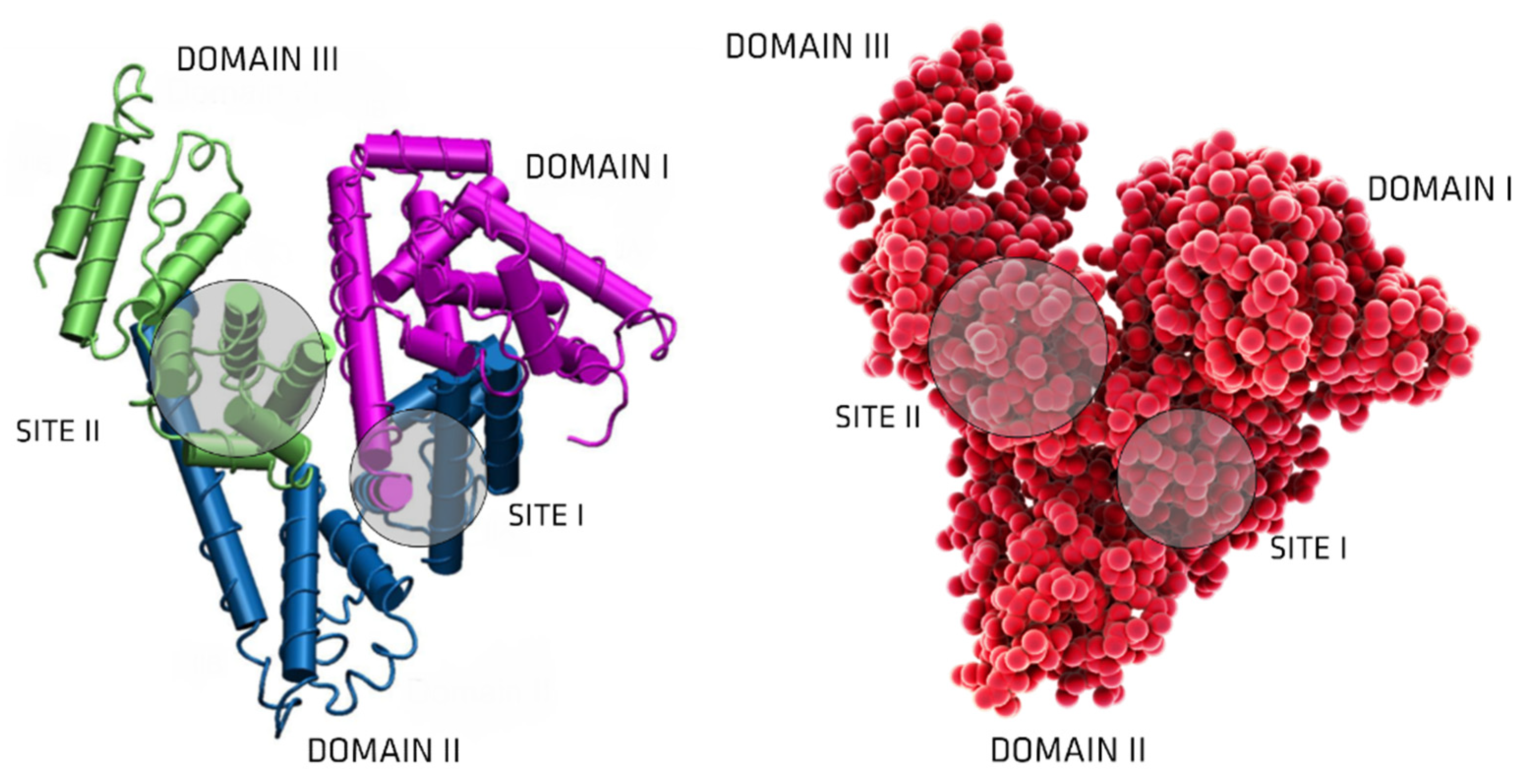
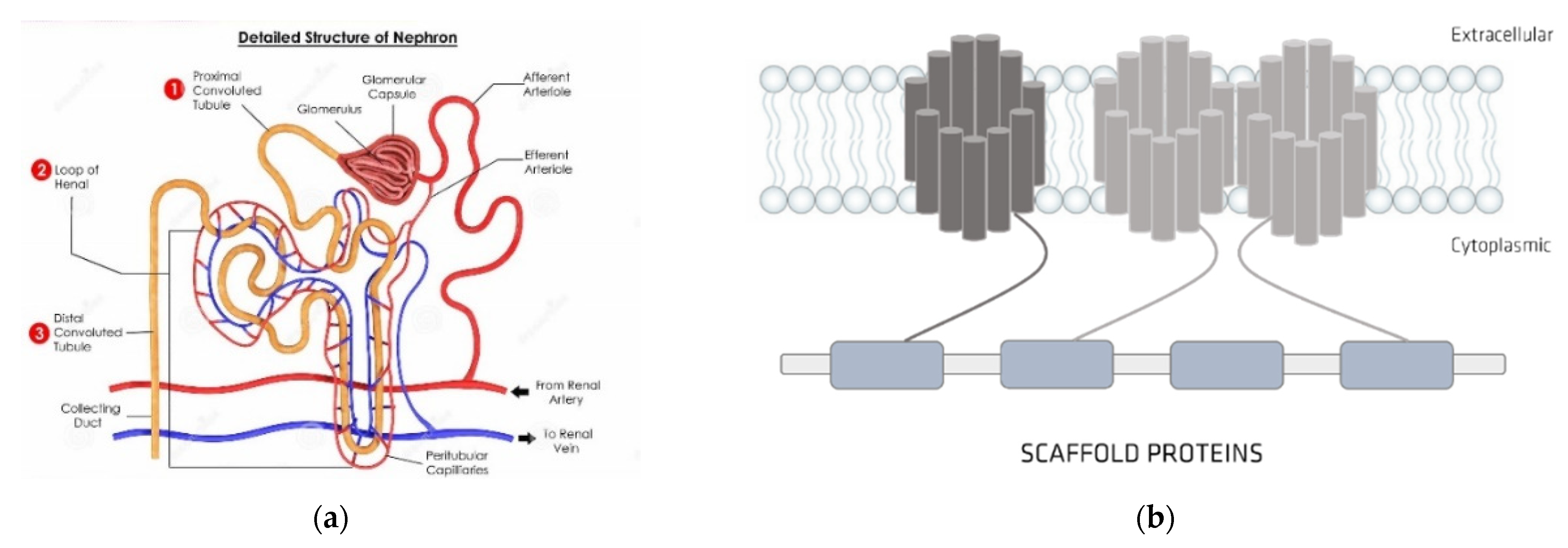
3. Albumin: Structure and Physiological Functions
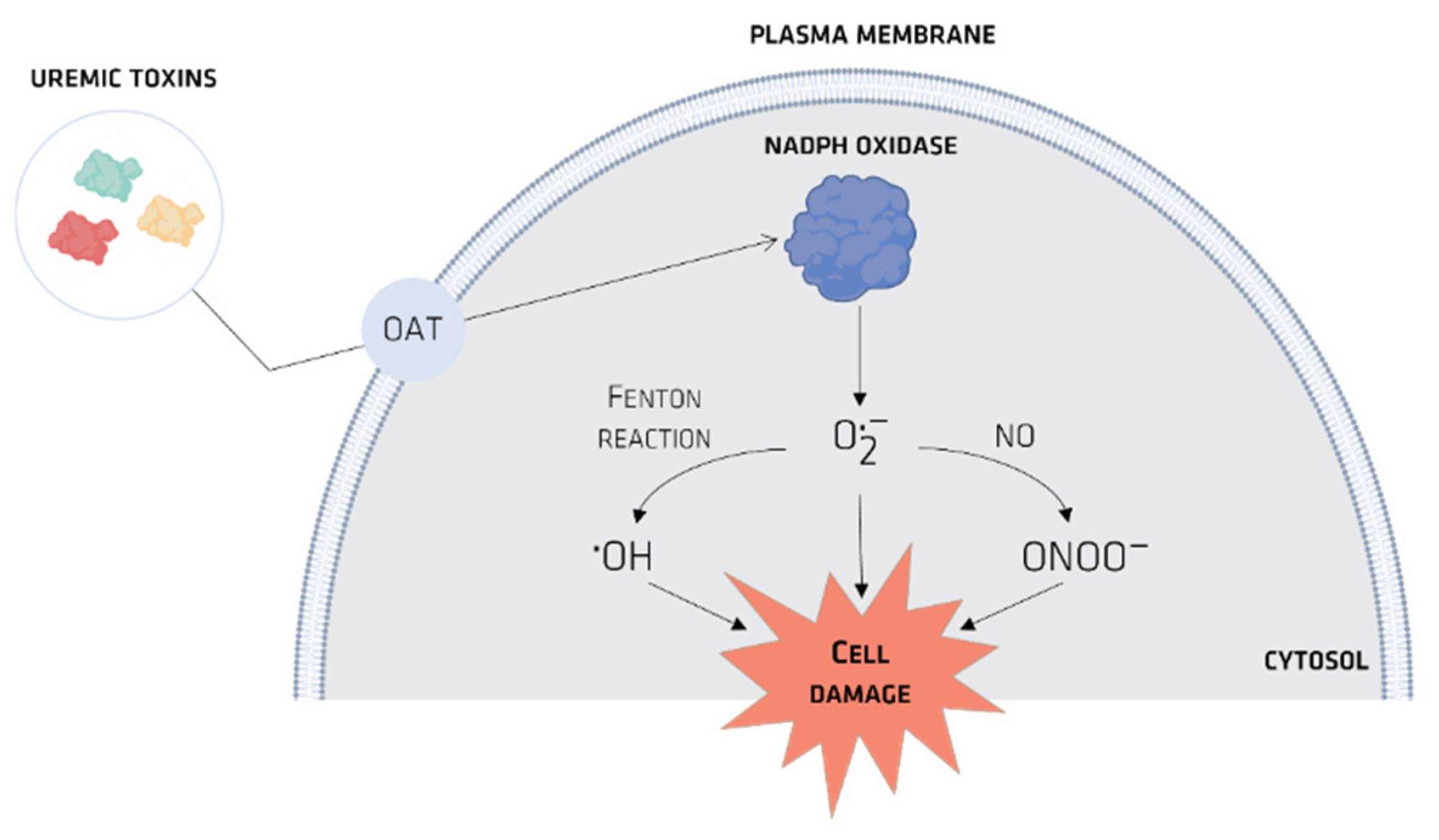
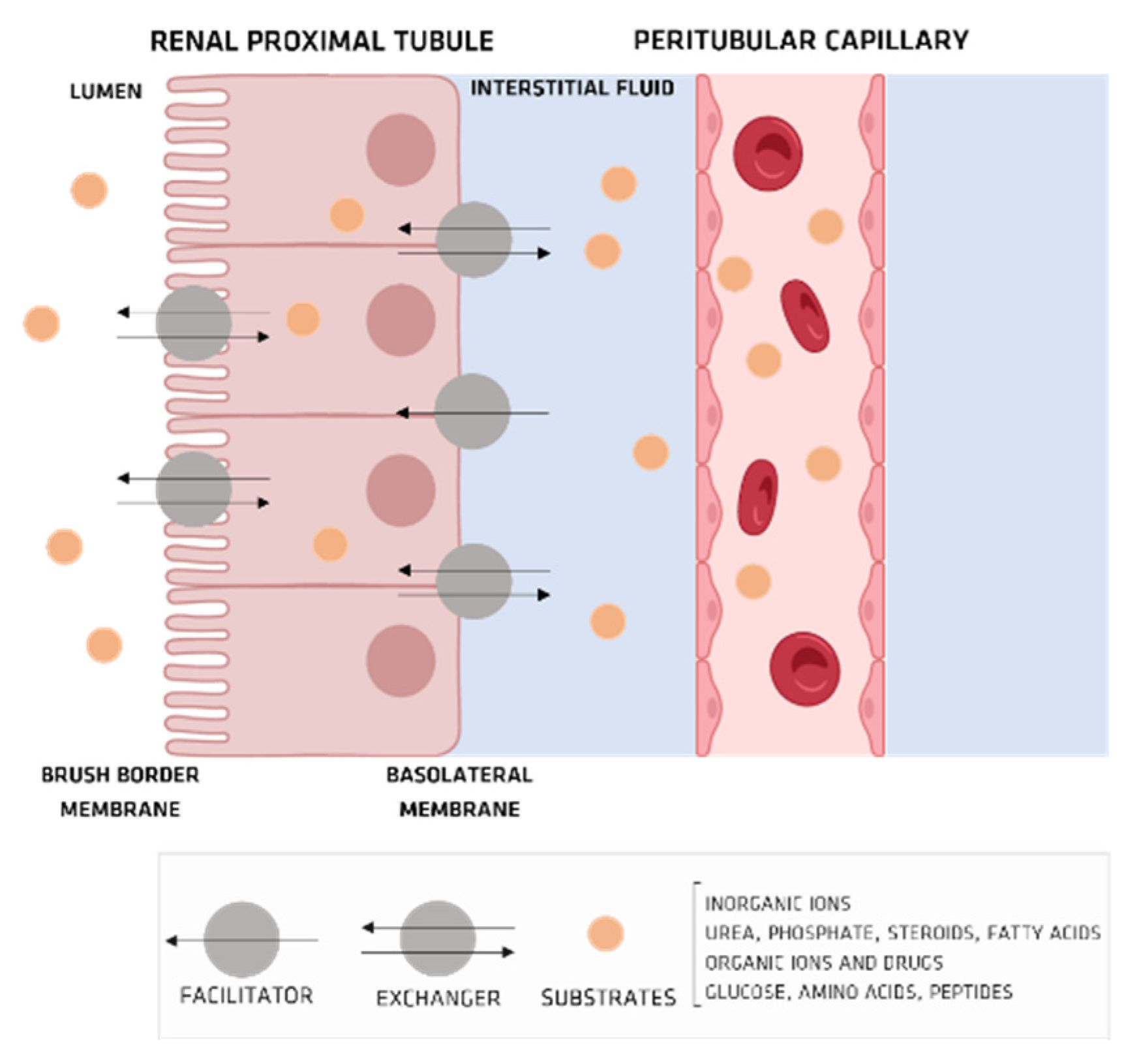
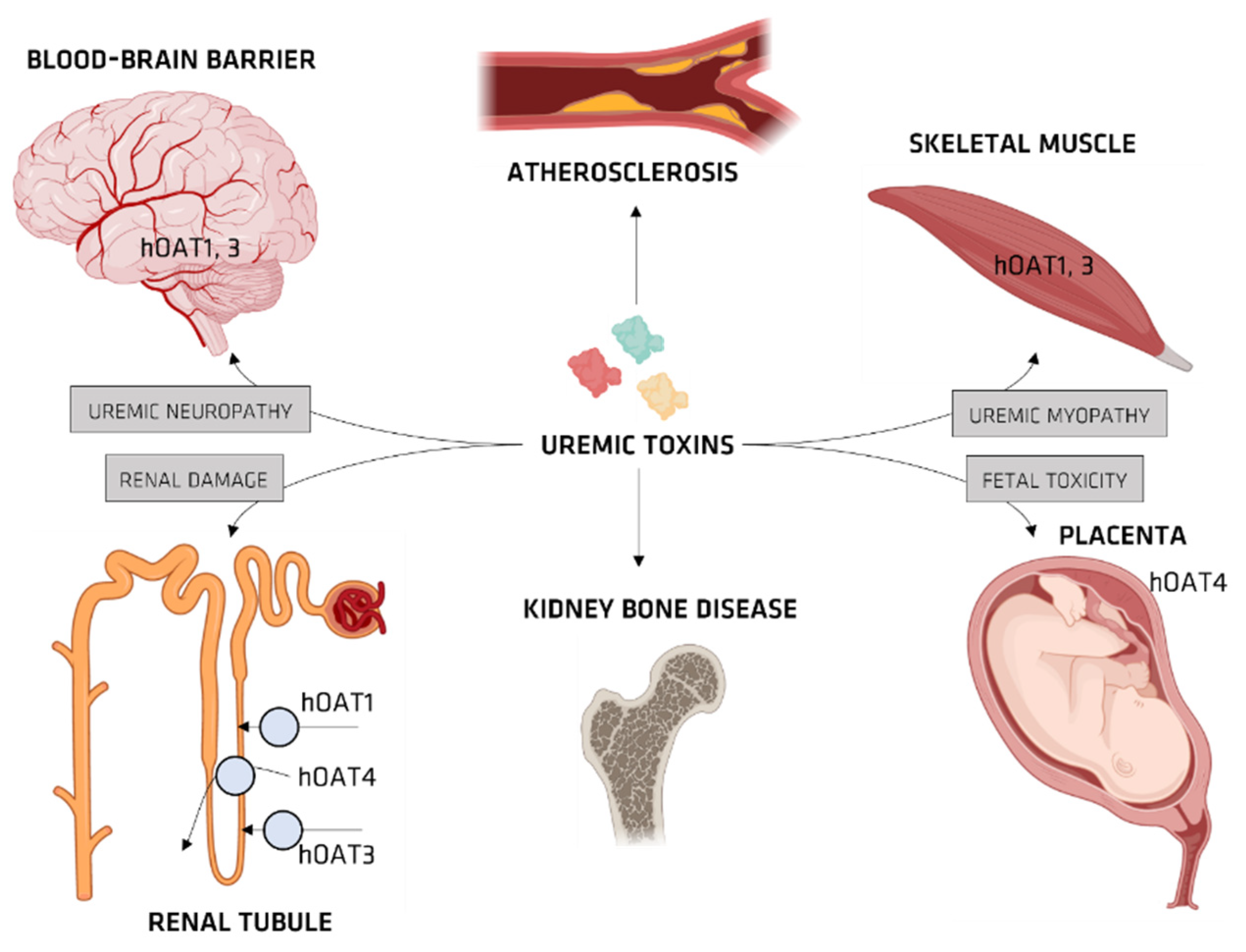
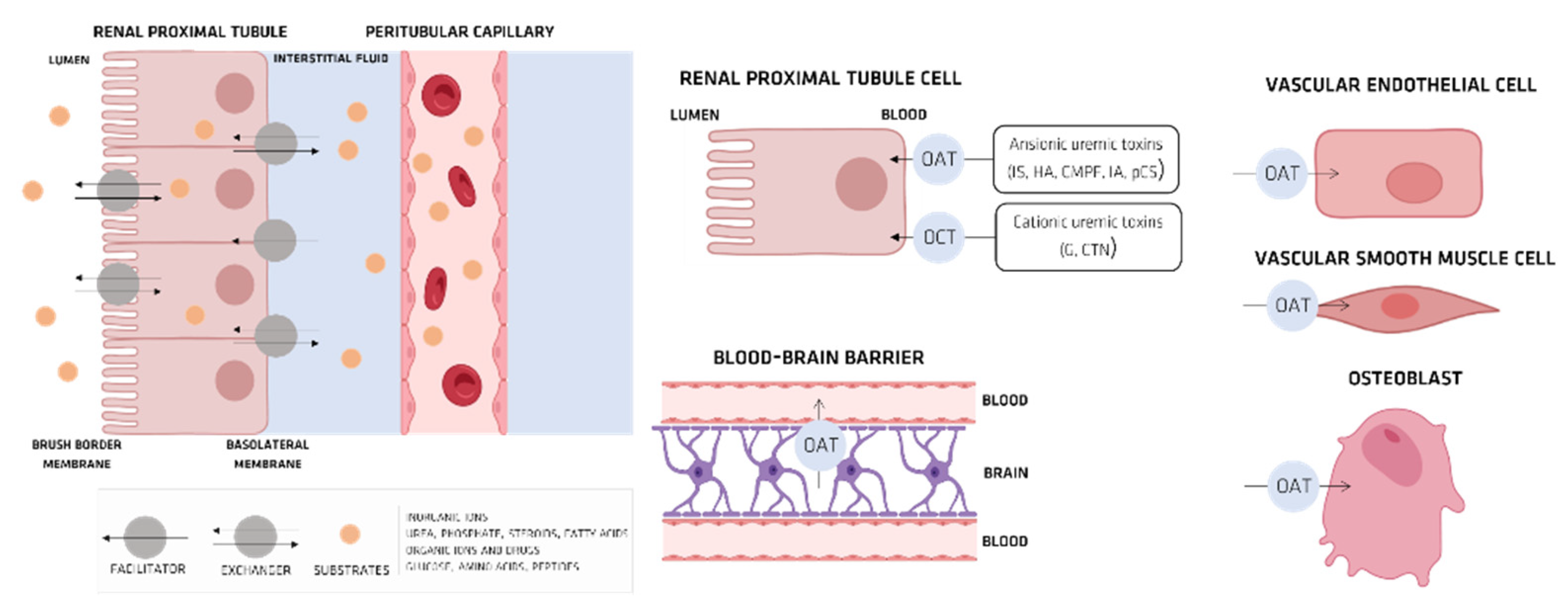
4. Organic Anionic UTs
Organic Ionic Transporters
5. Pharmacokinetics of UTs
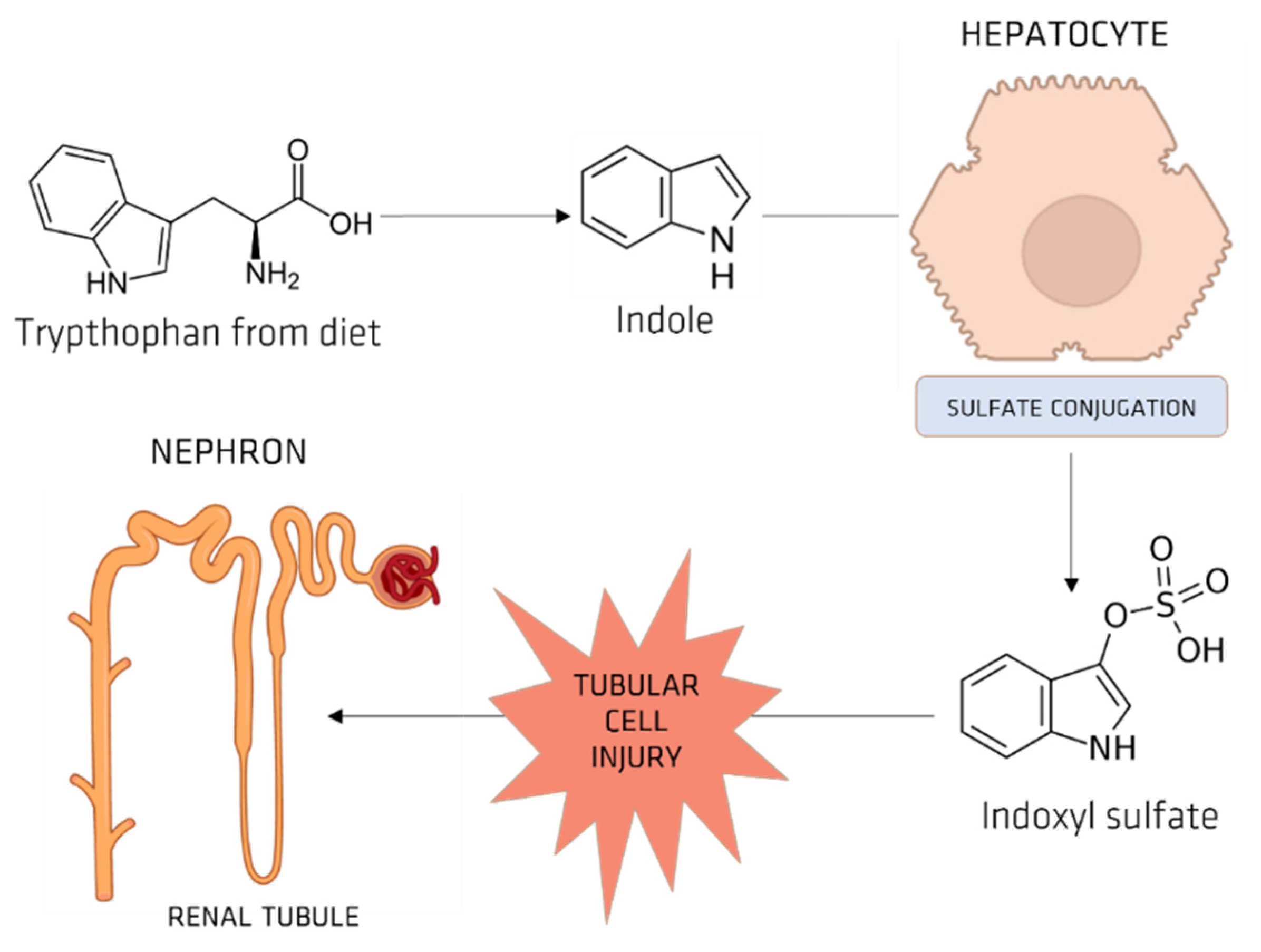
5.1. Indoxyl Sulfate (IS)
OAT and IS
5.2. Carboxy-4-methyl-5-propyl-2-furanpropanoic Acid
5.3. P-Cresol Sulfate
5.4. Hippuric Acid
5.5. Guanidino Compounds
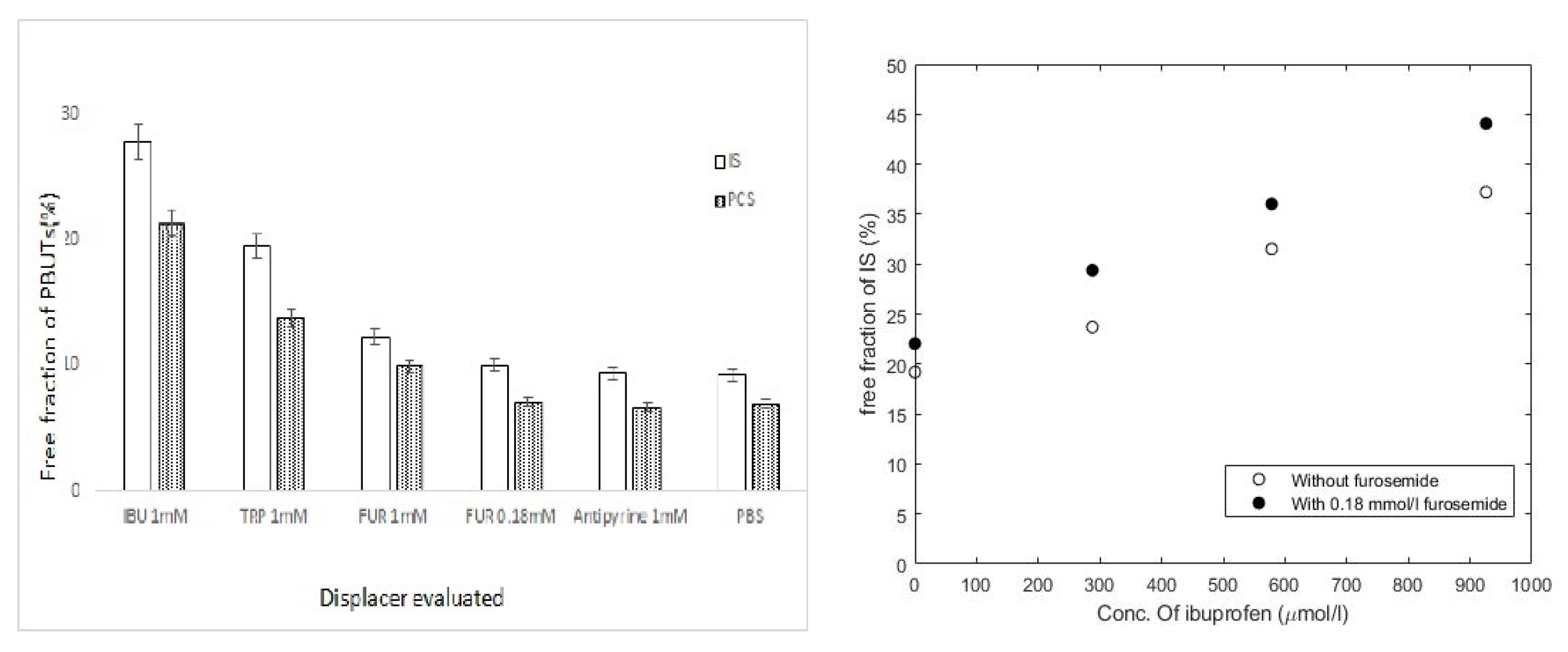
6. UTs Binding to Serum Albumin
7. Effect of UTs on Nonrenal Drug Clearance
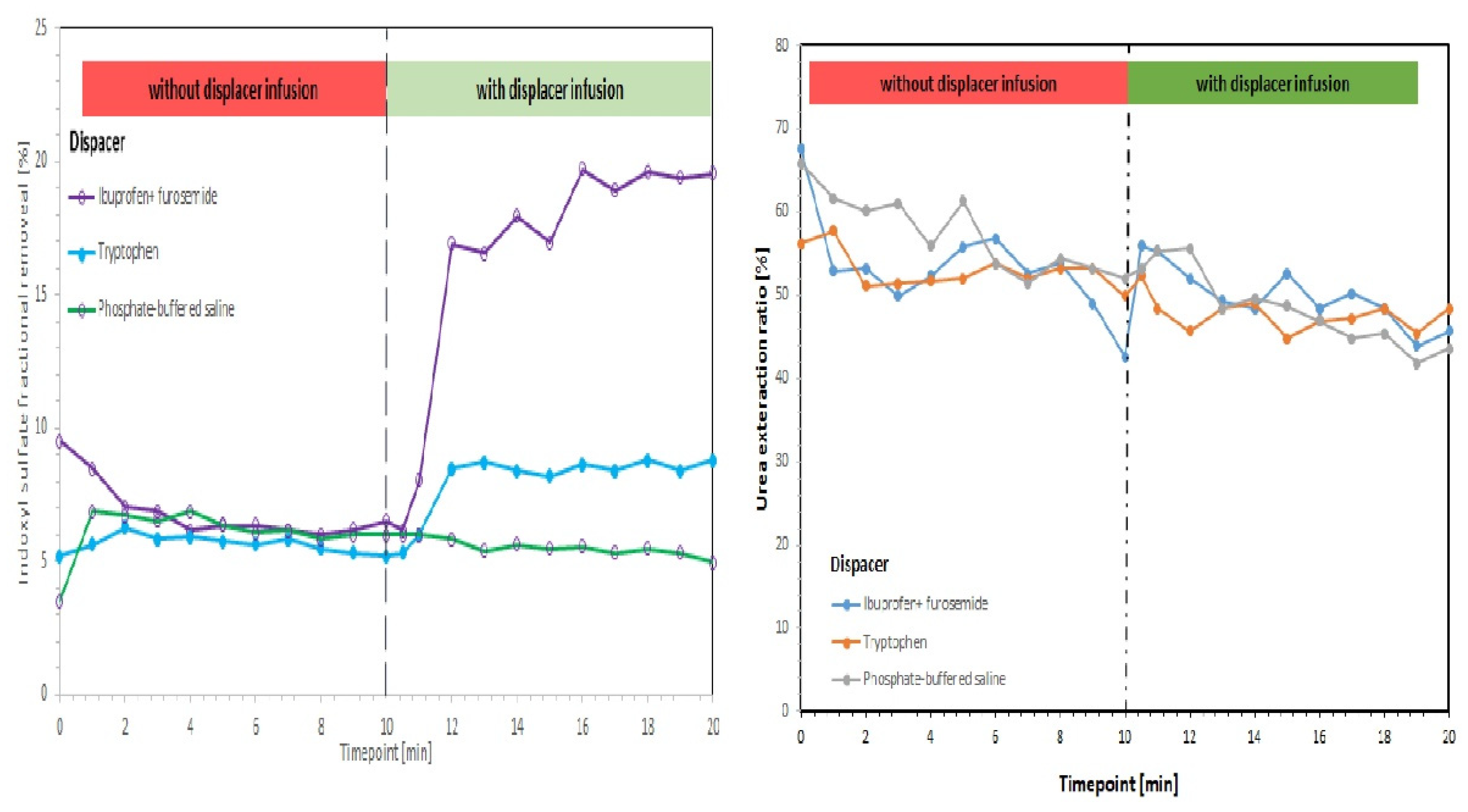
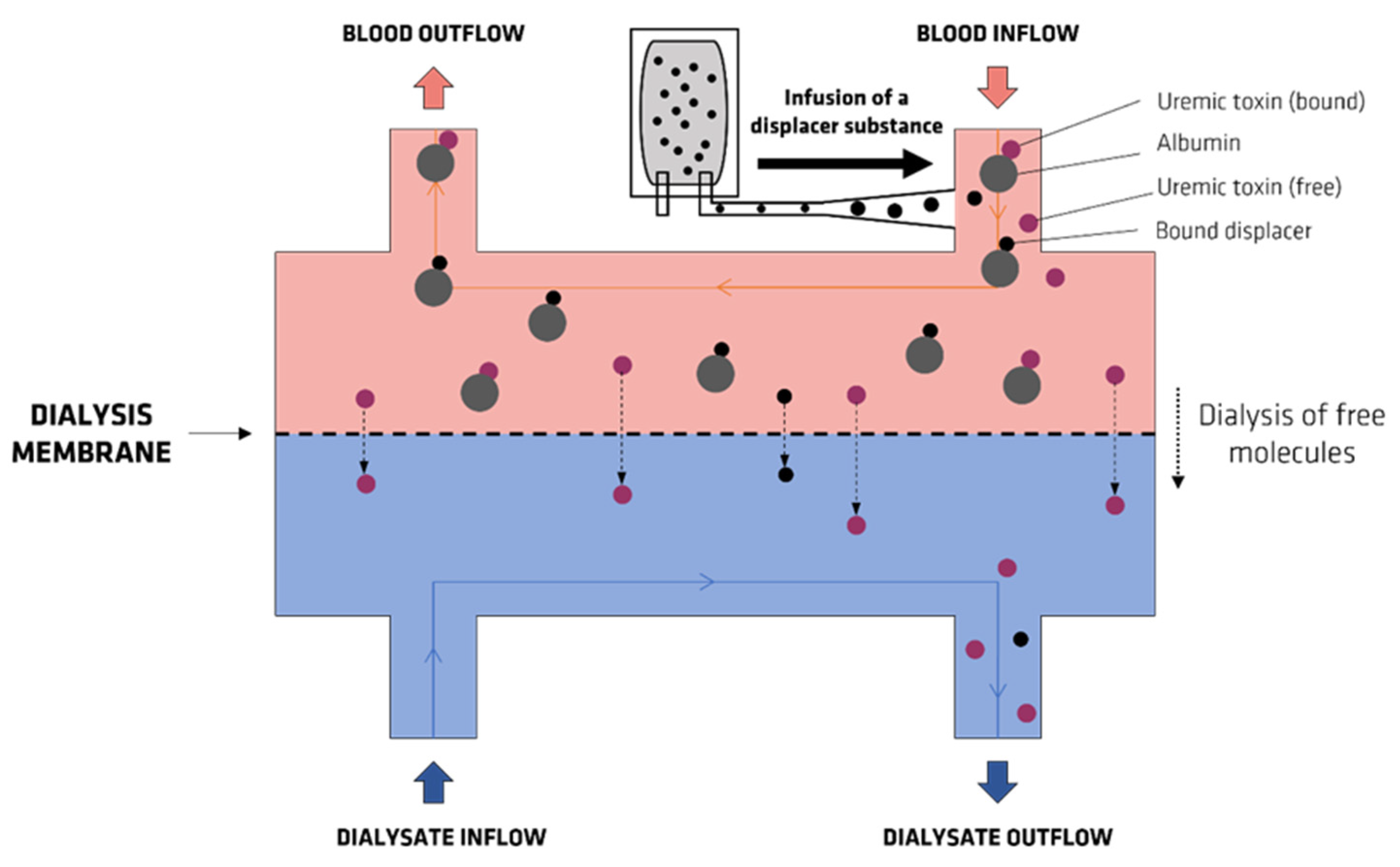
8. Strategies Aimed at the Removal of UTs
9. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Available online: https://www.sciencedirect.com/science/article/pii/B978141605185500002X (accessed on 1 September 2021).
- WHO Ageing-and-Health. Available online: https://www.who.int/news-room/fact-sheets/detail/ageing-and-health (accessed on 1 September 2021).
- Chawla, L.S.; Kimmel, P.L. Acute kidney injury and chronic kidney disease: An integrated clinical syndrome. Kidney Int. 2012, 82, 516–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishani, A.; Xue, J.L.; Himmelfarb, J.; Eggers, P.W.; Kimmel, P.L.; Molitoris, B.A.; Collins, A.J. Acute kidney injury increases risk of ESRD among elderly. J. Am. Soc. Nephrol. 2009, 20, 223–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C. Clinical Evaluation of Kidney Function. In Primer on kidney Diseases; NEJM Group: Boston, MA, USA, 2009. [Google Scholar]
- Watanabe, H.; Miyamoto, Y.; Otagiri, M.; Maruyama, T. Update on the Pharmacokinetics and Redox Properties of Protein-Bound Uremic Toxins. J. Pharm. Sci. 2011, 100, 3682–3695. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, E.S.; Tevlin, M.T.; Wang, Y.B.; Chiang, T.M.; Cardenas, R.; Myers, L.K.; Acchiardo, S.R. Hemodialysis hypotension: Interaction of inhibitors, iNOS, and the interdialytic period. Am. J. Med. Sci. 1999, 317, 9–21. [Google Scholar] [CrossRef]
- Leone, A.; Moncada, S.; Vallance, P.; Calver, A.; Collier, J. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 1992, 339, 572–575. [Google Scholar] [CrossRef]
- De Deyn, P.; Marescau, B.; Lornoy, W.; Becaus, I.; Lowenthal, A. Guanidino compounds in uraemic dialysed patients. Clin. Chim. Acta 1986, 157, 143–150. [Google Scholar] [CrossRef]
- De Deyn, P.P.; Macdonald, R.L. Guanidino compounds that are increased in cerebrospinal fluid and brain of uremic patients inhibit GABA and glycine responses on mouse neurons in cell culture. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1990, 28, 627–633. [Google Scholar] [CrossRef] [Green Version]
- Wengle, B.; Hellström, K. Volatile phenols in serum of uraemic patients. Clin. Sci. 1972, 43, 493–498. [Google Scholar] [CrossRef]
- De Deyn, P.P.; Robitaille, P.; Vanasse, M.; Qureshi, I.A.; Marescau, B. Serum guanidino compound levels in uremic pediatric patients treated with hemodialysis or continuous cycle peritoneal dialysis. Nephron 1995, 69, 411–417. [Google Scholar] [CrossRef]
- Tanaka, A.; Takaihashi, Y.; Mizokuchi, M.; Shimada, N.; Koide, H. Plasma, urinary, and erythrocyte concentrations of guanidino compounds in patients with chronic renal failure. Ren. Fail. 1999, 21, 499–514. [Google Scholar] [CrossRef]
- Marescau, B.; Nagels, G.; Possemiers, I.; De Broe, M.E.; Becaus, I.; Billiouw, J.-M.; Lornoy, W.; De Deyn, P.P. Guanidino compounds in serum and urine of nondialyzed patients with chronic renal insufficiency. Metabolism 1997, 46, 1024–1031. [Google Scholar] [CrossRef]
- Faria, M.; de Pinho, M.N. Challenges of reducing protein-bound uremic toxin levels in chronic kidney disease and end stage renal disease. Transl. Res. 2021, 229, 115–134. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Ise, M. Indoxyl sulfate, a circulating uremic toxin, stimulates the progression of glomerular sclerosis. J. Lab. Clin. Med. 1994, 124, 96–104. [Google Scholar] [PubMed]
- Farrell, P.C.; Gotch, F.A.; Peters, J.H.; Berridge jr, B.J.; Lam, M. Binding of hippurate in normal plasma and in uremic plasma pre-and postdialysis. Nephron 1978, 20, 40–46. [Google Scholar] [CrossRef]
- Ludwig, G.D.; Senesky, D.; Bluemle, L.W., Jr.; Elkinton, J.R. Indoles in uremia: Identification by countercurrent distribution and paper chromatography. Am. J. Clin. Nutr. 1968, 21, 436–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iitaka, M.; Kawasaki, S.; Sakurai, S.; Hara, Y.; Kuriyama, R.; Yamanaka, K.; Kitahama, S.; Miura, S.; Kawakami, Y.; Katayama, S. Serum substances that interfere with thyroid hormone assays in patients with chronic renal failure. Clin. Endocrinol. 1998, 48, 739–746. [Google Scholar] [CrossRef]
- Hida, M.; Aiba, Y.; Sawamura, S.; Suzuki, N.; Satoh, T.; Koga, Y. Inhibition of the accumulation of uremic toxins in the blood and their precursors in the feces after oral administration of Lebenin®, a lactic acid bacteria preparation, to uremic patients undergoing hemodialysis. Nephron 1996, 74, 349–355. [Google Scholar] [CrossRef]
- Aguilera, A.; Codoceo, R.; Selgas, R.; Garcia, P.; Picornell, M.; Diaz, C.; Sanchez, C.; Bajo, M.-A. Anorexigen (TNF-alpha, cholecystokinin) and orexigen (neuropeptide Y) plasma levels in peritoneal dialysis (PD) patients: Their relationship with nutritional parameters. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Ren. Assoc. 1998, 13, 1476–1483. [Google Scholar]
- Tsukamoto, Y.; Hanaoka, M.; Matsuo, T.; Saruta, T.; Nomura, M.; Takahashi, Y. Effect of 22-oxacalcitriol on bone histology of hemodialyzed patients with severe secondary hyperparathyroidism. Am. J. kidney Dis. 2000, 35, 458–464. [Google Scholar] [CrossRef]
- Kabanda, A.; Jadoul, M.; Pochet, J.M.; Lauwerys, R.; de Strihou, C.V.; Bernard, A. Determinants of the serum concentrations of low molecular weight proteins in patients on maintenance hemodialysis. Kidney Int. 1994, 45, 1689–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Ascenzi, P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005, 57, 787–796. [Google Scholar] [CrossRef]
- Murray, C.W.; Hartshorn, M.J. 4.31—New Applications for Structure-Based Drug Design; Taylor, J.B., Triggle, D.J., Eds.; Elsevier: Oxford, UK, 2007; pp. 775–806. ISBN 978-0-08-045044-5. [Google Scholar]
- Meijers, B.K.I.; Bammens, B.; De Moor, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. Free p-cresol is associated with cardiovascular disease in hemodialysis patients. Am. J. kidney Dis. 2008, 51, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Raff, A.C.; Meyer, T.W.; Hostetter, T.H. New insights into uremic toxicity. Curr. Opin. Nephrol. Hypertens. 2008, 17, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, S.; Asaba, H.; Takanaga, H.; Deguchi, T.; Hosoya, K.; Otagiri, M.; Terasaki, T. Role of blood–brain barrier organic anion transporter 3 (OAT3) in the efflux of indoxyl sulfate, a uremic toxin: Its involvement in neurotransmitter metabolite clearance from the brain. J. Neurochem. 2002, 83, 57–66. [Google Scholar] [CrossRef]
- Nomura, S.; Sasaki, T.; Kitano, Y.; Osawa, G.; Niederstadt, C.; Lerche, L.; Steinhoff, J. Hypothesis: Is accumulation of a furan dicarboxylic acid (3-carboxy-4-methyl-5-propyl-2-furanpropanoic acid) related to the neurological abnormalities in patients with renal failure? Nephron 1996, 73, 169–173. [Google Scholar]
- Adijiang, A.; Goto, S.; Uramoto, S.; Nishijima, F.; Niwa, T. Indoxyl sulphate promotes aortic calcification with expression of osteoblast-specific proteins in hypertensive rats. Nephrol. Dial. Transplant. 2008, 23, 1892–1901. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, Y.; Yamato, H.; Nii-Kono, T.; Fujieda, A.; Uchida, M.; Hosokawa, A.; Motojima, M.; Fukagawa, M. Administration of oral charcoal adsorbent (AST-120) suppresses low-turnover bone progression in uraemic rats. Nephrol. Dial. Transplant. 2006, 21, 2768–2774. [Google Scholar] [CrossRef] [Green Version]
- Nii-Kono, T.; Iwasaki, Y.; Uchida, M.; Fujieda, A.; Hosokawa, A.; Motojima, M.; Yamato, H.; Kurokawa, K.; Fukagawa, M. Indoxyl sulfate induces skeletal resistance to parathyroid hormone in cultured osteoblastic cells. Kidney Int. 2007, 71, 738–743. [Google Scholar] [CrossRef] [Green Version]
- Schepers, E.; Glorieux, G.; Dou, L.; Cerini, C.; Gayrard, N.; Louvet, L.; Maugard, C.; Preus, P.; Rodriguez-Ortiz, M.; Argiles, A. Guanidino compounds as cause of cardiovascular damage in chronic kidney disease: An in vitro evaluation. Blood Purif. 2010, 30, 277–287. [Google Scholar] [CrossRef]
- Enomoto, A.; Endou, H. Roles of organic anion transporters (OATs) and a urate transporter (URAT1) in the pathophysiology of human disease. Clin. Exp. Nephrol. 2005, 9, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Niwa, T. Roles of organic anion transporters in the progression of chronic renal failure. Ther. Apher. Dial. 2007, 11, S27–S31. [Google Scholar] [CrossRef] [PubMed]
- Anzai, N.; Miyazaki, H.; Noshiro, R.; Khamdang, S.; Chairoungdua, A.; Shin, H.-J.; Enomoto, A.; Sakamoto, S.; Hirata, T.; Tomita, K. The multivalent PDZ domain-containing protein PDZK1 regulates transport activity of renal urate-anion exchanger URAT1 via its C terminus. J. Biol. Chem. 2004, 279, 45942–45950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, H.; Anzai, N.; Ekaratanawong, S.; Sakata, T.; Shin, H.J.; Jutabha, P.; Hirata, T.; He, X.; Nonoguchi, H.; Tomita, K. Modulation of renal apical organic anion transporter 4 function by two pdz domain–containing proteins. J. Am. Soc. Nephrol. 2005, 16, 3498–3506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.-H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppe, L.; Mafra, D.; Fouque, D. Probiotics and chronic kidney disease. Kidney Int. 2015, 88, 958–966. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.-C.; Tomino, Y.; Lu, K.-C. Impacts of indoxyl sulfate and p-cresol sulfate on chronic kidney disease and mitigating effects of AST-120. Toxins 2018, 10, 367. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Tsuruoka, S.; Ioka, T.; Ando, H.; Ito, C.; Akimoto, T.; Fujimura, A.; Asano, Y.; Kusano, E. Indoxyl sulfate stimulates proliferation of rat vascular smooth muscle cells. Kidney Int. 2006, 69, 1780–1785. [Google Scholar] [CrossRef] [Green Version]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef]
- Gelasco, A.K.; Raymond, J.R. Indoxyl sulfate induces complex redox alterations in mesangial cells. Am. J. Physiol. Physiol. 2006, 290, F1551–F1558. [Google Scholar] [CrossRef] [Green Version]
- Shimoishi, K.; Anraku, M.; Kitamura, K.; Tasaki, Y.; Taguchi, K.; Hashimoto, M.; Fukunaga, E.; Maruyama, T.; Otagiri, M. An oral adsorbent, AST-120 protects against the progression of oxidative stress by reducing the accumulation of indoxyl sulfate in the systemic circulation in renal failure. Pharm. Res. 2007, 24, 1283–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owada, S.; Goto, S.; Bannai, K.; Hayashi, H.; Nishijima, F.; Niwa, T. Indoxyl sulfate reduces superoxide scavenging activity in the kidneys of normal and uremic rats. Am. J. Nephrol. 2008, 28, 446–454. [Google Scholar] [CrossRef]
- Taki, K.; Tsuruta, Y.; Niwa, T. Indoxyl sulfate and atherosclerotic risk factors in hemodialysis patients. Am. J. Nephrol. 2007, 27, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, T.; Ohtsuki, S.; Otagiri, M.; Takanaga, H.; Asaba, H.; Mori, S.; Terasaki, T. Major role of organic anion transporter 3 in the transport of indoxyl sulfate in the kidney. Kidney Int. 2002, 61, 1760–1768. [Google Scholar] [CrossRef] [Green Version]
- Deguchi, T.; Nakamura, M.; Tsutsumi, Y.; Suenaga, A.; Otagiri, M. Pharmacokinetics and tissue distribution of uraemic indoxyl sulphate in rats. Biopharm. Drug Dispos. 2003, 24, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Kusuhara, H.; Sugiyama, D.; Ito, K.; Ueda, S.; Endou, H.; Sugiyama, Y. Functional involvement of rat organic anion transporter 3 (rOat3; Slc22a8) in the renal uptake of organic anions. J. Pharmacol. Exp. Ther. 2002, 300, 746–753. [Google Scholar] [CrossRef] [Green Version]
- Nagata, Y.; Kusuhara, H.; Endou, H.; Sugiyama, Y. Expression and functional characterization of rat organic anion transporter 3 (rOat3) in the choroid plexus. Mol. Pharmacol. 2002, 61, 982–988. [Google Scholar] [CrossRef] [Green Version]
- Kusuhara, H.; Sekine, T.; Utsunomiya-Tate, N.; Tsuda, M.; Kojima, R.; Cha, S.H.; Sugiyama, Y.; Kanai, Y.; Endou, H. Molecular cloning and characterization of a new multispecific organic anion transporter from rat brain. J. Biol. Chem. 1999, 274, 13675–13680. [Google Scholar] [CrossRef] [Green Version]
- Enomoto, A.; Takeda, M.; Tojo, A.; Sekine, T.; Cha, S.H.; Khamdang, S.; Takayama, F.; Aoyama, I.; Nakamura, S.; Endou, H.; et al. Role of organic anion transporters in the tubular transport of indoxyl sulfate and the induction of its nephrotoxicity. J. Am. Soc. Nephrol. 2002, 13, 1711–1720. [Google Scholar] [CrossRef] [Green Version]
- Deguchi, T.; Kusuhara, H.; Takadate, A.; Endou, H.; Otagiri, M.; Sugiyama, Y. Characterization of uremic toxin transport by organic anion transporters in the kidney. Kidney Int. 2004, 65, 162–174. [Google Scholar] [CrossRef] [Green Version]
- Enomoto, A.; Takeda, M.; Taki, K.; Takayama, F.; Noshiro, R.; Niwa, T.; Endou, H. Interactions of human organic anion as well as cation transporters with indoxyl sulfate. Eur. J. Pharmacol. 2003, 466, 13–20. [Google Scholar] [CrossRef]
- Müting, D. Studies on the pathogenesis of uremia comparatie determinaions of glucuronic acid, indican, free and bound phenols in the serum, cerebrospinal fluid, and urine of renal diseases with and without uremia. Clin. Chim. Acta 1965, 12, 551–554. [Google Scholar] [CrossRef]
- Spiteller, M.; Spiteller, G. Separation and characterization of acidic urine constituents (author’s transl). J. Chromatogr. 1979, 164, 253–317. [Google Scholar] [CrossRef]
- Niwa, T. Organic acids and the uremic syndrome: Protein metabolite hypothesis in the progression of chronic renal failure. Seminars in Nephrology; NIH Natinal Library of Medicine: Bethesda, MD, USA, 1996; Volume 16, pp. 167–182.
- Tsutsumi, Y.; Deguchi, T.; Takano, M.; Takadate, A.; Lindup, W.E.; Otagiri, M. Renal disposition of a furan dicarboxylic acid and other uremic toxins in the rat. J. Pharmacol. Exp. Ther. 2002, 303, 880–887. [Google Scholar] [CrossRef] [Green Version]
- Deguchi, T.; Kouno, Y.; Terasaki, T.; Takadate, A.; Otagiri, M. Differential contributions of rOat1 (Slc22a6) and rOat3 (Slc22a8) to the in vivo renal uptake of uremic toxins in rats. Pharm. Res. 2005, 22, 619–627. [Google Scholar] [CrossRef]
- Deguchi, T.; Isozaki, K.; Yousuke, K.; Terasaki, T.; Otagiri, M. Involvement of organic anion transporters in the efflux of uremic toxins across the blood–brain barrier. J. Neurochem. 2006, 96, 1051–1059. [Google Scholar] [CrossRef]
- Martinez, A.W.; Recht, N.S.; Hostetter, T.H.; Meyer, T.W. Removal of P-cresol sulfate by hemodialysis. J. Am. Soc. Nephrol. 2005, 16, 3430–3436. [Google Scholar] [CrossRef] [Green Version]
- Bammens, B.; Evenepoel, P.; Keuleers, H.; Verbeke, K.; Vanrenterghem, Y. Free serum concentrations of the protein-bound retention solute p-cresol predict mortality in hemodialysis patients. Kidney Int. 2006, 69, 1081–1087. [Google Scholar] [CrossRef]
- Schepers, E.; Meert, N.; Glorieux, G.; Goeman, J.; Van der Eycken, J.; Vanholder, R. P-cresylsulphate, the main in vivo metabolite of p-cresol, activates leucocyte free radical production. Nephrol. Dial. Transplant. 2007, 22, 592–596. [Google Scholar] [CrossRef]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef]
- de Loor, H.; Bammens, B.; Evenepoel, P.; De Preter, V.; Verbeke, K. Gas chromatographic–mass spectrometric analysis for measurement of p-cresol and its conjugated metabolites in uremic and normal serum. Clin. Chem. 2005, 51, 1535–1538. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, T.; Takemoto, M.; Uehara, N.; Lindup, W.E.; Suenaga, A.; Otagiri, M. Renal clearance of endogenous hippurate correlates with expression levels of renal organic anion transporters in uremic rats. J. Pharmacol. Exp. Ther. 2005, 314, 932–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schools, A.C.; De Vries, P.; Thiemann, R.; Hazejager, W.A.; Visser, S.L.; Oe, P.L. Biochemical and neurophysiological parameters in hemodialyzed patients with chronic renal failure. Clin. Chim. Acta 1989, 185, 91–107. [Google Scholar] [CrossRef]
- De Deyn, P.P.; Marescau, B.; Cuykens, J.J.; Van Gorp, L.; Lowenthal, A.; De Potter, W.P. Guanidino compounds in serum and cerebrospinal fluid of non-dialyzed patients with renal insufficiency. Clin. Chim. Acta 1987, 167, 81–88. [Google Scholar] [CrossRef]
- De Deyn, P.P.; Vanholder, R.; Eloot, S.; Glorieux, G. Progress in uremic toxin research: Guanidino compounds as uremic (neuro) toxins. In Seminars in Dialysis; Blackwell Publishing Ltd.: Oxford, UK, 2009; Volume 22, pp. 340–345. [Google Scholar]
- Urakami, Y.; Kimura, N.; Okuda, M.; Inui, K. Creatinine transport by basolateral organic cation transporter hOCT2 in the human kidney. Pharm. Res. 2004, 21, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Masuda, S.; Tanihara, Y.; Ueo, H.; Okuda, M.; Katsura, T.; Inui, K. Metformin is a superior substrate for renal organic cation transporter OCT2 rather than hepatic OCT1. Drug Metab. Pharmacokinet. 2005, 20, 379–386. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Toyohara, T.; Akiyama, Y.; Takeuchi, Y.; Mishima, E.; Suzuki, C.; Ito, S.; Soga, T.; Abe, T. Transcriptional Regulation of Organic Anion Transporting Polypeptide SLCO4C1 as a New Therapeutic Modality to Prevent Chronic Kidney Disease. J. Pharm. Sci. 2011, 100, 3696–3707. [Google Scholar] [CrossRef]
- Mabuchi, H.; Nakahashi, H. Isolation and characterization of an endogenous drug-binding inhibitor present in uremic serum. Nephron 1986, 44, 277–281. [Google Scholar] [CrossRef]
- Mabuchi, H.; Nakahashi, H. Profiling of endogenous ligand solutes that bind to serum proteins in sera of patients with uremia. Nephron 1986, 43, 110–116. [Google Scholar] [CrossRef]
- Niwa, T.; Takeda, N.; Maeda, K.; Shibata, M.; Tatematsu, A. Accumulation of furancarboxylic acids in uremic serum as inhibitors of drug binding. Clin. Chim. Acta 1988, 173, 127–138. [Google Scholar] [CrossRef]
- SAKAI, T.; Takadate, A.; Otagiri, M. Characterization of binding site of uremic toxins on human serum albumin. Biol. Pharm. Bull. 1995, 18, 1755–1761. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Schuchardt, M.; Tölle, M.; van der Giet, M.; Zidek, W.; Dzubiella, J.; Ballauff, M. Interaction of human serum albumin with uremic toxins: A thermodynamic study. RSC Adv. 2017, 7, 27913–27922. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, S.; Ameen, F.; ur Rehman, S.; Sarwar, T.; Tabish, M. Studying the interaction of drug/ligand with serum albumin. J. Mol. Liq. 2021, 336, 116200. [Google Scholar] [CrossRef]
- Ishtikhar, M.; Rabbani, G.; Khan, R.H. Interaction of 5-fluoro-5′-deoxyuridine with human serum albumin under physiological and non-physiological condition: A biophysical investigation. Colloids Surf. B Biointerfaces 2014, 123, 469–477. [Google Scholar] [CrossRef]
- Paul, B.K.; Ghosh, N.; Mukherjee, S. Interplay of multiple interaction forces: Binding of norfloxacin to human serum albumin. J. Phys. Chem. B 2015, 119, 13093–13102. [Google Scholar] [CrossRef]
- Tesseromatis, C.; Alevizou, A. The role of the protein-binding on the mode of drug action as well the interactions with other drugs. Eur. J. Drug Metab. Pharmacokinet. 2008, 33, 225–230. [Google Scholar] [CrossRef]
- Tsutsumi, Y.; Maruyama, T.; Takadate, A.; Shimada, H.; Otagiri, M. Decreased bilirubin-binding capacity in uremic serum caused by an accumulation of furan dicarboxylic acid. Nephron 2000, 85, 60–64. [Google Scholar] [CrossRef]
- Tsutsumi, Y.; Maruyama, T.; Takadate, A.; Goto, M.; Matsunaga, H.; Otagiri, M. Interaction between two dicarboxylate endogenous substances, bilirubin and an uremic toxin, 3-carboxy-4-methyl-5-propyl-2-furanpropanoic acid, on human serum albumin. Pharm. Res. 1999, 16, 916–923. [Google Scholar] [CrossRef]
- Meijers, B.K.I.; De Loor, H.; Bammens, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. p-Cresyl sulfate and indoxyl sulfate in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1932–1938. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-J.; Wu, C.-J.; Pan, C.-F.; Chen, Y.-C.; Sun, F.-J.; Chen, H.-H. Serum protein-bound uraemic toxins and clinical outcomes in haemodialysis patients. Nephrol. Dial. Transplant. 2010, 25, 3693–3700. [Google Scholar] [CrossRef] [Green Version]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; (EUTox, E.U.T.W.G. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [Green Version]
- Jourde-Chiche, N.; Dou, L.; Cerini, C.; Dignat-George, F.; Vanholder, R.; Brunet, P. PROGRESS IN UREMIC TOXIN RESEARCH: Protein-Bound Toxins—Update 2009. In Seminars in Dialysis; Blackwell Publishing Ltd.: Oxford, UK, 2009; Volume 22, pp. 334–339. [Google Scholar]
- Liabeuf, S.; Barreto, D.V.; Barreto, F.C.; Meert, N.; Glorieux, G.; Schepers, E.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol. Dial. Transplant. 2010, 25, 1183–1191. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Temple, R.; Xiao, S.; Zhang, L.; Lesko, L.J. When to conduct a renal impairment study during drug development: US Food and Drug Administration perspective. Clin. Pharmacol. Ther. 2009, 86, 475–479. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, L.; Abraham, S.; Apparaju, S.; Wu, T.; Strong, J.M.; Xiao, S.; Atkinson Jr, A.J.; Thummel, K.E.; Leeder, J.S. Assessment of the impact of renal impairment on systemic exposure of new molecular entities: Evaluation of recent new drug applications. Clin. Pharmacol. Ther. 2009, 85, 305–311. [Google Scholar] [CrossRef]
- Maheshwari, V.; Tao, X.; Thijssen, S.; Kotanko, P. Removal of Protein-Bound Uremic Toxins Using Binding Competitors in Hemodialysis: A Narrative Review. Toxins 2021, 13, 622. [Google Scholar] [CrossRef]
- Tao, X.; Thijssen, S.; Kotanko, P.; Ho, C.-H.; Henrie, M.; Stroup, E.; Handelman, G. Improved dialytic removal of protein-bound uraemic toxins with use of albumin binding competitors: An in vitro human whole blood study. Sci. Rep. 2016, 6, 23389. [Google Scholar] [CrossRef] [Green Version]
- Madero, M.; Cano, K.B.; Campos, I.; Tao, X.; Maheshwari, V.; Brown, J.; Cornejo, B.; Handelman, G.; Thijssen, S.; Kotanko, P. Removal of protein-bound uremic toxins during hemodialysis using a binding competitor. Clin. J. Am. Soc. Nephrol. 2019, 14, 394–402. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.; Xu, X.; Cao, W.; Shen, Z.; Wang, N.; Leng, J.; Zou, N.; Shang, E.; Zhu, Z. Improved dialysis removal of protein-bound uremic toxins by salvianolic acids. Phytomedicine 2019, 57, 166–173. [Google Scholar] [CrossRef]
- Van Biesen, W.; Eloot, S. Enhanced Removal of Protein-Bound Uremic Toxins Using Displacers: Road to Success? Clin. J. Am. Soc. Nephrol. 2019, 14, 324–326. [Google Scholar] [CrossRef] [Green Version]
- Pham, P.C.; Khaing, K.; Sievers, T.M.; Pham, P.M.; Miller, J.M.; Pham, S.V.; Pham, P.A.; Pham, P.T. 2017 update on pain management in patients with chronic kidney disease. Clin. Kidney J. 2017, 10, 688–697. [Google Scholar] [CrossRef]
- Faria, M.; Moreira, C.; Eusébio, T.; Brogueira, P.; de Pinho, M.N. Hybrid flat sheet cellulose acetate/silicon dioxide ultrafiltration membranes for uremic blood purification. Cellulose 2020, 27, 3847–3869. [Google Scholar] [CrossRef]
- Janeca, A.; Rodrigues, F.S.C.; Gonçalves, M.C.; Faria, M. Novel Cellulose Acetate-Based Monophasic Hybrid Membranes for Improved Blood Purification Devices: Characterization under Dynamic Conditions. Membranes 2021, 11, 825. [Google Scholar] [CrossRef]
- Andrade, M.C.; Pereira, J.C.; de Almeida, N.; Marques, P.; Faria, M.; Gonçalves, M.C. Improving hydraulic permeability, mechanical properties, and chemical functionality of cellulose acetate-based membranes by co-polymerization with tetraethyl orthosilicate and 3-(aminopropyl) triethoxysilane. Carbohydr. Polym. 2021, 261, 117813. [Google Scholar] [CrossRef]
- Mendes, G.; Faria, M.; Carvalho, A.; Gonçalves, M.C.; de Pinho, M.N. Structure of water in hybrid cellulose acetate-silica ultrafiltration membranes and permeation properties. Carbohydr. Polym. 2018, 189, 342–351. [Google Scholar] [CrossRef]
- De Pascale, M.; Faria, M.; Boi, C.; Semiao, V.; de Pinho, M.N.; Pekguleryuz, M.O. The effect of ultrafiltration transmembrane permeation on the flow field in a surrogate system of an artificial kidney. Exp. Results 2021, 2, E16. [Google Scholar] [CrossRef]
- Pavlenko, D.; Van Geffen, E.; Van Steenbergen, M.J.; Glorieux, G.; Vanholder, R.; Gerritsen, K.G.F.; Stamatialis, D. New low-flux mixed matrix membranes that offer superior removal of protein-bound toxins from human plasma. Sci. Rep. 2016, 6, 34429. [Google Scholar] [CrossRef]
- Tijink, M.S.L.; Wester, M.; Sun, J.; Saris, A.; Bolhuis-Versteeg, L.A.M.; Saiful, S.; Joles, J.A.; Borneman, Z.; Wessling, M.; Stamatialis, D.F. A novel approach for blood purification: Mixed-matrix membranes combining diffusion and adsorption in one step. Acta Biomater. 2012, 8, 2279–2287. [Google Scholar] [CrossRef]
- Kim, D.; Stamatialis, D. High flux mixed matrix membrane with low albumin leakage for blood plasma detoxification. J. Memb. Sci. 2020, 609, 118187. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MW (Da) | Average CU (mg/L) | Average CN (mg/L) | Maximum CU (CMAX) (mg/L) | Group | References | |
|---|---|---|---|---|---|---|
| ADMA mg/L | 202 | 1.6 ± 1.2/10 | 0.2 ± 0.06/6 | 7.3 | Guanidines | [7,8,9] |
| Creatine mg/L | 131 | 134.0 ± 30.3/29 | 9.7 ± 3.3/24 | 235.8 | Guanidines | [7,10] |
| Creatinine mg/L | 113 | 136.0 ± 46.0/19746 | <12.0/23 | 240.0 | Guanidines | [7,11,12] |
| Guanidine µg/L | 59 | 172.9 ± 83.8/13 | <11.8/16 | 800.0 | Guanidines | [7,13,14] |
| SDMA µg/L | 202 | 640.3 ± 212.1/38 | 76.1 ± 21.0/66 | 1232.2 | Guanidines | [7,15] |
| Urea g/L | 60 | 2.3 ± 1.1/16 | <0.4/23 | 4.6 | [7,12] |
| MW (Da) | Average CU (mg/L) | Average CN (mg/L) | Maximum CU (CMAX) (mg/L) | Group | References | |
|---|---|---|---|---|---|---|
| CMPF mg/L | 240 | 61.0 ± 16.5/15 | 7.7 ± 3.3/7 | 94.0 | [7,16,17] | |
| HA mg/L | 179 | 247.0 ± 112.0/7 | <5.0 | 471.0 | Hippurates | [7,16,18] |
| IA µg/L | 175 | 875.0 ± 560.0/42 | 17.5 ± 17.5/7 | 9076.9 | Indoles | [7,16,19,20] |
| IS mg/L | 251 | 53.0 ± 91.5/20 | 0.6 ± 5.4/40 | 236.0 | Indoles | [7,17] |
| PCS mg/L | 108 | 20.1 ± 10.3/20 | 0.6 ± 1.0/12 | 40.7 | Phenols | [7,16,21] |
| MW (Da) | Average CU (mg/L) | Average CN (mg/L) | Maximum CU (CMAX) (mg/L) | Group | References | |
|---|---|---|---|---|---|---|
| Leptin µg/L | 16,000 | 72.0 ± 60.6/8 | 8.4 ± 6.7/56 | 490.0 | Peptides | [7] |
| Neuropeptide Y ng/L | 4272 | 64.9 ± 25.5/19 | <80.0 | 115.9 | Peptides | [7,22] |
| Parathyroid hormone µg/L | 9225 | 1.2 ± 0.6/10 | <0.06 | 2.4 | Peptides | [7,23] |
| Retinol-binding protein mg/L | 21,200 | 192.0 ± 78.0/112 | <80.0 | 369.2 | Peptides | [7,24] |
| Anionic UTS | Intracellular UPTAKE by | Albumin Binding | Ease Removal of HD | ROS Production | Effect on Nonrenal Clearance |
|---|---|---|---|---|---|
| IS | OAT1, OAT3 | High affinity (site II) | Partially | Yes | Inhibition of rCYP3A, hCYP2C9, hCYP1A, hCYP3A, AHR ligand |
| CMPF | OAT1, OAT3 | High affinity (site I) | Difficult | Yes? | Inhibition of rOatp2, rOatp1a4 |
| PCS | OAT1, OAT3 | High affinity (site II) | Partially | Yes | - |
| HA | OAT1, OAT3 | High affinity (site II) | Partially | unknown | - |
| IA | OAT1, OAT3 | High affinity (site II) | Partially | Yes | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zare, F.; Janeca, A.; Jokar, S.M.; Faria, M.; Gonçalves, M.C. Interaction of Human Serum Albumin with Uremic Toxins: The Need of New Strategies Aiming at Uremic Toxins Removal. Membranes 2022, 12, 261. https://doi.org/10.3390/membranes12030261
Zare F, Janeca A, Jokar SM, Faria M, Gonçalves MC. Interaction of Human Serum Albumin with Uremic Toxins: The Need of New Strategies Aiming at Uremic Toxins Removal. Membranes. 2022; 12(3):261. https://doi.org/10.3390/membranes12030261
Chicago/Turabian StyleZare, Fahimeh, Adriana Janeca, Seyyed M. Jokar, Mónica Faria, and Maria Clara Gonçalves. 2022. "Interaction of Human Serum Albumin with Uremic Toxins: The Need of New Strategies Aiming at Uremic Toxins Removal" Membranes 12, no. 3: 261. https://doi.org/10.3390/membranes12030261